
Multiomic Investigations into Lung Health and Disease


Abstract
:1. Introduction
2. Insights into Cell Biology Using Multi-Omics
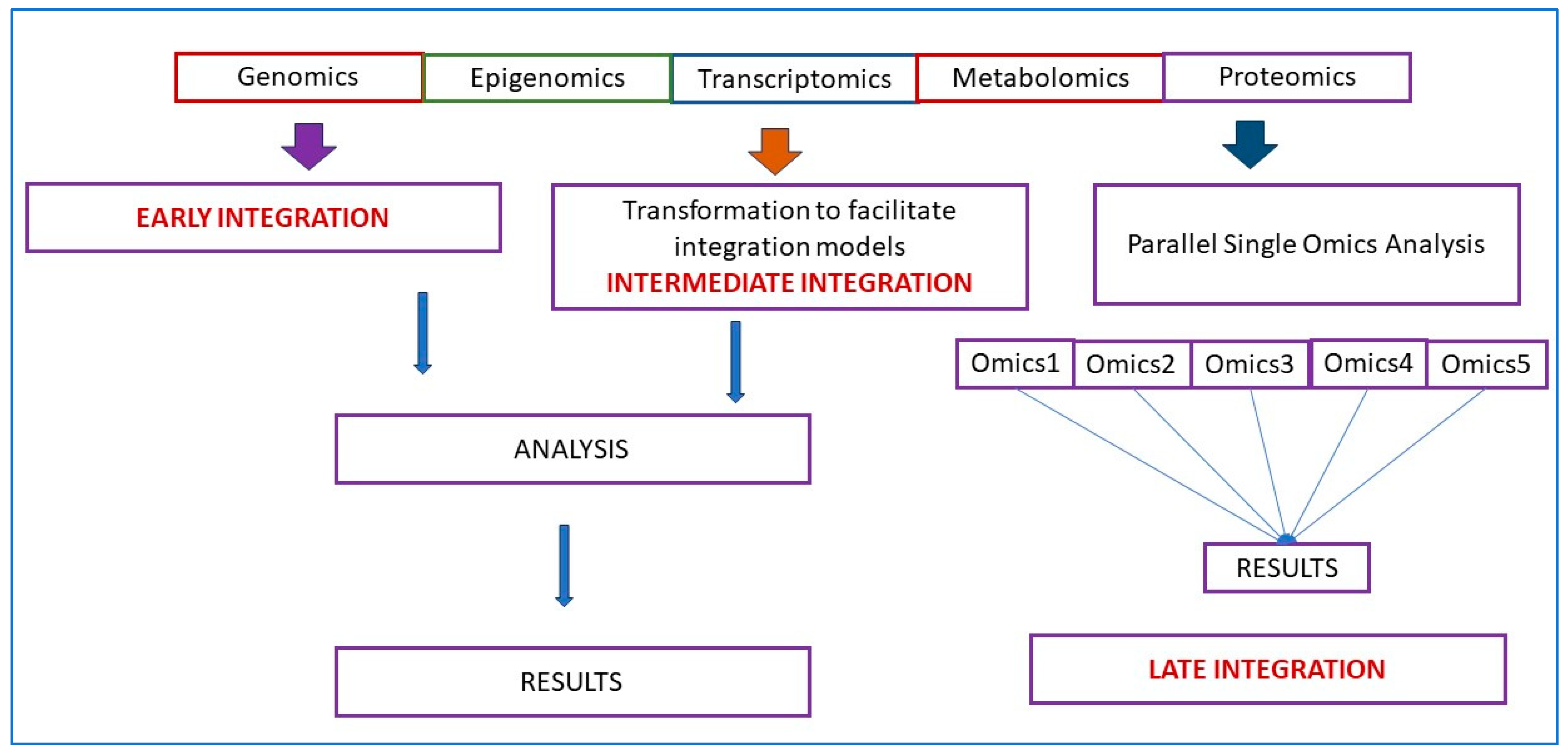
3. Integration of Multiomics Data
4. Lung Multiomics Models
5. Multiomics Insight into Clinical Disease
5.1. Cystic Fibrosis
5.2. Chronic Obstructive Pulmonary Disease (COPD)
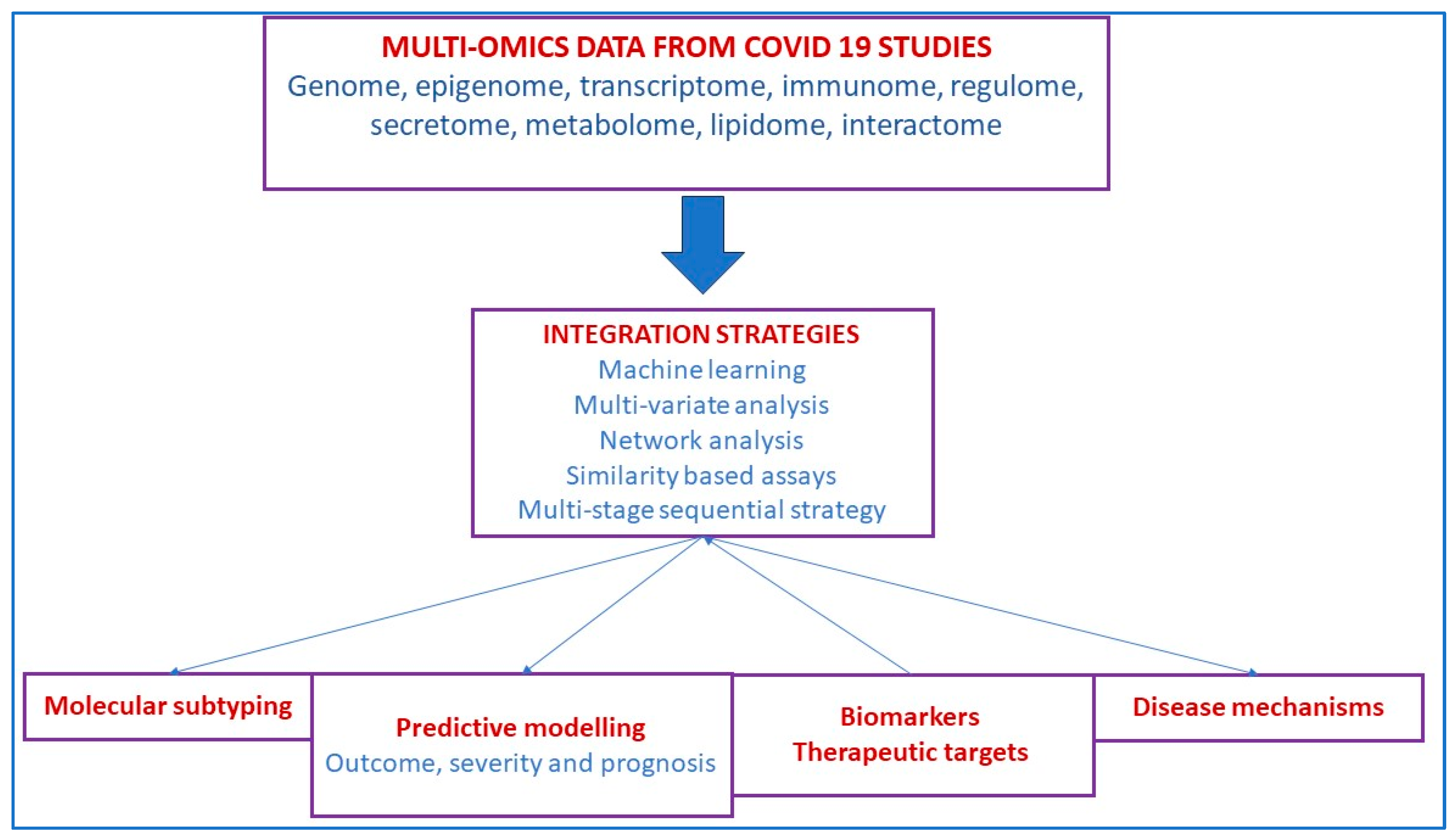
5.3. SARS-CoV-2 Infection
5.4. Lung Cancer and Lung Metastases
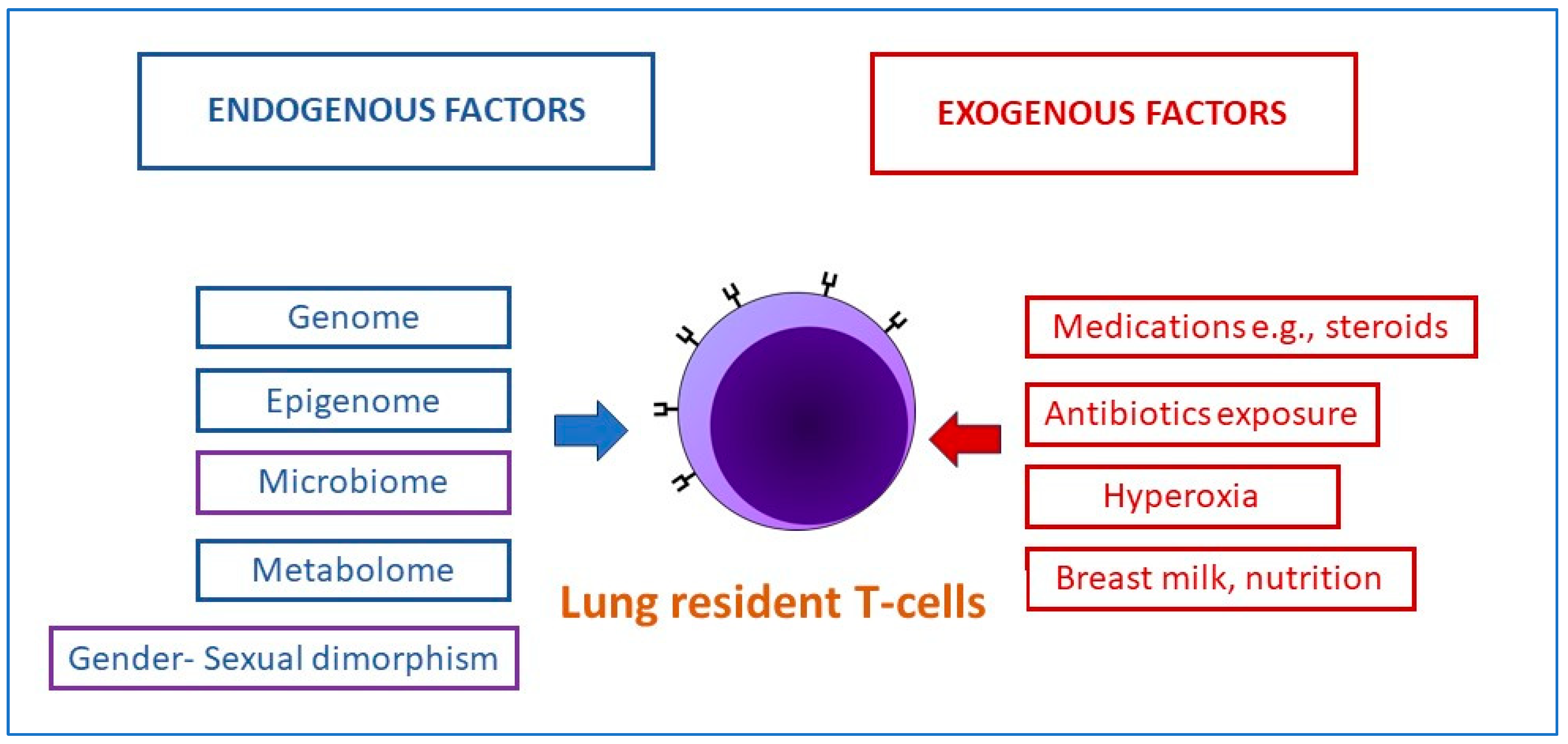
5.5. Bronchopulmonary Dysplasia in Preterm Infants
5.6. Pulmonary Hypertension
6. Societal and Ethical Issues Related to the Use of Multiomics and Machine Learning in Healthcare [150]
7. Summary
Funding
Data Availability Statement
Conflicts of Interest
References
- Humbert, M.V.; Spalluto, C.M.; Bell, J.; Blume, C.; Conforti, F.; Davies, E.R.; Dean, L.S.N.; Elkington, P.; Haitchi, H.M.; Jackson, C.; et al. Towards an artificial human lung: Modelling organ-like complexity to aid mechanistic understanding. Eur. Respir. J. 2022, 60, 2200455. [Google Scholar] [CrossRef]
- GBD Chronic Respiratory Disease Collaborators. Prevalence and attributable health burden of chronic respiratory diseases, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet Respir. Med. 2020, 8, 585–596. [Google Scholar] [CrossRef]
- Atzrodt, C.L.; Maknojia, I.; McCarthy, R.D.P.; Oldfield, T.M.; Po, J.; Ta, K.T.L.; Stepp, H.E.; Clements, T.P. A Guide to COVID-19: A global pandemic caused by the novel coronavirus SARS-CoV-2. FEBS J. 2020, 287, 3633–3650. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.J.; Einarsson, G.G.; Gilpin, D.F.; Tunney, M.M. Multi-Omics Approaches: The Key to Improving Respiratory Health in People with Cystic Fibrosis? Front. Pharmacol. 2020, 11, 569821. [Google Scholar] [CrossRef] [PubMed]
- MacEachern, S.J.; Forkert, N.D. Machine learning for precision medicine. Genome 2021, 64, 416–425. [Google Scholar] [CrossRef]
- Mathema, V.B.; Sen, P.; Lamichhane, S.; Orešič, M.; Khoomrung, S. Deep learning facilitates multi-data type analysis and predictive biomarker discovery in cancer precision medicine. Comput. Struct. Biotechnol. J. 2023, 21, 1372–1382. [Google Scholar] [CrossRef]
- Bellman, R. Dynamic programming. Science 1966, 153, 34–37. [Google Scholar] [CrossRef]
- Kaiser, J. NIH’s ‘precision nutrition’ bet aims for individualized diets. Science 2021, 371, 552. [Google Scholar] [CrossRef]
- Tanaka, I.; Furukawa, T.; Morise, M. The current issues and future perspective of artificial intelligence for developing new treatment strategy in non-small cell lung cancer: Harmonization of molecular cancer biology and artificial intelligence. Cancer Cell Int. 2021, 21, 454. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, Z.P. Robust biomarker discovery for hepatocellular carcinoma from high-throughput data by multiple feature selection methods. BMC Med. Genom. 2021, 14, 112. [Google Scholar] [CrossRef]
- Gevaert, O.; De Smet, F.; Timmerman, D.; Moreau, Y.; De Moor, B. Predicting the prognosis of breast cancer by integrating clinical and microarray data with Bayesian networks. Bioinformatics 2006, 22, e184–e190. [Google Scholar] [CrossRef]
- Zhao, M.; Tang, Y.; Kim, H.; Hasegawa, K. Machine Learning with K-Means Dimensional Reduction for Predicting Survival Outcomes in Patients with Breast Cancer. Cancer Inf. 2018, 17, 1176935118810215. [Google Scholar] [CrossRef]
- Vantaku, V.; Dong, J.; Ambati, C.R.; Perera, D.; Donepudi, S.R.; Amara, C.S.; Putluri, V.; Ravi, S.S.; Robertson, M.J.; Piyarathna, D.W.B.; et al. Multi-omics Integration Analysis Robustly Predicts High-Grade Patient Survival and Identifies CPT1B Effect on Fatty Acid Metabolism in Bladder Cancer. Clin. Cancer Res. 2019, 25, 3689–3701. [Google Scholar] [CrossRef] [PubMed]
- Tong, D.; Tian, Y.; Zhou, T.; Ye, Q.; Li, J.; Ding, K.; Li, J. Improving prediction performance of colon cancer prognosis based on the integration of clinical and multi-omics data. BMC Med. Inf. Decis. Mak. 2020, 20, 22. [Google Scholar] [CrossRef] [PubMed]
- Tong, L.; Mitchel, J.; Chatlin, K.; Wang, M.D. Deep learning based feature-level integration of multi-omics data for breast cancer patients survival analysis. BMC Med. Inf. Decis. Mak. 2020, 20, 225. [Google Scholar] [CrossRef]
- Zhao, Y.J.; Wu, L.Y.; Pang, J.S.; Liao, W.; Chen, Y.J.; He, Y.; Yang, H. Integrated multi-omics analysis of the clinical relevance and potential regulatory mechanisms of splicing factors in hepatocellular carcinoma. Bioengineered 2021, 12, 3978–3992. [Google Scholar] [CrossRef]
- Zafari, N.; Bathaei, P.; Velayati, M.; Khojasteh-Leylakoohi, F.; Khazaei, M.; Fiuji, H.; Nassiri, M.; Hassanian, S.M.; Ferns, G.A.; Nazari, E.; et al. Integrated analysis of multi-omics data for the discovery of biomarkers and therapeutic targets for colorectal cancer. Comput. Biol. Med. 2023, 155, 106639. [Google Scholar] [CrossRef]
- Azimzadeh, O.; Sievert, W.; Sarioglu, H.; Merl-Pham, J.; Yentrapalli, R.; Bakshi, M.V.; Janik, D.; Ueffing, M.; Atkinson, M.J.; Multhoff, G.; et al. Integrative proteomics and targeted transcriptomics analyses in cardiac endothelial cells unravel mechanisms of long-term radiation-induced vascular dysfunction. J. Proteome Res. 2015, 14, 1203–1219. [Google Scholar] [CrossRef]
- Wang, D.D.; Hu, F.B. Precision nutrition for prevention and management of type 2 diabetes. Lancet Diabetes Endocrinol. 2018, 6, 416–426. [Google Scholar] [CrossRef]
- Wang, L.; Wang, X.; Chen, A.; Jin, X.; Che, H. Prediction of Type 2 Diabetes Risk and Its Effect Evaluation Based on the XGBoost Model. Healthcare 2020, 8, 247. [Google Scholar] [CrossRef]
- Wang, A.; Chiou, J.; Poirion, O.B.; Buchanan, J.; Valdez, M.J.; Verheyden, J.M.; Hou, X.; Kudtarkar, P.; Narendra, S.; Newsome, J.M.; et al. Single-cell multiomic profiling of human lungs reveals cell-type-specific and age-dynamic control of SARS-CoV2 host genes. eLife 2020, 9, e62522. [Google Scholar] [CrossRef]
- Pliner, H.A.; Packer, J.S.; McFaline-Figueroa, J.L.; Cusanovich, D.A.; Daza, R.M.; Aghamirzaie, D.; Srivatsan, S.; Qiu, X.; Jackson, D.; Minkina, A.; et al. Cicero Predicts cis-Regulatory DNA Interactions from Single-Cell Chromatin Accessibility Data. Mol. Cell 2018, 71, 858–871.e858. [Google Scholar] [CrossRef]
- He, P.; Lim, K.; Sun, D.; Pett, J.P.; Jeng, Q.; Polanski, K.; Dong, Z.; Bolt, L.; Richardson, L.; Mamanova, L.; et al. A human fetal lung cell atlas uncovers proximal-distal gradients of differentiation and key regulators of epithelial fates. Cell 2022, 185, 4841–4860.e4825. [Google Scholar] [CrossRef] [PubMed]
- Efremova, M.; Vento-Tormo, M.; Teichmann, S.A.; Vento-Tormo, R. CellPhoneDB: Inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat. Protoc. 2020, 15, 1484–1506. [Google Scholar] [CrossRef] [PubMed]
- Daly, J.L.; Danson, C.M.; Lewis, P.A.; Zhao, L.; Riccardo, S.; Di Filippo, L.; Cacchiarelli, D.; Lee, D.; Cross, S.J.; Heesom, K.J.; et al. Multi-omic approach characterises the neuroprotective role of retromer in regulating lysosomal health. Nat. Commun. 2023, 14, 3086. [Google Scholar] [CrossRef] [PubMed]
- Lareau, C.A.; Dubois, S.M.; Buquicchio, F.A.; Hsieh, Y.H.; Garg, K.; Kautz, P.; Nitsch, L.; Praktiknjo, S.D.; Maschmeyer, P.; Verboon, J.M.; et al. Single-cell multi-omics of mitochondrial DNA disorders reveals dynamics of purifying selection across human immune cells. Nat. Genet. 2023, 55, 1198–1209. [Google Scholar] [CrossRef]
- Amarasekera, S.S.C.; Hock, D.H.; Lake, N.J.; Calvo, S.E.; Grønborg, S.W.; Krzesinski, E.I.; Amor, D.J.; Fahey, M.C.; Simons, C.; Wibrand, F.; et al. Multi-omics identifies large mitoribosomal subunit instability caused by pathogenic MRPL39 variants as a cause of pediatric onset mitochondrial disease. Hum. Mol. Genet. 2023, 32, 2441–2454. [Google Scholar] [CrossRef]
- Mirza, B.; Wang, W.; Wang, J.; Choi, H.; Chung, N.C.; Ping, P. Machine Learning and Integrative Analysis of Biomedical Big Data. Genes 2019, 10, 87. [Google Scholar] [CrossRef] [PubMed]
- Reel, P.S.; Reel, S.; Pearson, E.; Trucco, E.; Jefferson, E. Using machine learning approaches for multi-omics data analysis: A review. Biotechnol. Adv. 2021, 49, 107739. [Google Scholar] [CrossRef] [PubMed]
- Picard, M.; Scott-Boyer, M.P.; Bodein, A.; Périn, O.; Droit, A. Integration strategies of multi-omics data for machine learning analysis. Comput. Struct. Biotechnol. J. 2021, 19, 3735–3746. [Google Scholar] [CrossRef] [PubMed]
- Hoadley, K.A.; Yau, C.; Wolf, D.M.; Cherniack, A.D.; Tamborero, D.; Ng, S.; Leiserson, M.D.M.; Niu, B.; McLellan, M.D.; Uzunangelov, V.; et al. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell 2014, 158, 929–944. [Google Scholar] [CrossRef] [PubMed]
- De Cecco, L.; Giannoccaro, M.; Marchesi, E.; Bossi, P.; Favales, F.; Locati, L.D.; Licitra, L.; Pilotti, S.; Canevari, S. Integrative miRNA-Gene Expression Analysis Enables Refinement of Associated Biology and Prediction of Response to Cetuximab in Head and Neck Squamous Cell Cancer. Genes 2017, 8, 35. [Google Scholar] [CrossRef] [PubMed]
- Zheng, P.Z.; Wang, K.K.; Zhang, Q.Y.; Huang, Q.H.; Du, Y.Z.; Zhang, Q.H.; Xiao, D.K.; Shen, S.H.; Imbeaud, S.; Eveno, E.; et al. Systems analysis of transcriptome and proteome in retinoic acid/arsenic trioxide-induced cell differentiation/apoptosis of promyelocytic leukemia. Proc. Natl. Acad. Sci. USA 2005, 102, 7653–7658. [Google Scholar] [CrossRef] [PubMed]
- Bahado-Singh, R.O.; Sonek, J.; McKenna, D.; Cool, D.; Aydas, B.; Turkoglu, O.; Bjorndahl, T.; Mandal, R.; Wishart, D.; Friedman, P.; et al. Artificial intelligence and amniotic fluid multiomics: Prediction of perinatal outcome in asymptomatic women with short cervix. Ultrasound Obs. Gynecol. 2019, 54, 110–118. [Google Scholar] [CrossRef]
- Lee, K.S.; Ahn, K.H. Artificial Neural Network Analysis of Spontaneous Preterm Labor and Birth and Its Major Determinants. J. Korean Med. Sci. 2019, 34, e128. [Google Scholar] [CrossRef]
- Cai, Z.; Poulos, R.C.; Liu, J.; Zhong, Q. Machine learning for multi-omics data integration in cancer. iScience 2022, 25, 103798. [Google Scholar] [CrossRef]
- Zitnik, M.; Nguyen, F.; Wang, B.; Leskovec, J.; Goldenberg, A.; Hoffman, M.M. Machine Learning for Integrating Data in Biology and Medicine: Principles, Practice, and Opportunities. Inf. Fusion 2019, 50, 71–91. [Google Scholar] [CrossRef] [PubMed]
- Argelaguet, R.; Velten, B.; Arnol, D.; Dietrich, S.; Zenz, T.; Marioni, J.C.; Buettner, F.; Huber, W.; Stegle, O. Multi-Omics Factor Analysis-a framework for unsupervised integration of multi-omics data sets. Mol. Syst. Biol. 2018, 14, e8124. [Google Scholar] [CrossRef]
- Cavill, R.; Jennen, D.; Kleinjans, J.; Briedé, J.J. Transcriptomic and metabolomic data integration. Brief. Bioinform. 2016, 17, 891–901. [Google Scholar] [CrossRef] [PubMed]
- Costello, J.C.; Heiser, L.M.; Georgii, E.; Gönen, M.; Menden, M.P.; Wang, N.J.; Bansal, M.; Ammad-ud-din, M.; Hintsanen, P.; Khan, S.A.; et al. A community effort to assess and improve drug sensitivity prediction algorithms. Nat. Biotechnol. 2014, 32, 1202–1212. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Mezlini, A.M.; Demir, F.; Fiume, M.; Tu, Z.; Brudno, M.; Haibe-Kains, B.; Goldenberg, A. Similarity network fusion for aggregating data types on a genomic scale. Nat. Methods 2014, 11, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Greenbaum, J.; Luttrell, J.T.; Zhou, W.; Wu, C.; Shen, H.; Gong, P.; Zhang, C.; Deng, H.W. A Review of Integrative Imputation for Multi-Omics Datasets. Front. Genet. 2020, 11, 570255. [Google Scholar] [CrossRef] [PubMed]
- Agamah, F.E.; Bayjanov, J.R.; Niehues, A.; Njoku, K.F.; Skelton, M.; Mazandu, G.K.; Ederveen, T.H.A.; Mulder, N.; Chimusa, E.R.; Hoen, P.A.C. Computational approaches for network-based integrative multi-omics analysis. Front. Mol. Biosci. 2022, 9, 967205. [Google Scholar] [CrossRef]
- Ardini-Poleske, M.E.; Clark, R.F.; Ansong, C.; Carson, J.P.; Corley, R.A.; Deutsch, G.H.; Hagood, J.S.; Kaminski, N.; Mariani, T.J.; Potter, S.S.; et al. LungMAP: The Molecular Atlas of Lung Development Program. Am. J. Physiol. Lung Cell Mol. Physiol. 2017, 313, L733–L740. [Google Scholar] [CrossRef]
- Luo, Y.; Hitz, B.C.; Gabdank, I.; Hilton, J.A.; Kagda, M.S.; Lam, B.; Myers, Z.; Sud, P.; Jou, J.; Lin, K.; et al. New developments on the Encyclopedia of DNA Elements (ENCODE) data portal. Nucleic Acids Res. 2020, 48, D882–D889. [Google Scholar] [CrossRef] [PubMed]
- Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; Ziller, M.J.; et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330. [Google Scholar] [CrossRef]
- Edwards, N.J.; Oberti, M.; Thangudu, R.R.; Cai, S.; McGarvey, P.B.; Jacob, S.; Madhavan, S.; Ketchum, K.A. The CPTAC Data Portal: A Resource for Cancer Proteomics Research. J. Proteome Res. 2015, 14, 2707–2713. [Google Scholar] [CrossRef]
- Tarazona, S.; Arzalluz-Luque, A.; Conesa, A. Undisclosed, unmet and neglected challenges in multi-omics studies. Nat. Comput. Sci. 2021, 1, 395–402. [Google Scholar] [CrossRef]
- Gruenert, D.C.; Willems, M.; Cassiman, J.J.; Frizzell, R.A. Established cell lines used in cystic fibrosis research. J. Cyst. Fibros. 2004, 3 (Suppl. S2), 191–196. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Birch, N.P.; Suresh, V. An Optimised Human Cell Culture Model for Alveolar Epithelial Transport. PLoS ONE 2016, 11, e0165225. [Google Scholar] [CrossRef]
- Hermanns, M.I.; Unger, R.E.; Kehe, K.; Peters, K.; Kirkpatrick, C.J. Lung epithelial cell lines in coculture with human pulmonary microvascular endothelial cells: Development of an alveolo-capillary barrier in vitro. Lab. Investig. 2004, 84, 736–752. [Google Scholar] [CrossRef] [PubMed]
- Kiełbus, M.; Czapiński, J.; Kałafut, J.; Woś, J.; Stepulak, A.; Rivero-Müller, A. Genetically Engineered Lung Cancer Cells for Analyzing Epithelial-Mesenchymal Transition. Cells 2019, 8, 1644. [Google Scholar] [CrossRef]
- Kallunki, T.; Barisic, M.; Jäättelä, M.; Liu, B. How to Choose the Right Inducible Gene Expression System for Mammalian Studies? Cells 2019, 8, 796. [Google Scholar] [CrossRef] [PubMed]
- Ling, A.; Gruener, R.F.; Fessler, J.; Huang, R.S. More than fishing for a cure: The promises and pitfalls of high throughput cancer cell line screens. Pharmacol. Ther. 2018, 191, 178–189. [Google Scholar] [CrossRef]
- Kitaeva, K.V.; Rutland, C.S.; Rizvanov, A.A.; Solovyeva, V.V. Cell Culture Based in vitro Test Systems for Anticancer Drug Screening. Front. Bioeng. Biotechnol. 2020, 8, 322. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.H.; Li, H.; Jia, Y.; Mak, P.I.; Martins, R.; Liu, Y.; Vong, C.M.; Wong, H.C.; Wong, P.K.; Wang, H.; et al. Drug screening of cancer cell lines and human primary tumors using droplet microfluidics. Sci. Rep. 2017, 7, 9109. [Google Scholar] [CrossRef]
- Wilson, J.D.K. The lungs at the frontlines of immunity. Nat. Immunol. 2015, 16, 17. [Google Scholar] [CrossRef]
- van der Vaart, J.; Clevers, H. Airway organoids as models of human disease. J. Intern. Med. 2021, 289, 604–613. [Google Scholar] [CrossRef]
- McCauley, K.B.; Hawkins, F.; Serra, M.; Thomas, D.C.; Jacob, A.; Kotton, D.N. Efficient Derivation of Functional Human Airway Epithelium from Pluripotent Stem Cells via Temporal Regulation of Wnt Signaling. Cell Stem Cell 2017, 20, 844–857.e6. [Google Scholar] [CrossRef] [PubMed]
- McCauley, K.B.; Hawkins, F.; Kotton, D.N. Derivation of Epithelial-Only Airway Organoids from Human Pluripotent Stem Cells. Curr. Protoc. Stem Cell Biol. 2018, 45, e51. [Google Scholar] [CrossRef]
- Kumar, P.A.; Hu, Y.; Yamamoto, Y.; Hoe, N.B.; Wei, T.S.; Mu, D.; Sun, Y.; Joo, L.S.; Dagher, R.; Zielonka, E.M.; et al. Distal airway stem cells yield alveoli in vitro and during lung regeneration following H1N1 influenza infection. Cell 2011, 147, 525–538. [Google Scholar] [CrossRef]
- Usui, S.; Shimizu, T.; Kishioka, C.; Fujita, K.; Sakakura, Y. Secretory cell differentiation and mucus secretion in cultures of human nasal epithelial cells: Use of a monoclonal antibody to study human nasal mucin. Ann. Otol. Rhinol. Laryngol. 2000, 109, 271–277. [Google Scholar] [CrossRef]
- Sachs, N.; Papaspyropoulos, A.; Zomer-van Ommen, D.D.; Heo, I.; Böttinger, L.; Klay, D.; Weeber, F.; Huelsz-Prince, G.; Iakobachvili, N.; Amatngalim, G.D.; et al. Long-term expanding human airway organoids for disease modeling. EMBO J. 2019, 38, e100300. [Google Scholar] [CrossRef] [PubMed]
- Chiu, M.C.; Li, C.; Liu, X.; Yu, Y.; Huang, J.; Wan, Z.; Xiao, D.; Chu, H.; Cai, J.P.; Zhou, B.; et al. A bipotential organoid model of respiratory epithelium recapitulates high infectivity of SARS-CoV-2 Omicron variant. Cell Discov. 2022, 8, 57. [Google Scholar] [CrossRef] [PubMed]
- Bluhmki, T.; Traub, S.; Müller, A.K.; Bitzer, S.; Schruf, E.; Bammert, M.T.; Leist, M.; Gantner, F.; Garnett, J.P.; Heilker, R. Functional human iPSC-derived alveolar-like cells cultured in a miniaturized 96-Transwell air-liquid interface model. Sci. Rep. 2021, 11, 17028. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Li, Y.; Shi, F.; Liu, H. Human lung organoid: Models for respiratory biology and diseases. Dev. Biol. 2023, 494, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Liu, F.; Liang, X.; Duan, L.; Li, Q.; Pan, G.; Ma, C.; Liu, M.; Li, M.; Wang, P.; et al. iPSC-Derived Airway Epithelial Cells: Progress, Promise, and Challenges. Stem Cells 2023, 41, 1–10. [Google Scholar] [CrossRef]
- Lu, T.; Cao, Y.; Zhao, P.; Shen, S.; Xi, Y. Organoid: A powerful tool to study lung regeneration and disease. Cell Regen. 2021, 10, 21. [Google Scholar] [CrossRef]
- Chen, J.; Na, F. Organoid technology and applications in lung diseases: Models, mechanism research and therapy opportunities. Front. Bioeng. Biotechnol. 2022, 10, 1066869. [Google Scholar] [CrossRef]
- van der Sanden, S.M.G.; Sachs, N.; Koekkoek, S.M.; Koen, G.; Pajkrt, D.; Clevers, H.; Wolthers, K.C. Enterovirus 71 infection of human airway organoids reveals VP1-145 as a viral infectivity determinant. Emerg. Microbes Infect. 2018, 7, 84. [Google Scholar] [CrossRef]
- Zhou, J.; Li, C.; Sachs, N.; Chiu, M.C.; Wong, B.H.; Chu, H.; Poon, V.K.; Wang, D.; Zhao, X.; Wen, L.; et al. Differentiated human airway organoids to assess infectivity of emerging influenza virus. Proc. Natl. Acad. Sci. USA 2018, 115, 6822–6827. [Google Scholar] [CrossRef]
- Heo, I.; Dutta, D.; Schaefer, D.A.; Iakobachvili, N.; Artegiani, B.; Sachs, N.; Boonekamp, K.E.; Bowden, G.; Hendrickx, A.P.A.; Willems, R.J.L.; et al. Modelling Cryptosporidium infection in human small intestinal and lung organoids. Nat. Microbiol. 2018, 3, 814–823. [Google Scholar] [CrossRef]
- Hui, K.P.Y.; Ching, R.H.H.; Chan, S.K.H.; Nicholls, J.M.; Sachs, N.; Clevers, H.; Peiris, J.S.M.; Chan, M.C.W. Tropism, replication competence, and innate immune responses of influenza virus: An analysis of human airway organoids and ex-vivo bronchus cultures. Lancet Respir. Med. 2018, 6, 846–854. [Google Scholar] [CrossRef]
- Wilkinson, D.C.; Alva-Ornelas, J.A.; Sucre, J.M.; Vijayaraj, P.; Durra, A.; Richardson, W.; Jonas, S.J.; Paul, M.K.; Karumbayaram, S.; Dunn, B.; et al. Development of a Three-Dimensional Bioengineering Technology to Generate Lung Tissue for Personalized Disease Modeling. Stem Cells Transl. Med. 2017, 6, 622–633. [Google Scholar] [CrossRef]
- Miller, A.J.; Dye, B.R.; Ferrer-Torres, D.; Hill, D.R.; Overeem, A.W.; Shea, L.D.; Spence, J.R. Generation of lung organoids from human pluripotent stem cells in vitro. Nat. Protoc. 2019, 14, 518–540. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Li, J.; Dang, S.; Schurmann, P.; Dost, A.; Moye, A.; Paschini, M.; Bhetariya, P.; Bronson, R.; Sui, S.H. Organoid modeling reveals the tumorigenic potential of the alveolar progenitor cell state. Res. Sq. 2023. [Google Scholar] [CrossRef]
- Heinrich, V.A.; Uvalle, C.; Manni, M.L.; Li, K.; Mullett, S.J.; Donepudi, S.R.; Clader, J.; Fitch, A.; Ellgass, M.; Cechova, V.; et al. Meta-omics profiling of the gut-lung axis illuminates metabolic networks and host-microbial interactions associated with elevated lung elastance in a murine model of obese allergic asthma. Front. Microbiomes 2023, 2, 1153691. [Google Scholar] [CrossRef]
- Wilke, M.; Buijs-Offerman, R.M.; Aarbiou, J.; Colledge, W.H.; Sheppard, D.N.; Touqui, L.; Bot, A.; Jorna, H.; de Jonge, H.R.; Scholte, B.J. Mouse models of cystic fibrosis: Phenotypic analysis and research applications. J. Cyst. Fibros. 2011, 10 (Suppl. S2), S152–S171. [Google Scholar] [CrossRef] [PubMed]
- Walters, D.M.; Kleeberger, S.R. Mouse models of bleomycin-induced pulmonary fibrosis. Curr. Protoc. Pharmacol. 2008, 40, 5–46. [Google Scholar] [CrossRef] [PubMed]
- Tashiro, J.; Rubio, G.A.; Limper, A.H.; Williams, K.; Elliot, S.J.; Ninou, I.; Aidinis, V.; Tzouvelekis, A.; Glassberg, M.K. Exploring Animal Models that Resemble Idiopathic Pulmonary Fibrosis. Front. Med. 2017, 4, 118. [Google Scholar] [CrossRef] [PubMed]
- Lemaitre, J.; Naninck, T.; Delache, B.; Creppy, J.; Huber, P.; Holzapfel, M.; Bouillier, C.; Contreras, V.; Martinon, F.; Kahlaoui, N.; et al. Non-human primate models of human respiratory infections. Mol. Immunol. 2021, 135, 147–164. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.C.; Berns, A. Mouse models for lung cancer. Mol. Oncol. 2013, 7, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Baron, R.M.; Choi, A.J.; Owen, C.A.; Choi, A.M. Genetically manipulated mouse models of lung disease: Potential and pitfalls. Am. J. Physiol. Lung Cell Mol. Physiol. 2012, 302, L485–L497. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Deutsch, G.H.; Wert, S.E. Comprehensive anatomic ontologies for lung development: A comparison of alveolar formation and maturation within mouse and human lung. J. Biomed. Semant. 2019, 10, 18. [Google Scholar] [CrossRef] [PubMed]
- Hou, K.; Wu, Z.X.; Chen, X.Y.; Wang, J.Q.; Zhang, D.; Xiao, C.; Zhu, D.; Koya, J.B.; Wei, L.; Li, J.; et al. Microbiota in health and diseases. Signal Transduct. Target. Ther. 2022, 7, 135. [Google Scholar] [CrossRef]
- Gibson, R.L.; Burns, J.L.; Ramsey, B.W. Pathophysiology and management of pulmonary infections in cystic fibrosis. Am. J. Respir. Crit. Care Med. 2003, 168, 918–951. [Google Scholar] [CrossRef] [PubMed]
- Rowe, S.M.; Heltshe, S.L.; Gonska, T.; Donaldson, S.H.; Borowitz, D.; Gelfond, D.; Sagel, S.D.; Khan, U.; Mayer-Hamblett, N.; Van Dalfsen, J.M.; et al. Clinical mechanism of the cystic fibrosis transmembrane conductance regulator potentiator ivacaftor in G551D-mediated cystic fibrosis. Am. J. Respir. Crit. Care Med. 2014, 190, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Nichols, D.; Chmiel, J.; Berger, M. Chronic inflammation in the cystic fibrosis lung: Alterations in inter- and intracellular signaling. Clin. Rev. Allergy Immunol. 2008, 34, 146–162. [Google Scholar] [CrossRef]
- Keiser, N.W.; Birket, S.E.; Evans, I.A.; Tyler, S.R.; Crooke, A.K.; Sun, X.; Zhou, W.; Nellis, J.R.; Stroebele, E.K.; Chu, K.K.; et al. Defective innate immunity and hyperinflammation in newborn cystic fibrosis transmembrane conductance regulator-knockout ferret lungs. Am. J. Respir. Cell Mol. Biol. 2015, 52, 683–694. [Google Scholar] [CrossRef]
- O’Connor, J.B.; Mottlowitz, M.M.; Wagner, B.D.; Boyne, K.L.; Stevens, M.J.; Robertson, C.E.; Harris, J.K.; Laguna, T.A. Divergence of bacterial communities in the lower airways of CF patients in early childhood. PLoS ONE 2021, 16, e0257838. [Google Scholar] [CrossRef]
- Twomey, K.B.; Alston, M.; An, S.Q.; O’Connell, O.J.; McCarthy, Y.; Swarbreck, D.; Febrer, M.; Dow, J.M.; Plant, B.J.; Ryan, R.P. Microbiota and metabolite profiling reveal specific alterations in bacterial community structure and environment in the cystic fibrosis airway during exacerbation. PLoS ONE 2013, 8, e82432. [Google Scholar] [CrossRef] [PubMed]
- Quinn, R.A.; Phelan, V.V.; Whiteson, K.L.; Garg, N.; Bailey, B.A.; Lim, Y.W.; Conrad, D.J.; Dorrestein, P.C.; Rohwer, F.L. Microbial, host and xenobiotic diversity in the cystic fibrosis sputum metabolome. ISME J. 2016, 10, 1483–1498. [Google Scholar] [CrossRef]
- O’Connor, J.B.; Mottlowitz, M.; Kruk, M.E.; Mickelson, A.; Wagner, B.D.; Harris, J.K.; Wendt, C.H.; Laguna, T.A. Network Analysis to Identify Multi-Omic Correlations in the Lower Airways of Children with Cystic Fibrosis. Front. Cell Infect. Microbiol. 2022, 12, 805170. [Google Scholar] [CrossRef] [PubMed]
- Montuschi, P.; Paris, D.; Melck, D.; Lucidi, V.; Ciabattoni, G.; Raia, V.; Calabrese, C.; Bush, A.; Barnes, P.J.; Motta, A. NMR spectroscopy metabolomic profiling of exhaled breath condensate in patients with stable and unstable cystic fibrosis. Thorax 2012, 67, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Monge, M.E.; Pérez, J.J.; Dwivedi, P.; Zhou, M.; McCarty, N.A.; Stecenko, A.A.; Fernández, F.M. Ion mobility and liquid chromatography/mass spectrometry strategies for exhaled breath condensate glucose quantitation in cystic fibrosis studies. Rapid Commun. Mass. Spectrom. 2013, 27, 2263–2271. [Google Scholar] [CrossRef] [PubMed]
- Wolak, J.E.; Esther, C.R., Jr.; O’Connell, T.M. Metabolomic analysis of bronchoalveolar lavage fluid from cystic fibrosis patients. Biomarkers 2009, 14, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Esther, C.R., Jr.; Coakley, R.D.; Henderson, A.G.; Zhou, Y.H.; Wright, F.A.; Boucher, R.C. Metabolomic Evaluation of Neutrophilic Airway Inflammation in Cystic Fibrosis. Chest 2015, 148, 507–515. [Google Scholar] [CrossRef]
- Esther, C.R., Jr.; Turkovic, L.; Rosenow, T.; Muhlebach, M.S.; Boucher, R.C.; Ranganathan, S.; Stick, S.M. Metabolomic biomarkers predictive of early structural lung disease in cystic fibrosis. Eur. Respir. J. 2016, 48, 1612–1621. [Google Scholar] [CrossRef]
- Zemanick, E.T.; Wagner, B.D.; Robertson, C.E.; Stevens, M.J.; Szefler, S.J.; Accurso, F.J.; Sagel, S.D.; Harris, J.K. Assessment of airway microbiota and inflammation in cystic fibrosis using multiple sampling methods. Ann. Am. Thorac. Soc. 2015, 12, 221–229. [Google Scholar] [CrossRef]
- Jorth, P.; Ehsan, Z.; Rezayat, A.; Caldwell, E.; Pope, C.; Brewington, J.J.; Goss, C.H.; Benscoter, D.; Clancy, J.P.; Singh, P.K. Direct Lung Sampling Indicates That Established Pathogens Dominate Early Infections in Children with Cystic Fibrosis. Cell Rep. 2019, 27, 1190–1204.e3. [Google Scholar] [CrossRef] [PubMed]
- Raghuvanshi, R.; Vasco, K.; Vázquez-Baeza, Y.; Jiang, L.; Morton, J.T.; Li, D.; Gonzalez, A.; DeRight Goldasich, L.; Humphrey, G.; Ackermann, G.; et al. High-Resolution Longitudinal Dynamics of the Cystic Fibrosis Sputum Microbiome and Metabolome through Antibiotic Therapy. mSystems 2020, 5, e00292.20. [Google Scholar] [CrossRef] [PubMed]
- Hahn, A.; Whiteson, K.; Davis, T.J.; Phan, J.; Sami, I.; Koumbourlis, A.C.; Freishtat, R.J.; Crandall, K.A.; Bean, H.D. Longitudinal Associations of the Cystic Fibrosis Airway Microbiome and Volatile Metabolites: A Case Study. Front. Cell Infect. Microbiol. 2020, 10, 174. [Google Scholar] [CrossRef]
- Hoen, A.G.; Li, J.; Moulton, L.A.; O’Toole, G.A.; Housman, M.L.; Koestler, D.C.; Guill, M.F.; Moore, J.H.; Hibberd, P.L.; Morrison, H.G.; et al. Associations between Gut Microbial Colonization in Early Life and Respiratory Outcomes in Cystic Fibrosis. J. Pediatr. 2015, 167, 138–147.e3. [Google Scholar] [CrossRef] [PubMed]
- LiPuma, J.J. Assessing Airway Microbiota in Cystic Fibrosis: What More Should Be Done? J. Clin. Microbiol. 2015, 53, 2006–2007. [Google Scholar] [CrossRef] [PubMed]
- Tracy, M.; Cogen, J.; Hoffman, L.R. The pediatric microbiome and the lung. Curr. Opin. Pediatr. 2015, 27, 348–355. [Google Scholar] [CrossRef]
- Prevaes, S.M.; de Winter-de Groot, K.M.; Janssens, H.M.; de Steenhuijsen Piters, W.A.; Tramper-Stranders, G.A.; Wyllie, A.L.; Hasrat, R.; Tiddens, H.A.; van Westreenen, M.; van der Ent, C.K.; et al. Development of the Nasopharyngeal Microbiota in Infants with Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2016, 193, 504–515. [Google Scholar] [CrossRef] [PubMed]
- Serkova, N.J.; Standiford, T.J.; Stringer, K.A. The emerging field of quantitative blood metabolomics for biomarker discovery in critical illnesses. Am. J. Respir. Crit. Care Med. 2011, 184, 647–655. [Google Scholar] [CrossRef]
- Shi, W.J.; Zhuang, Y.; Russell, P.H.; Hobbs, B.D.; Parker, M.M.; Castaldi, P.J.; Rudra, P.; Vestal, B.; Hersh, C.P.; Saba, L.M.; et al. Unsupervised discovery of phenotype-specific multi-omics networks. Bioinformatics 2019, 35, 4336–4343. [Google Scholar] [CrossRef]
- Quinn, R.A.; Adem, S.; Mills, R.H.; Comstock, W.; DeRight Goldasich, L.; Humphrey, G.; Aksenov, A.A.; Melnik, A.V.; da Silva, R.; Ackermann, G.; et al. Neutrophilic proteolysis in the cystic fibrosis lung correlates with a pathogenic microbiome. Microbiome 2019, 7, 23. [Google Scholar] [CrossRef] [PubMed]
- Agustí, A.; Celli, B.R.; Criner, G.J.; Halpin, D.; Anzueto, A.; Barnes, P.; Bourbeau, J.; Han, M.K.; Martinez, F.J.; Montes de Oca, M.; et al. Global Initiative for Chronic Obstructive Lung Disease 2023 Report: GOLD Executive Summary. Eur. Respir. J. 2023, 61, 2300239. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Chen, B.; Yang, Y.; Yi, X.; Wei, M.; Ecklu-Mensah, G.; Buschmann, M.M.; Liu, H.; Gao, J.; Liang, W.; et al. Multi-omics analyses of airway host-microbe interactions in chronic obstructive pulmonary disease identify potential therapeutic interventions. Nat. Microbiol. 2022, 7, 1361–1375. [Google Scholar] [CrossRef]
- Rhodes, C.J.; Sweatt, A.J.; Maron, B.A. Harnessing Big Data to Advance Treatment and Understanding of Pulmonary Hypertension. Circ. Res. 2022, 130, 1423–1444. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yang, Y.; Yan, Z.; Liu, H.; Chen, B.; Liang, Z.; Wang, F.; Miller, B.E.; Tal-Singer, R.; Yi, X.; et al. Multi-omic meta-analysis identifies functional signatures of airway microbiome in chronic obstructive pulmonary disease. ISME J. 2020, 14, 2748–2765. [Google Scholar] [CrossRef] [PubMed]
- Sandri, B.J.; Kaplan, A.; Hodgson, S.W.; Peterson, M.; Avdulov, S.; Higgins, L.; Markowski, T.; Yang, P.; Limper, A.H.; Griffin, T.J.; et al. Multi-omic molecular profiling of lung cancer in COPD. Eur. Respir. J. 2018, 52, 1702665. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Hafiz, M.; Najafi, M.; Helmi, S.; Pratte, K.A.; Zhuang, Y.; Liu, W.; Kechris, K.J.; Bowler, R.P.; Lange, L.; Banaei-Kashani, F. Significant Subgraph Detection in Multi-omics Networks for Disease Pathway Identification. Front. Big Data 2022, 5, 894632. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Wu, Y.; Xiao, T.; Qi, F.; Fan, L.; Zhang, S.; Zhou, J.; He, Y.; Gao, X.; Zeng, H.; et al. Multiomics approach reveals the ubiquitination-specific processes hijacked by SARS-CoV-2. Signal Transduct. Target. Ther. 2022, 7, 312. [Google Scholar] [CrossRef]
- Unterman, A.; Sumida, T.S.; Nouri, N.; Yan, X.; Zhao, A.Y.; Gasque, V.; Schupp, J.C.; Asashima, H.; Liu, Y.; Cosme, C., Jr.; et al. Single-cell multi-omics reveals dyssynchrony of the innate and adaptive immune system in progressive COVID-19. Nat. Commun. 2022, 13, 440. [Google Scholar] [CrossRef] [PubMed]
- Li, C.X.; Gao, J.; Zhang, Z.; Chen, L.; Li, X.; Zhou, M.; Wheelock, Å.M. Multiomics integration-based molecular characterizations of COVID-19. Brief. Bioinform. 2022, 23, bbab485. [Google Scholar] [CrossRef]
- Wu, L.; Zhu, J.; Liu, D.; Sun, Y.; Wu, C. An integrative multiomics analysis identifies putative causal genes for COVID-19 severity. Genet. Med. 2021, 23, 2076–2086. [Google Scholar] [CrossRef]
- Cantwell, A.M.; Singh, H.; Platt, M.; Yu, Y.; Lin, Y.H.; Ikeno, Y.; Hubbard, G.; Xiang, Y.; Gonzalez-Juarbe, N.; Dube, P.H. Kinetic Multi-omic Analysis of Responses to SARS-CoV-2 Infection in a Model of Severe COVID-19. J. Virol. 2021, 95, e0101021. [Google Scholar] [CrossRef]
- Wilk, A.J.; Lee, M.J.; Wei, B.; Parks, B.; Pi, R.; Martínez-Colón, G.J.; Ranganath, T.; Zhao, N.Q.; Taylor, S.; Becker, W.; et al. Multi-omic profiling reveals widespread dysregulation of innate immunity and hematopoiesis in COVID-19. J. Exp. Med. 2021, 218, e20210582. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012, 489, 519–525. [Google Scholar] [CrossRef] [PubMed]
- CGARN. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Creighton, C.J.; Nagaraja, A.K.; Hanash, S.M.; Matzuk, M.M.; Gunaratne, P.H. A bioinformatics tool for linking gene expression profiling results with public databases of microRNA target predictions. RNA 2008, 14, 2290–2296. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, X. miRDB: An online database for prediction of functional microRNA targets. Nucleic Acids Res. 2020, 48, D127–D131. [Google Scholar] [CrossRef] [PubMed]
- Vlachos, I.S.; Paraskevopoulou, M.D.; Karagkouni, D.; Georgakilas, G.; Vergoulis, T.; Kanellos, I.; Anastasopoulos, I.L.; Maniou, S.; Karathanou, K.; Kalfakakou, D.; et al. DIANA-TarBase v7.0: Indexing more than half a million experimentally supported miRNA:mRNA interactions. Nucleic Acids Res. 2015, 43, D153–D159. [Google Scholar] [CrossRef]
- Wang, X.J.; Gao, J.; Wang, Z.; Yu, Q. Identification of a Potentially Functional microRNA-mRNA Regulatory Network in Lung Adenocarcinoma Using a Bioinformatics Analysis. Front. Cell Dev. Biol. 2021, 9, 641840. [Google Scholar] [CrossRef]
- Liu, Y.; He, L.; Wang, W. Systematic assessment of microRNAs associated with lung cancer and physical exercise. Front. Oncol. 2022, 12, 917667. [Google Scholar] [CrossRef]
- Campbell, J.D.; Yau, C.; Bowlby, R.; Liu, Y.; Brennan, K.; Fan, H.; Taylor, A.M.; Wang, C.; Walter, V.; Akbani, R.; et al. Genomic, Pathway Network, and Immunologic Features Distinguishing Squamous Carcinomas. Cell Rep. 2018, 23, 194–212.e6. [Google Scholar] [CrossRef]
- Thaiparambil, J.; Dong, J.; Grimm, S.L.; Perera, D.; Ambati, C.S.R.; Putluri, V.; Robertson, M.J.; Patel, T.D.; Mistretta, B.; Gunaratne, P.H.; et al. Integrative metabolomics and transcriptomics analysis reveals novel therapeutic vulnerabilities in lung cancer. Cancer Med. 2023, 12, 584–596. [Google Scholar] [CrossRef]
- Ho, W.J.; Erbe, R.; Danilova, L.; Phyo, Z.; Bigelow, E.; Stein-O’Brien, G.; Thomas, D.L., 2nd; Charmsaz, S.; Gross, N.; Woolman, S.; et al. Multi-omic profiling of lung and liver tumor microenvironments of metastatic pancreatic cancer reveals site-specific immune regulatory pathways. Genome Biol. 2021, 22, 154. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Xu, M.; Li, X.; Gaynor, S.; Zhou, H.; Li, Z.; Bossé, Y.; Lam, S.; Tsao, M.S.; Tardon, A.; et al. Integration of multiomic annotation data to prioritize and characterize inflammation and immune-related risk variants in squamous cell lung cancer. Genet. Epidemiol. 2021, 45, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.C.; Reuben, A.; Hu, X.; McGranahan, N.; Chen, R.; Jalali, A.; Negrao, M.V.; Hubert, S.M.; Tang, C.; Wu, C.C.; et al. Multiomics profiling of primary lung cancers and distant metastases reveals immunosuppression as a common characteristic of tumor cells with metastatic plasticity. Genome Biol. 2020, 21, 271. [Google Scholar] [CrossRef]
- Jensen, E.A.; Dysart, K.; Gantz, M.G.; McDonald, S.; Bamat, N.A.; Keszler, M.; Kirpalani, H.; Laughon, M.M.; Poindexter, B.B.; Duncan, A.F.; et al. The Diagnosis of Bronchopulmonary Dysplasia in Very Preterm Infants. An Evidence-based Approach. Am. J. Respir. Crit. Care Med. 2019, 200, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Perl, A.K.; Li, R.; Bell, S.M.; Sajti, E.; Kalinichenko, V.V.; Kalin, T.V.; Misra, R.S.; Deshmukh, H.; Clair, G.; et al. A census of the lung: CellCards from LungMAP. Dev. Cell 2022, 57, 112–145.e2. [Google Scholar] [CrossRef] [PubMed]
- Lal, C.V.; Kandasamy, J.; Dolma, K.; Ramani, M.; Kumar, R.; Wilson, L.; Aghai, Z.; Barnes, S.; Blalock, J.E.; Gaggar, A.; et al. Early airway microbial metagenomic and metabolomic signatures are associated with development of severe bronchopulmonary dysplasia. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 315, L810–L815. [Google Scholar] [CrossRef] [PubMed]
- Lal, C.V.; Olave, N.; Travers, C.; Rezonzew, G.; Dolma, K.; Simpson, A.; Halloran, B.; Aghai, Z.; Das, P.; Sharma, N.; et al. Exosomal microRNA predicts and protects against severe bronchopulmonary dysplasia in extremely premature infants. JCI Insight 2018, 3, e93994. [Google Scholar] [CrossRef] [PubMed]
- Lal, C.V.; Travers, C.; Aghai, Z.H.; Eipers, P.; Jilling, T.; Halloran, B.; Carlo, W.A.; Keeley, J.; Rezonzew, G.; Kumar, R.; et al. The Airway Microbiome at Birth. Sci. Rep. 2016, 6, 31023. [Google Scholar] [CrossRef] [PubMed]
- Pammi, M.; Lal, C.V.; Wagner, B.D.; Mourani, P.M.; Lohmann, P.; Luna, R.A.; Sisson, A.; Shivanna, B.; Hollister, E.B.; Abman, S.H.; et al. Airway Microbiome and Development of Bronchopulmonary Dysplasia in Preterm Infants: A Systematic Review. J. Pediatr. 2019, 204, 126–133.e2. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Coarfa, C.; Dong, X.; Jiang, W.; Hayward-Piatkovskyi, B.; Gleghorn, J.P.; Lingappan, K. MicroRNA-30a as a candidate underlying sex-specific differences in neonatal hyperoxic lung injury: Implications for BPD. Am. J. Physiol. Lung Cell Mol. Physiol. 2019, 316, L144–L156. [Google Scholar] [CrossRef] [PubMed]
- Coarfa, C.; Zhang, Y.; Maity, S.; Perera, D.N.; Jiang, W.; Wang, L.; Couroucli, X.; Moorthy, B.; Lingappan, K. Sexual dimorphism of the pulmonary transcriptome in neonatal hyperoxic lung injury: Identification of angiogenesis as a key pathway. Am. J. Physiol. Lung Cell Mol. Physiol. 2017, 313, L991–L1005. [Google Scholar] [CrossRef] [PubMed]
- El Saie, A.; Fu, C.; Grimm, S.L.; Robertson, M.J.; Hoffman, K.; Putluri, V.; Ambati, C.S.R.; Putluri, N.; Shivanna, B.; Coarfa, C.; et al. Metabolome and microbiome multi-omics integration from a murine lung inflammation model of bronchopulmonary dysplasia. Pediatr. Res. 2022, 92, 1580–1589. [Google Scholar] [CrossRef]
- Toldi, G.; Hummler, H.; Pillay, T. T Lymphocytes, Multi-Omic Interactions and Bronchopulmonary Dysplasia. Front. Pediatr. 2021, 9, 694034. [Google Scholar] [CrossRef]
- Maron, B.A. Revised Definition of Pulmonary Hypertension and Approach to Management: A Clinical Primer. J. Am. Heart Assoc. 2023, 12, e029024. [Google Scholar] [CrossRef]
- Chen, J.; Zhou, D.; Miao, J.; Zhang, C.; Li, X.; Feng, H.; Xing, Y.; Zhang, Z.; Bao, C.; Lin, Z.; et al. Microbiome and metabolome dysbiosis of the gut-lung axis in pulmonary hypertension. Microbiol. Res. 2022, 265, 127205. [Google Scholar] [CrossRef]
- Konigsberg, I.R.; Borie, R.; Walts, A.D.; Cardwell, J.; Rojas, M.; Metzger, F.; Hauck, S.M.; Fingerlin, T.E.; Yang, I.V.; Schwartz, D.A. Molecular Signatures of Idiopathic Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2021, 65, 430–441. [Google Scholar] [CrossRef] [PubMed]
- Titz, B.; Szostak, J.; Sewer, A.; Phillips, B.; Nury, C.; Schneider, T.; Dijon, S.; Lavrynenko, O.; Elamin, A.; Guedj, E.; et al. Multi-omics systems toxicology study of mouse lung assessing the effects of aerosols from two heat-not-burn tobacco products and cigarette smoke. Comput. Struct. Biotechnol. J. 2020, 18, 1056–1073. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Wong, B.; Rhodes, C.J.; Kurt, Z.; Schwantes-An, T.H.; Mickler, E.A.; Gräf, S.; Eyries, M.; Lutz, K.A.; Pauciulo, M.W.; et al. Integrative Multiomics to Dissect the Lung Transcriptional Landscape of Pulmonary Arterial Hypertension. bioRxiv 2023. [Google Scholar] [CrossRef]
- Lamb, J.; Crawford, E.D.; Peck, D.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Lerner, J.; Brunet, J.P.; Subramanian, A.; Ross, K.N.; et al. The Connectivity Map: Using gene-expression signatures to connect small molecules, genes, and disease. Science 2006, 313, 1929–1935. [Google Scholar] [CrossRef] [PubMed]
- Pammi, M.; Aghaeepour, N.; Neu, J. Multiomics, artificial intelligence, and precision medicine in perinatology. Pediatr. Res. 2023, 93, 308–315. [Google Scholar] [CrossRef]
- Vellido, A. Societal Issues Concerning the Application of Artificial Intelligence in Medicine. Kidney Dis. 2019, 5, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Morley, J.; Machado, C.C.V.; Burr, C.; Cowls, J.; Joshi, I.; Taddeo, M.; Floridi, L. The ethics of AI in health care: A mapping review. Soc. Sci. Med. 2020, 260, 113172. [Google Scholar] [CrossRef] [PubMed]
- Oliva, A.; Grassi, S.; Vetrugno, G.; Rossi, R.; Della Morte, G.; Pinchi, V.; Caputo, M. Management of Medico-Legal Risks in Digital Health Era: A Scoping Review. Front. Med. 2021, 8, 821756. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| “Omic” Technology | Description |
|---|---|
| Genome | Genomics focuses on identifying genetic variations associated with disease, response to treatment, or prognosis. Genome-wide associations (GWAS) have successfully explained complex phenotypes in human diseases (GWAS catalogue https://www.ebi.ac.uk/gwas/home (accessed 18 August 2023)). |
| Epigenome | Epigenomics focuses on the genome-wide characterization of reversible modifications of DNA or DNA-associated proteins, such as DNA methylation or histone acetylation, which are major regulators of gene transcription and cellular fate. Those modifications can be influenced both by genetic and environmental factors, can be long-lasting, and are sometimes heritable. |
| Transcriptome | Transcriptomics focuses on genome-wide mRNA transcription qualitatively (which transcripts are present, identification of novel splice sites, RNA editing sites) and quantitatively (how much of each transcript is expressed). A small amount of RNA is transcribed for protein synthesis, and a much larger amount is encoded for other purposes, which may be implicated in disease. |
| Proteome | Proteomics quantifies peptide abundance, modification, and interaction. Specific peptides may be helpful in diagnosis, monitoring or prognostication of disease and may function as disease biomarkers. Mass spectroscopy has revolutionized the field of proteomics not only for quantifying peptides but also for identifying functionality mediated by post-translational modifications, including proteolysis, glycosylation, phosphorylation, nitrosylation, and ubiquitination. |
| Metabolome | Metabolomics quantifies multiple small molecules, including amino acids, fatty acids, carbohydrates, or other products of cellular metabolic functions. Metabolite levels and relative ratios reflect metabolic function, and out-of-normal range perturbations often indicate disease. |
| Microbiome | Microbiomics focuses on the abundance and composition of microbioal communities in humans and their association with health and disease. Human skin, mucosal surfaces, and the gut are colonized by microorganisms, including bacteria, viruses, and fungi, collectively known as the microbiota (and their genes constituting the microbiome). |
| Challenges in Multi-Omics Analysis | Possible Solutions |
|---|---|
|
|
|
|
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blutt, S.E.; Coarfa, C.; Neu, J.; Pammi, M. Multiomic Investigations into Lung Health and Disease. Microorganisms 2023, 11, 2116. https://doi.org/10.3390/microorganisms11082116
Blutt SE, Coarfa C, Neu J, Pammi M. Multiomic Investigations into Lung Health and Disease. Microorganisms. 2023; 11(8):2116. https://doi.org/10.3390/microorganisms11082116
Chicago/Turabian StyleBlutt, Sarah E., Cristian Coarfa, Josef Neu, and Mohan Pammi. 2023. "Multiomic Investigations into Lung Health and Disease" Microorganisms 11, no. 8: 2116. https://doi.org/10.3390/microorganisms11082116