The Phylogeny, Metabolic Potentials, and Environmental Adaptation of an Anaerobe, Abyssisolibacter sp. M8S5, Isolated from Cold Seep Sediments of the South China Sea

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Description and Strain Isolation
2.2. DNA Extraction, Genome Sequencing, and Assembly
2.3. Gene Annotation and Metabolic Pathway Reconstruction
2.4. Phylogenetic Tree Construction
2.5. Comparative Genomic Analyses
2.6. 16S rRNA gene Distribution Analysis of Strain M8S5
3. Results and Discussion
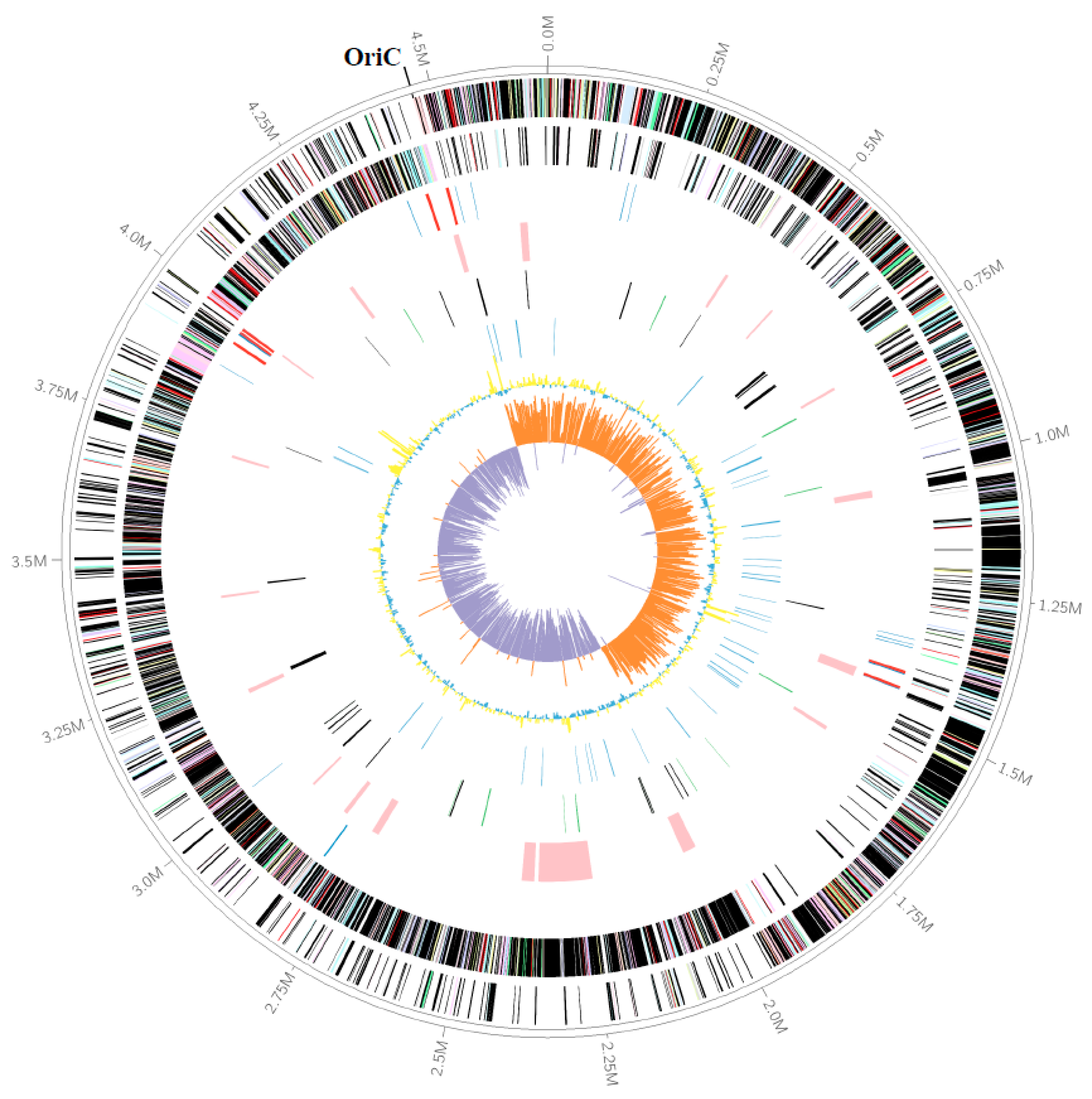
3.1. The Genomic Features of Strain M8S5
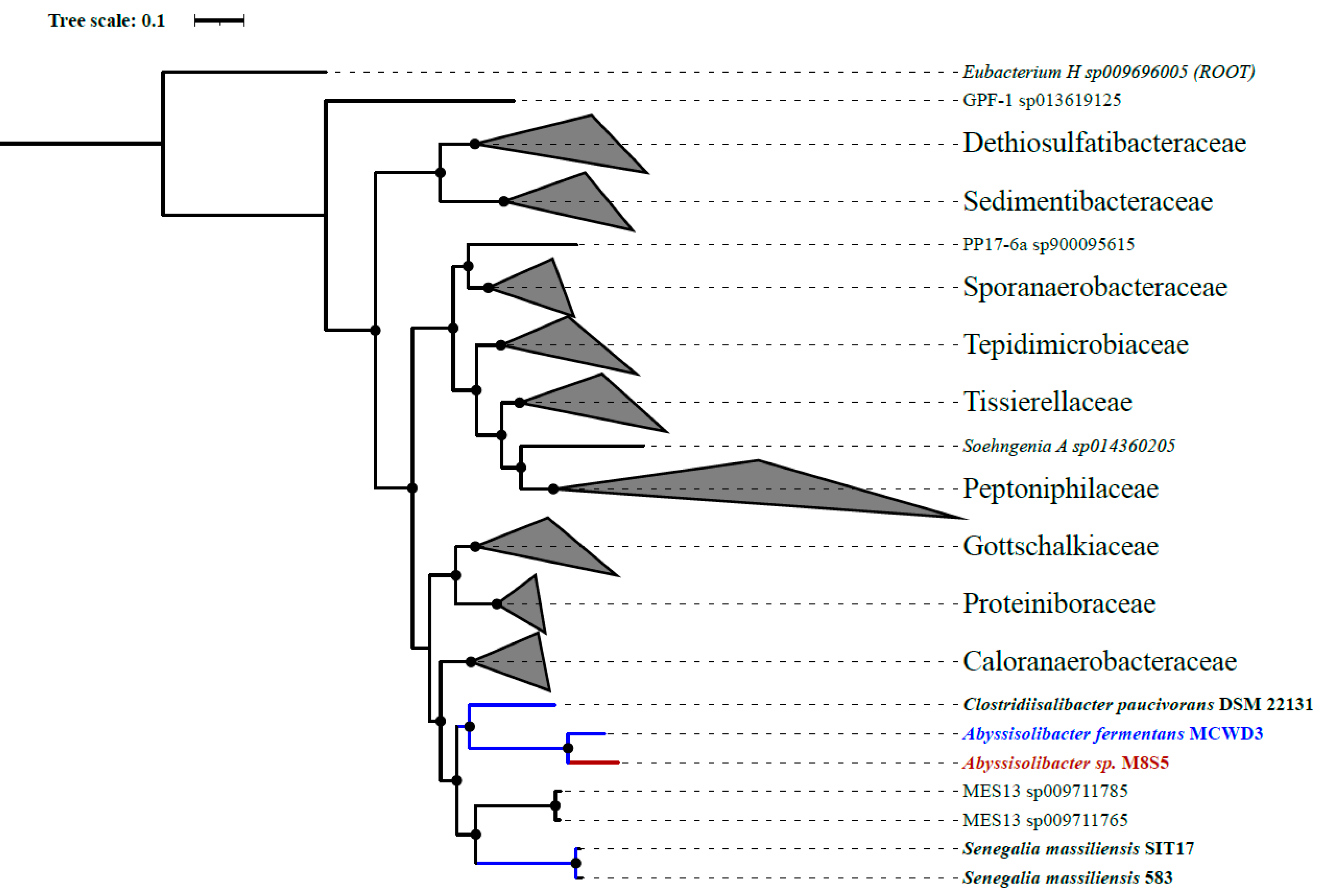
3.2. The Phylogeny of Strain M8S5
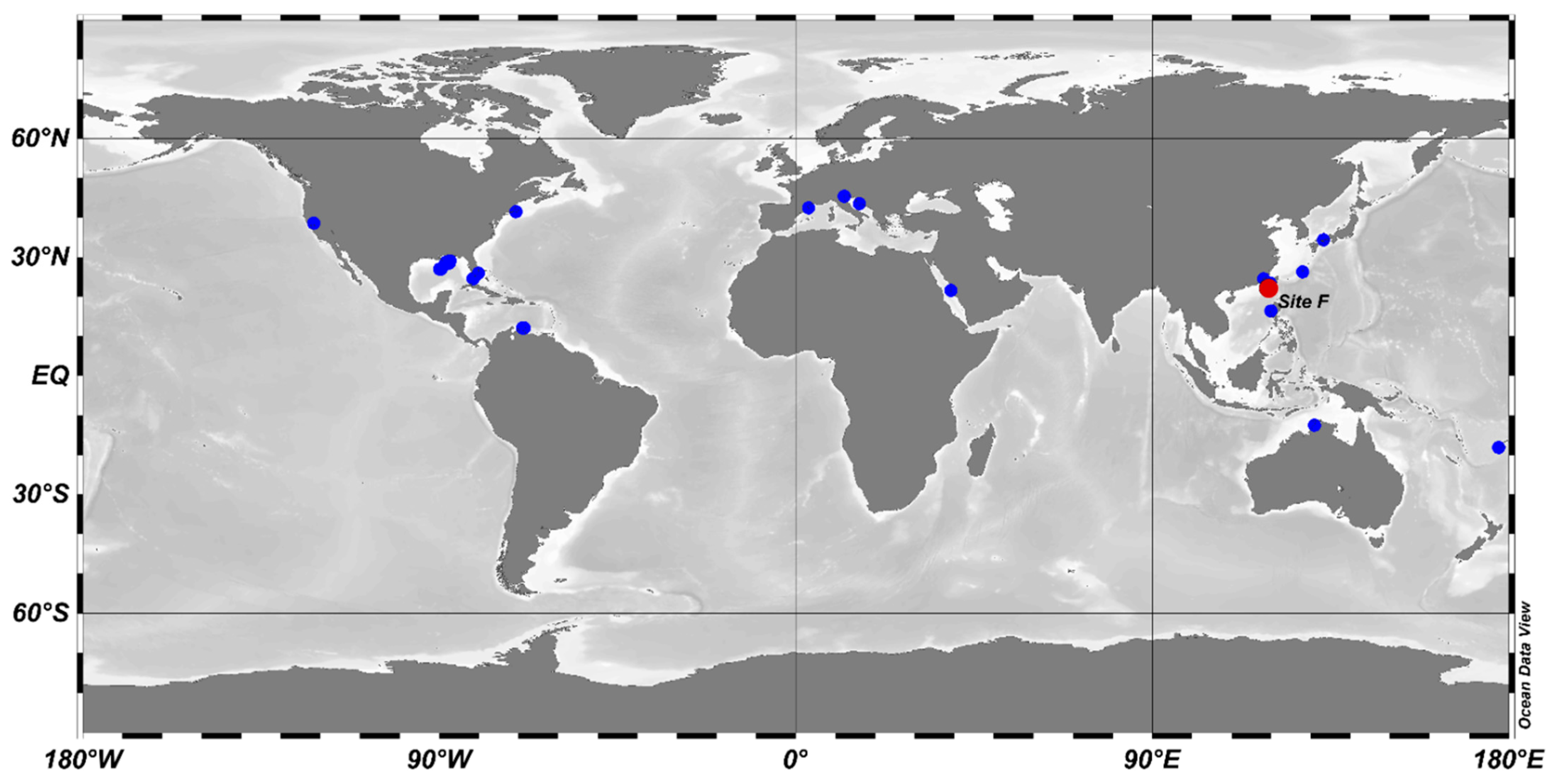
3.3. The Environmental Distribution of Abyssisolibacter Species
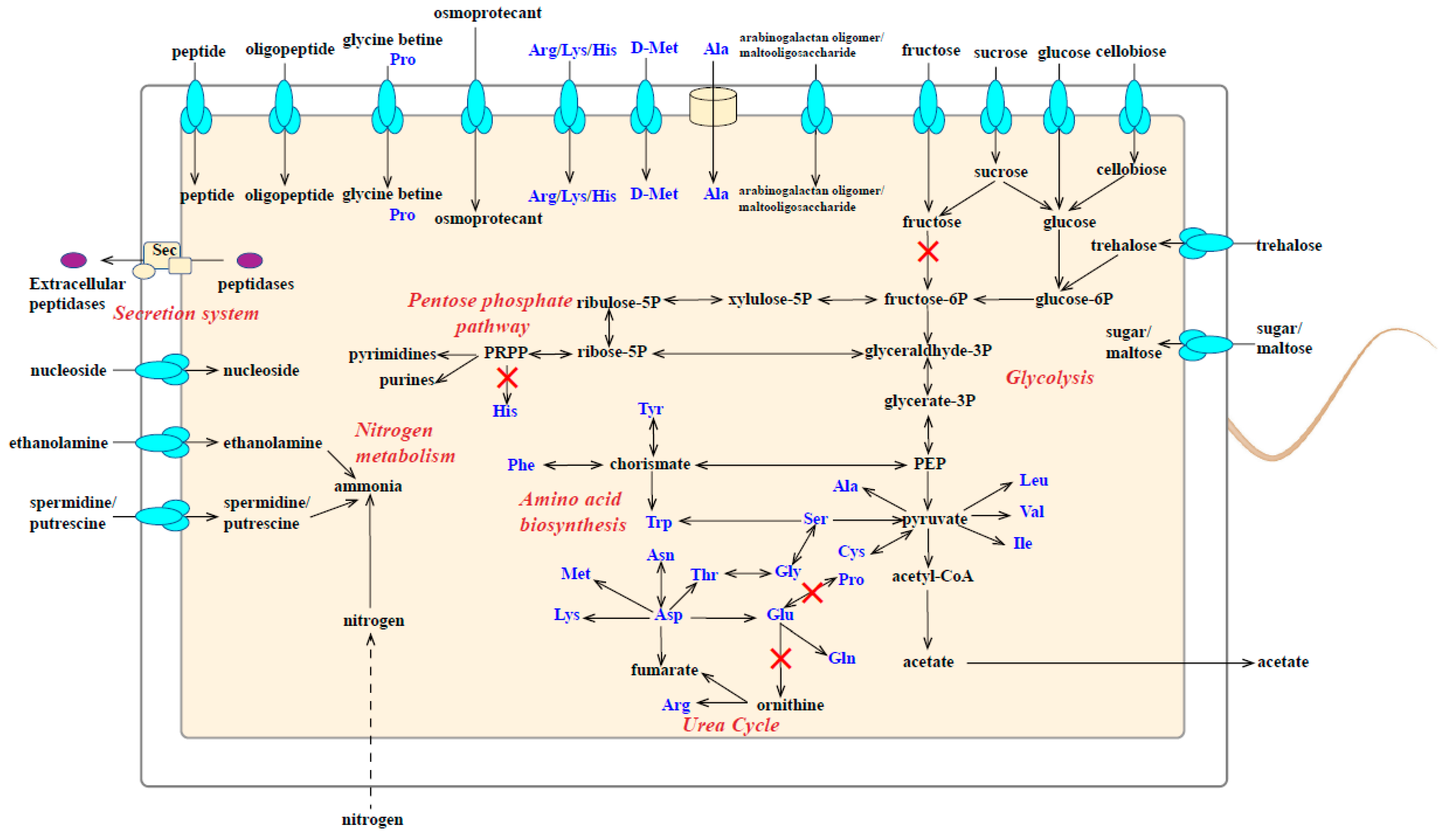
3.4. Metabolic Characteristics of Strain M8S5
- (1)
- Carbohydrate metabolism
- (2)
- Amino acid metabolism and extracellular protein degradation
- (3)
- d-amino acid metabolism
- (4)
- Nitrogen metabolism
- (5)
- Nucleic acid metabolism
- (6)
- Glycine reductase in substrate-level phosphorylation
3.5. Genes Involved in High-Pressure and Low-Temperature Adaptation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Wang, X.; Guan, H.; Qiu, J.-W.; Xu, T.; Peckmann, J.; Chen, D.; Feng, D. Macro-ecology of cold seeps in the South China Sea. Geosyst. Geoenviron. 2022, 1, 100081. [Google Scholar] [CrossRef]
- Feng, D.; Qiu, J.-W.; Hu, Y.; Peckmann, J.; Guan, H.; Tong, H.; Chen, C.; Chen, J.; Gong, S.; Li, N.; et al. Cold seep systems in the South China Sea: An overview. J. Asian Earth Sci. 2018, 168, 3–16. [Google Scholar] [CrossRef]
- Dong, X.; Rattray, J.E.; Campbell, D.C.; Webb, J.; Chakraborty, A.; Adebayo, O.; Matthews, S.; Li, C.; Fowler, M.; Morrison, N.M.; et al. Thermogenic hydrocarbon biodegradation by diverse depth-stratified microbial populations at a Scotian Basin cold seep. Nat. Commun. 2020, 11, 5825. [Google Scholar] [CrossRef]
- Niu, M.; Fan, X.; Zhuang, G.; Liang, Q.; Wang, F. Methane-metabolizing microbial communities in sediments of the Haima cold seep area, northwest slope of the South China Sea. FEMS Microbiol. Ecol. 2017, 93, fix101. [Google Scholar] [CrossRef]
- Cui, H.; Su, X.; Chen, F.; Holland, M.; Yang, S.; Liang, J.; Su, P.; Dong, H.; Hou, W. Microbial diversity of two cold seep systems in gas hydrate-bearing sediments in the South China Sea. Mar. Environ. Res. 2019, 144, 230–239. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, X.; Shi, L.-D.; Li, J.; Xiao, X.; Shao, Z.; Dong, X. Unexpected genetic and microbial diversity for arsenic cycling in deep sea cold seep sediments. NPJ Biofilms Microbiomes 2023, 9, 13. [Google Scholar] [CrossRef] [PubMed]
- Cong, M.; Pang, X.; Zhao, K.; Song, Y.; Liu, Y.; Wang, J. Deep-Sea Natural Products from Extreme Environments: Cold Seeps and Hydrothermal Vents. Mar. Drugs 2022, 20, 404. [Google Scholar] [CrossRef] [PubMed]
- Lapham, L.L.; Chanton, J.P.; Martens, C.S.; Sleeper, K.; Woolsey, J.R. Microbial activity in surficial sediments overlying acoustic wipeout zones at a Gulf of Mexico cold seep. Geochem. Geophys. Geosyst. 2008, 9, Q06001. [Google Scholar] [CrossRef]
- Carlier, A.; Ritt, B.; Rodrigues, C.F.; Sarrazin, J.; Olu, K.; Grall, J.; Clavier, J. Heterogeneous energetic pathways and carbon sources on deep eastern Mediterranean cold seep communities. Mar. Biol. 2010, 157, 2545–2565. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, Y.; Bougouffa, S.; Tian, R.; Cao, H.; Li, Y.; Cai, L.; Wong, Y.H.; Zhang, G.; Zhou, G.; et al. Synchronized dynamics of bacterial niche-specific functions during biofilm development in a cold seep brine pool. Environ. Microbiol. 2015, 17, 4089–4104. [Google Scholar] [CrossRef]
- Jiang, Q.; Jing, H.; Jiang, Q.; Zhang, Y. Insights into carbon-fixation pathways through metagonomics in the sediments of deep-sea cold seeps. Mar. Pollut. Bull. 2022, 176, 113458. [Google Scholar] [CrossRef]
- Ke, Z.; Li, R.; Chen, Y.; Chen, D.; Chen, Z.; Lian, X.; Tan, Y. A preliminary study of macrofaunal communities and their carbon and nitrogen stable isotopes in the Haima cold seeps, South China Sea. Deep Sea Res. Part I Oceanogr. Res. Pap. 2022, 184, 103774. [Google Scholar] [CrossRef]
- Galperin, M.Y.; Mekhedov, S.L.; Puigbo, P.; Smirnov, S.; Wolf, Y.I.; Rigden, D.J. Genomic determinants of sporulation in Bacilli and Clostridia: Towards the minimal set of sporulation-specific genes. Environ. Microbiol. 2012, 14, 2870–2890. [Google Scholar] [CrossRef] [PubMed]
- Azman, S.; Khadem, A.F.; van Lier, J.B.; Zeeman, G.; Plugge, C.M. Presence and Role of Anaerobic Hydrolytic Microbes in Conversion of Lignocellulosic Biomass for Biogas Production. Crit. Rev. Environ. Sci. Technol. 2015, 45, 2523–2564. [Google Scholar] [CrossRef]
- Zhang, X.; Tu, B.; Dai, L.-r.; Lawson, P.A.; Zheng, Z.-z.; Liu, L.-Y.; Deng, Y.; Zhang, H.; Cheng, L. Petroclostridium xylanilyticum gen. nov., sp. nov., a xylan-degrading bacterium isolated from an oilfield, and reclassification of clostridial cluster III members into four novel genera in a new Hungateiclostridiaceae fam. nov. Int. J. Syst. Evol. Microbiol. 2018, 68, 3197–3211. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Su, X.; Chen, F.; Wang, Y.; Jiao, L.; Dong, H.; Huang, Y.; Jiang, H. Microbial diversity in cold seep sediments from the northern South China Sea. Geosci. Front. 2012, 3, 301–316. [Google Scholar] [CrossRef]
- da Silva, M.A.C.; Cavalett, A.; Spinner, A.; Rosa, D.C.; Jasper, R.B.; Quecine, M.C.; Bonatelli, M.L.; Pizzirani-Kleiner, A.; Corção, G.; Lima, A.O.d.S. Phylogenetic identification of marine bacteria isolated from deep-sea sediments of the eastern South Atlantic Ocean. SpringerPlus 2013, 2, 127. [Google Scholar] [CrossRef]
- Cupit, C.; Lomstein, B.A.; Kjeldsen, K.U. Contrasting community composition of endospores and vegetative Firmicutes in a marine sediment suggests both endogenous and exogenous sources of endospore accumulation. Environ. Microbiol. Rep. 2019, 11, 352–360. [Google Scholar] [CrossRef]
- Li, Y.; Cao, W.; Wang, Y.; Ma, Q. Microbial diversity in the sediments of the southern Mariana Trench. J. Oceanol. Limnol. 2019, 37, 1024–1029. [Google Scholar] [CrossRef]
- Zhao, X.; Luo, H.; He, S.; Yang, B.; Wei, T.; Hu, Y.; Wang, Z.; Li, X. Vertical distribution of size-fractionated bacterial communities in the water column of the Atacama Trench. Reg. Stud. Mar. Sci. 2022, 55, 102470. [Google Scholar] [CrossRef]
- Su, H.; Wu, C.; Han, P.; Liu, Z.; Liang, M.; Zhang, Z.; Wang, Z.; Guo, G.; He, X.; Pang, J.; et al. The microbiome and its association with antibiotic resistance genes in the hadal biosphere at the Yap Trench. J. Hazard. Mater. 2022, 439, 129543. [Google Scholar] [CrossRef]
- Ramos, L.R.; Vollú, R.E.; Jurelevicius, D.; Rosado, A.S.; Seldin, L. Firmicutes in different soils of Admiralty Bay, King George Island, Antarctica. Polar Biol. 2019, 42, 2219–2226. [Google Scholar] [CrossRef]
- Govil, T.; Paste, M.; Samanta, D.; David, A.; Goh, K.M.; Li, X.; Salem, D.R.; Sani, R.K. Metagenomics and Culture Dependent Insights into the Distribution of Firmicutes across Two Different Sample Types Located in the Black Hills Region of South Dakota, USA. Microorganisms 2021, 9, 113. [Google Scholar] [CrossRef]
- Horn, D.J.V.; Okie, J.G.; Buelow, H.N.; Gooseff, M.N.; Barrett, J.E.; Takacs-Vesbach, C.D. Soil Microbial Responses to Increased Moisture and Organic Resources along a Salinity Gradient in a Polar Desert. Appl. Environ. Microbiol. 2014, 80, 3034–3043. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, T.; Li, J.; Wu, M.; Liu, G.; Zhang, W.; Zhang, B.; Zhang, S.; Zhang, G. High proportions of radiation-resistant strains in Culturable bacteria from the Taklimakan Desert. Biology 2022, 11, 501. [Google Scholar] [CrossRef]
- Frank, Y.; Banks, D.; Avakian, M.; Antsiferov, D.; Kadychagov, P.; Karnachuk, O. Firmicutes is an Important Component of Microbial Communities in Water-Injected and Pristine Oil Reservoirs, Western Siberia, Russia. Geomicrobiol. J. 2016, 33, 387–400. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, Y.; Zhao, L.; Li, Y.; Xie, S.; Liu, Y. Distribution of sediment bacterial and archaeal communities in plateau freshwater lakes. Appl. Microbiol. Biotechnol. 2015, 99, 3291–3302. [Google Scholar] [CrossRef]
- Magnusson, K.R.; Hauck, L.; Jeffrey, B.M.; Elias, V.; Humphrey, A.; Nath, R.; Perrone, A.; Bermudez, L.E. Relationships between diet-related changes in the gut microbiome and cognitive flexibility. Neuroscience 2015, 300, 128–140. [Google Scholar] [CrossRef]
- Gomes, B.C.; Adorno, M.A.T.; Okada, D.Y.; Delforno, T.P.; Lima Gomes, P.C.F.; Sakamoto, I.K.; Varesche, M.B.A. Analysis of a microbial community associated with polychlorinated biphenyl degradation in anaerobic batch reactors. Biodegradation 2014, 25, 797–810. [Google Scholar] [CrossRef]
- Hashmi, I.; Bindschedler, S.; Junier, P. Chapter 18—Firmicutes. In Beneficial Microbes in Agro-Ecology; Amaresan, N., Senthil Kumar, M., Annapurna, K., Kumar, K., Sankaranarayanan, A., Eds.; Academic Press: Cambridge, MA, USA, 2020; pp. 363–396. [Google Scholar]
- Fuentes, S.; Méndez, V.; Aguila, P.; Seeger, M. Bioremediation of petroleum hydrocarbons: Catabolic genes, microbial communities, and applications. Appl. Microbiol. Biotechnol. 2014, 98, 4781–4794. [Google Scholar] [CrossRef]
- Dworkin, M.M.; Falkow, S.; Rosenberg, E.; Schleifer, K.-H.; Stackebrandt, E. Prokaryotes: A Handbook on the Biology of Bacteria: Vol. 3: Archaea and Bacteria: Firmicutes, Actinomycetes; Springer: New York, NY, USA, 2007. [Google Scholar]
- Choi, Y.J.; Lee, S.Y. Microbial production of short-chain alkanes. Nature 2013, 502, 571–574. [Google Scholar] [CrossRef]
- Kim, W.; Lee, J.-H.; Kwon, K.K. Abyssisolibacter fermentans gen. nov. sp. nov., isolated from deep sub-seafloor sediment. J. Microbiol. 2016, 54, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Jing, H.; Wang, R.; Jiang, Q.; Zhang, Y.; Peng, X. Anaerobic methane oxidation coupled to denitrification is an important potential methane sink in deep-sea cold seeps. Sci. Total Environ. 2020, 748, 142459. [Google Scholar] [CrossRef] [PubMed]
- Feng, D.; Chen, D. Authigenic carbonates from an active cold seep of the northern South China Sea: New insights into fluid sources and past seepage activity. Deep Sea Res. Part II Top. Stud. Oceanogr. 2015, 122, 74–83. [Google Scholar] [CrossRef]
- Liang, Q.; Hu, Y.; Feng, D.; Peckmann, J.; Chen, L.; Yang, S.; Liang, J.; Tao, J.; Chen, D. Authigenic carbonates from newly discovered active cold seeps on the northwestern slope of the South China Sea: Constraints on fluid sources, formation environments, and seepage dynamics. Deep Sea Res. Part I Oceanogr. Res. Pap. 2017, 124, 31–41. [Google Scholar] [CrossRef]
- Zhao, Y.; Xu, T.; Law, Y.S.; Feng, D.; Li, N.; Xin, R.; Wang, H.; Ji, F.; Zhou, H.; Qiu, J.-W. Ecological characterization of cold-seep epifauna in the South China Sea. Deep Sea Res. Part I Oceanogr. Res. Pap. 2020, 163, 103361. [Google Scholar] [CrossRef]
- Liu, C.S.; Morita, S.; Liao, Y.-H.; Ku, C.K.; Machiyama, H.; Lin, S.; Soh, W. High-resolution seismic images of the Formosa Ridge off Southwestern Taiwan where “hydrothermal” chemosynthetic community is present at a cold seep site. In Proceedings of the 6th International Conference on Gas Hydrates (ICGH 2008), Vancouver, BC, Canada, 6–10 July 2008. [Google Scholar] [CrossRef]
- Lin, S.; Lim, Y.; Liu, C.S.; Yang, T.; Chen, Y.G.; Machiyama, H.; Soh, W. Formosa Ridge, a cold seep with densely populated chemosynthetic community in the passive margin, southwest of Taiwan. Geochim. Cosmochim. Acta 2007, 71. [Google Scholar]
- Zhang, H.; Wang, M.; Wang, H.; Chen, H.; Cao, L.; Zhong, Z.; Lian, C.; Zhou, L.; Li, C. Metagenome sequencing and 768 microbial genomes from cold seep in South China Sea. Sci. Data 2022, 9, 480. [Google Scholar] [CrossRef]
- Balch, W.E.; Fox, G.E.; Magrum, L.J.; Woese, C.R.; Wolfe, R.S. Methanogens: Reevaluation of a unique biological group. Microbiol. Rev. 1979, 43, 260–296. [Google Scholar] [CrossRef]
- Widdel, F.; Bak, F. Gram-negative mesotrophic sulfate-reducing bacteria. In The Prokaryotes; Balows, A., Trüper, H.G., Dworkin, M., Harder, W., Schleifer, K., Eds.; Springer: New York, NY, USA, 1992. [Google Scholar]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef]
- Tatusova, T.; DiCuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2016, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef] [PubMed]
- Tatusov, R.L.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Kiryutin, B.; Koonin, E.V.; Krylov, D.M.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; et al. The COG database: An updated version includes eukaryotes. BMC Bioinform. 2003, 4, 41. [Google Scholar] [CrossRef] [PubMed]
- Lombard, V.; Golaconda Ramulu, H.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2013, 42, D490–D495. [Google Scholar] [CrossRef] [PubMed]
- Bertelli, C.; Laird, M.R.; Williams, K.P.; Simon Fraser University Research Computing Group; Lau, B.Y.; Hoad, G.; Winsor, G.L.; Brinkman, F.S. IslandViewer 4: Expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017, 45, W30–W35. [Google Scholar] [CrossRef]
- Elbourne, L.D.H.; Tetu, S.G.; Hassan, K.A.; Paulsen, I.T. TransportDB 2.0: A database for exploring membrane transporters in sequenced genomes from all domains of life. Nucleic Acids Res. 2016, 45, D320–D324. [Google Scholar] [CrossRef]
- Rawlings, N.D.; Barrett, A.J.; Finn, R. Twenty years of the MEROPS database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 2015, 44, D343–D350. [Google Scholar] [CrossRef]
- Almagro Armenteros, J.J.; Tsirigos, K.D.; Sønderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef]
- Chaumeil, P.-A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk: A toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 2019, 36, 1925–1927. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Higgins, D.G. The Clustal Omega Multiple Alignment Package. In Multiple Sequence Alignment: Methods and Protocols; Katoh, K., Ed.; Springer: New York, NY, USA, 2021; pp. 3–16. [Google Scholar]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. barrnap 0.9: Rapid Ribosomal RNA Prediction. 2018. Available online: https://github.com/tseemann/barrnap (accessed on 5 April 2023).
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Kim, M.; Oh, H.-S.; Park, S.-C.; Chun, J. Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int. J. Syst. Evol. Microbiol. 2014, 64, 346–351. [Google Scholar] [CrossRef]
- Gaffney, J.; Embree, J.; Gilmore, S.; Embree, M. Chordicoccus furentiruminis, gen. nov., sp. nov., a novel succinic acid producing bacterium isolated from a steer on a high grain diet. Int. J. Syst. Evol. Microbiol. 2023, 73, 005751. [Google Scholar] [CrossRef]
- Richter, M.; Rosselló-Móra, R.; Oliver Glöckner, F.; Peplies, J. JSpeciesWS: A web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 2015, 32, 929–931. [Google Scholar] [CrossRef]
- Konstantinidis, K.T.; Tiedje, J.M. Towards a Genome-Based Taxonomy for Prokaryotes. J. Bacteriol. 2005, 187, 6258–6264. [Google Scholar] [CrossRef]
- Lagkouvardos, I.; Joseph, D.; Kapfhammer, M.; Giritli, S.; Horn, M.; Haller, D.; Clavel, T. IMNGS: A comprehensive open resource of processed 16S rRNA microbial profiles for ecology and diversity studies. Sci. Rep. 2016, 6, 33721. [Google Scholar] [CrossRef]
- Schlitzer, R. Ocean Data View. 2015. Available online: https://odv.awi.de/ (accessed on 24 May 2023).
- Vesth, T.; Ozen, A.; Andersen, S.C.; Kaas, R.S.; Lukjancenko, O.; Bohlin, J.; Nookaew, I.; Wassenaar, T.M.; Ussery, D.W. Veillonella, Firmicutes: Microbes disguised as Gram negatives. Stand. Genom. Sci. 2013, 9, 431–448. [Google Scholar] [CrossRef]
- Crost, E.H.; Tailford, L.E.; Le Gall, G.; Fons, M.; Henrissat, B.; Juge, N. Utilisation of Mucin Glycans by the Human Gut Symbiont Ruminococcus gnavus Is Strain-Dependent. PLoS ONE 2013, 8, e76341. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, N.D.; Barrett, A.J.; Bateman, A. MEROPS: The peptidase database. Nucleic Acids Res. 2009, 38, D227–D233. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lu, Y.; Nawaz, M.Z.; Xu, J. Comparative Genomics Reveals Evidence of Genome Reduction and High Extracellular Protein Degradation Potential in Kangiella. Front. Microbiol. 2018, 9, 01224. [Google Scholar] [CrossRef]
- Miyamoto, T.; Homma, H. D-Amino acid metabolism in bacteria. J. Biochem. 2021, 170, 5–13. [Google Scholar] [CrossRef]
- Kubota, T.; Kobayashi, T.; Nunoura, T.; Maruyama, F.; Deguchi, S. Enantioselective Utilization of D-Amino Acids by Deep-Sea Microorganisms. Front. Microbiol. 2016, 7, 00511. [Google Scholar] [CrossRef] [PubMed]
- Tempelaars, M.H.; Rodrigues, S.; Abee, T. Comparative Analysis of Antimicrobial Activities of Valinomycin and Cereulide, the Bacillus cereus Emetic Toxin. Appl. Environ. Microbiol. 2011, 77, 2755–2762. [Google Scholar] [CrossRef] [PubMed]
- Stepanauskas, R.; Laudon, H.; Jørgensen, N.O.G. High DON bioavailability in boreal streams during a spring flood. Limnol. Oceanogr. 2000, 45, 1298–1307. [Google Scholar] [CrossRef]
- Dekas, A.E.; Fike, D.A.; Chadwick, G.L.; Green-Saxena, A.; Fortney, J.; Connon, S.A.; Dawson, K.S.; Orphan, V.J. Widespread nitrogen fixation in sediments from diverse deep-sea sites of elevated carbon loading. Environ. Microbiol. 2018, 20, 4281–4296. [Google Scholar] [CrossRef]
- Dekas, A.E.; Chadwick, G.L.; Bowles, M.W.; Joye, S.B.; Orphan, V.J. Spatial distribution of nitrogen fixation in methane seep sediment and the role of the ANME archaea. Environ. Microbiol. 2014, 16, 3012–3029. [Google Scholar] [CrossRef]
- Tsoy, O.; Ravcheev, D.; Mushegian, A. Comparative Genomics of Ethanolamine Utilization. J. Bacteriol. 2009, 191, 7157–7164. [Google Scholar] [CrossRef]
- Garsin, D.A. Ethanolamine utilization in bacterial pathogens: Roles and regulation. Nat. Rev. Microbiol. 2010, 8, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Kaval, K.G.; Garsin, D.A. Ethanolamine Utilization in Bacteria. mBio 2018, 9, e00066-18. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.D.; Itoh, Y.; Nakada, Y.; Jiang, Y. Functional analysis and regulation of the divergent spuABCDEFGH-spuI operons for polyamine uptake and utilization in pseudomonas aeruginosa PAO1. J. Bacteriol. 2002, 184, 3765–3773. [Google Scholar] [CrossRef] [PubMed]
- Mou, X.; Vila-Costa, M.; Sun, S.; Zhao, W.; Sharma, S.; Moran, M.A. Metatranscriptomic signature of exogenous polyamine utilization by coastal bacterioplankton. Environ. Microbiol. Rep. 2011, 3, 798–806. [Google Scholar] [CrossRef]
- Lu, X.; Sun, S.; Hollibaugh, J.T.; Mou, X. Identification of polyamine-responsive bacterioplankton taxa in South Atlantic Bight. Environ. Microbiol. Rep. 2015, 7, 831–838. [Google Scholar] [CrossRef]
- Dong, X.; Zhang, C.; Peng, Y.; Zhang, H.-X.; Shi, L.-D.; Wei, G.; Hubert, C.R.J.; Wang, Y.; Greening, C. Phylogenetically and catabolically diverse diazotrophs reside in deep-sea cold seep sediments. Nat. Commun. 2022, 13, 4885. [Google Scholar] [CrossRef]
- Kapili, B.J.; Barnett, S.E.; Buckley, D.H.; Dekas, A.E. Evidence for phylogenetically and catabolically diverse active diazotrophs in deep-sea sediment. ISME J. 2020, 14, 971–983. [Google Scholar] [CrossRef]
- Andreesen, J.R. Glycine reductase mechanism. Curr. Opin. Chem. Biol. 2004, 8, 454–461. [Google Scholar] [CrossRef]
- Wagner, M.; Sonntag, D.; Grimm, R.; Pich, A.; Eckerskorn, C.; Söhling, B.; Andreesen, J.R. Substrate-specific selenoprotein B of glycine reductase from Eubacterium acidaminophilum. Eur. J. Biochem. 1999, 260, 38–49. [Google Scholar] [CrossRef]
- Oger, P.M.; Jebbar, M. The many ways of coping with pressure. Res. Microbiol. 2010, 161, 799–809. [Google Scholar] [CrossRef]
- Yancey, P.H. Organic osmolytes as compatible, metabolic and counteracting cytoprotectants in high osmolarity and other stresses. J. Exp. Biol. 2005, 208, 2819–2830. [Google Scholar] [CrossRef]
- Chattopadhyay, M.K. The cryoprotective effects of glycine betaine on bacteria. Trends Microbiol. 2002, 10, 311. [Google Scholar] [CrossRef]
- Martin, D.; Bartlett, D.H.; Roberts, M.F. Solute accumulation in the deep-sea bacterium Photobacterium profundum. Extremophiles 2002, 6, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Rath, H.; Sappa, P.K.; Hoffmann, T.; Gesell Salazar, M.; Reder, A.; Steil, L.; Hecker, M.; Bremer, E.; Mäder, U.; Völker, U. Impact of high salinity and the compatible solute glycine betaine on gene expression of Bacillus subtilis. Environ. Microbiol. 2020, 22, 3266–3286. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, Y.; Liu, Y.; Xie, Z.; Cao, J.; Zhang, H.; Liu, J.; Bao, T.; Sun, C.; Liu, B.; et al. The phylogeny and metabolic potentials of an n-alkane-degrading Venatorbacter bacterium isolated from deep-sea sediment of the Mariana Trench. Front. Microbiol. 2023, 14, 1108651. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wang, J.; Liu, J.; Zhang, H.; Bao, T.; Sun, C.; Fang, J.; Cao, J. Genomic Analysis of the Deep-Sea Bacterium Shewanella sp. MTB7 Reveals Backgrounds Related to Its Deep-Sea Environment Adaptation. Microorganisms 2023, 11, 798. [Google Scholar] [CrossRef]
- Ashraf, M.; Foolad, M.R. Roles of glycine betaine and proline in improving plant abiotic stress resistance. Environ. Exp. Bot. 2007, 59, 206–216. [Google Scholar] [CrossRef]
- Simonato, F.; Campanaro, S.; Lauro, F.M.; Vezzi, A.; D’Angelo, M.; Vitulo, N.; Valle, G.; Bartlett, D.H. Piezophilic adaptation: A genomic point of view. J. Biotechnol. 2006, 126, 11–25. [Google Scholar] [CrossRef]
- Jin, S.; Wang, Y.; Zhao, X. Cold-adaptive mechanism of psychrophilic bacteria in food and its application. Microb. Pathog. 2022, 169, 105652. [Google Scholar] [CrossRef]
- Xie, Z.; Jian, H.; Jin, Z.; Xiao, X. Enhancing the Adaptability of the Deep-Sea Bacterium Shewanella piezotolerans WP3 to High Pressure and Low Temperature by Experimental Evolution under H2O2 Stress. Appl. Environ. Microbiol. 2018, 84, e02342-17. [Google Scholar] [CrossRef]
- Fimlaid, K.A.; Shen, A. Diverse mechanisms regulate sporulation sigma factor activity in the Firmicutes. Curr. Opin. Microbiol. 2015, 24, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Browne, H.P.; Almeida, A.; Kumar, N.; Vervier, K.; Adoum, A.T.; Viciani, E.; Dawson, N.J.R.; Forster, S.C.; Cormie, C.; Goulding, D.; et al. Host adaptation in gut Firmicutes is associated with sporulation loss and altered transmission cycle. Genome Biol. 2021, 22, 204. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Items | M8S5 |
|---|---|
| Size (bp) | 4,683,267 |
| G+C content (%) | 30.3 |
| Coding sequence (%) | 83.76 |
| Total genes | 4376 |
| Protein-coding genes | 4250 |
| Pseudo genes | 24 |
| Genes assigned to COG | 1575 |
| rRNA operons | 7 |
| tRNA genes | 77 |
| ncRNA genes | 4 |
| Gene islands | 20 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Chen, S.; Wang, J.; Shao, B.; Fang, J.; Cao, J. The Phylogeny, Metabolic Potentials, and Environmental Adaptation of an Anaerobe, Abyssisolibacter sp. M8S5, Isolated from Cold Seep Sediments of the South China Sea. Microorganisms 2023, 11, 2156. https://doi.org/10.3390/microorganisms11092156
Liu Y, Chen S, Wang J, Shao B, Fang J, Cao J. The Phylogeny, Metabolic Potentials, and Environmental Adaptation of an Anaerobe, Abyssisolibacter sp. M8S5, Isolated from Cold Seep Sediments of the South China Sea. Microorganisms. 2023; 11(9):2156. https://doi.org/10.3390/microorganisms11092156
Chicago/Turabian StyleLiu, Ying, Songze Chen, Jiahua Wang, Baoying Shao, Jiasong Fang, and Junwei Cao. 2023. "The Phylogeny, Metabolic Potentials, and Environmental Adaptation of an Anaerobe, Abyssisolibacter sp. M8S5, Isolated from Cold Seep Sediments of the South China Sea" Microorganisms 11, no. 9: 2156. https://doi.org/10.3390/microorganisms11092156
APA StyleLiu, Y., Chen, S., Wang, J., Shao, B., Fang, J., & Cao, J. (2023). The Phylogeny, Metabolic Potentials, and Environmental Adaptation of an Anaerobe, Abyssisolibacter sp. M8S5, Isolated from Cold Seep Sediments of the South China Sea. Microorganisms, 11(9), 2156. https://doi.org/10.3390/microorganisms11092156