
Prevalence and Genetic Diversity of Cross-Assembly Phages in Wastewater Treatment Plants in Riyadh, Saudi Arabia


Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Viral Concentration
2.3. PCR Primer Design
2.4. DNA Extraction and PCR
2.5. Agarose Gel Electrophoresis
2.6. Amplicon Purification and Sequencing
2.7. Phylogenetic Analysis
2.8. Statistical Analysis
2.9. Prevalence of crAssphage in Different Sampling Areas
3. Results
3.1. Prevalence of crAssphage in Different Sampling Areas
3.2. Phylogenetic Analysis for the MCP Gene Sequences
3.3. Phylogenetic Analysis for the MP-PBD Gene Sequences
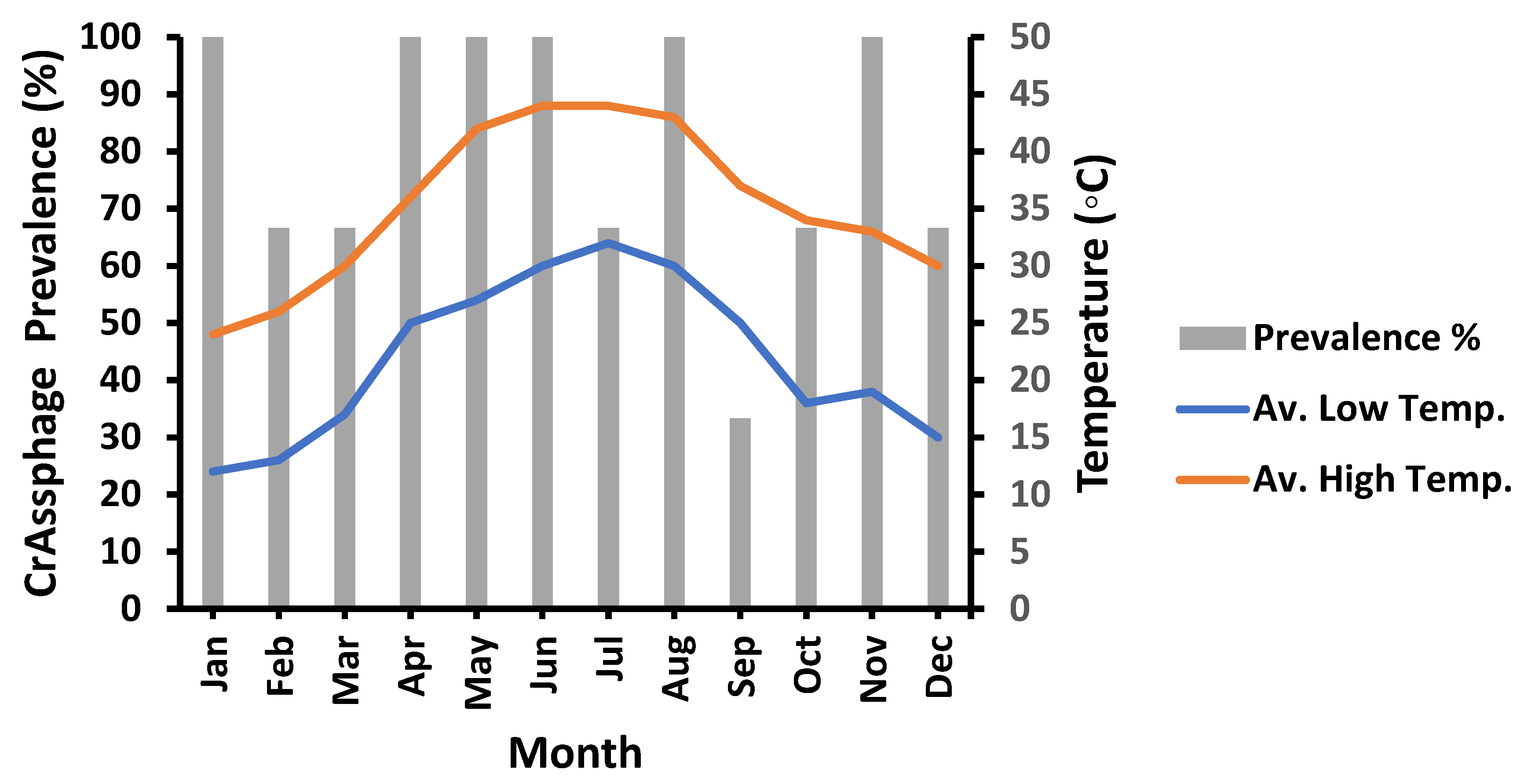
3.4. Meteorological Influences on crAssphage Prevalence
3.5. Temperature Variation’s Role on crAssphage Prevalence
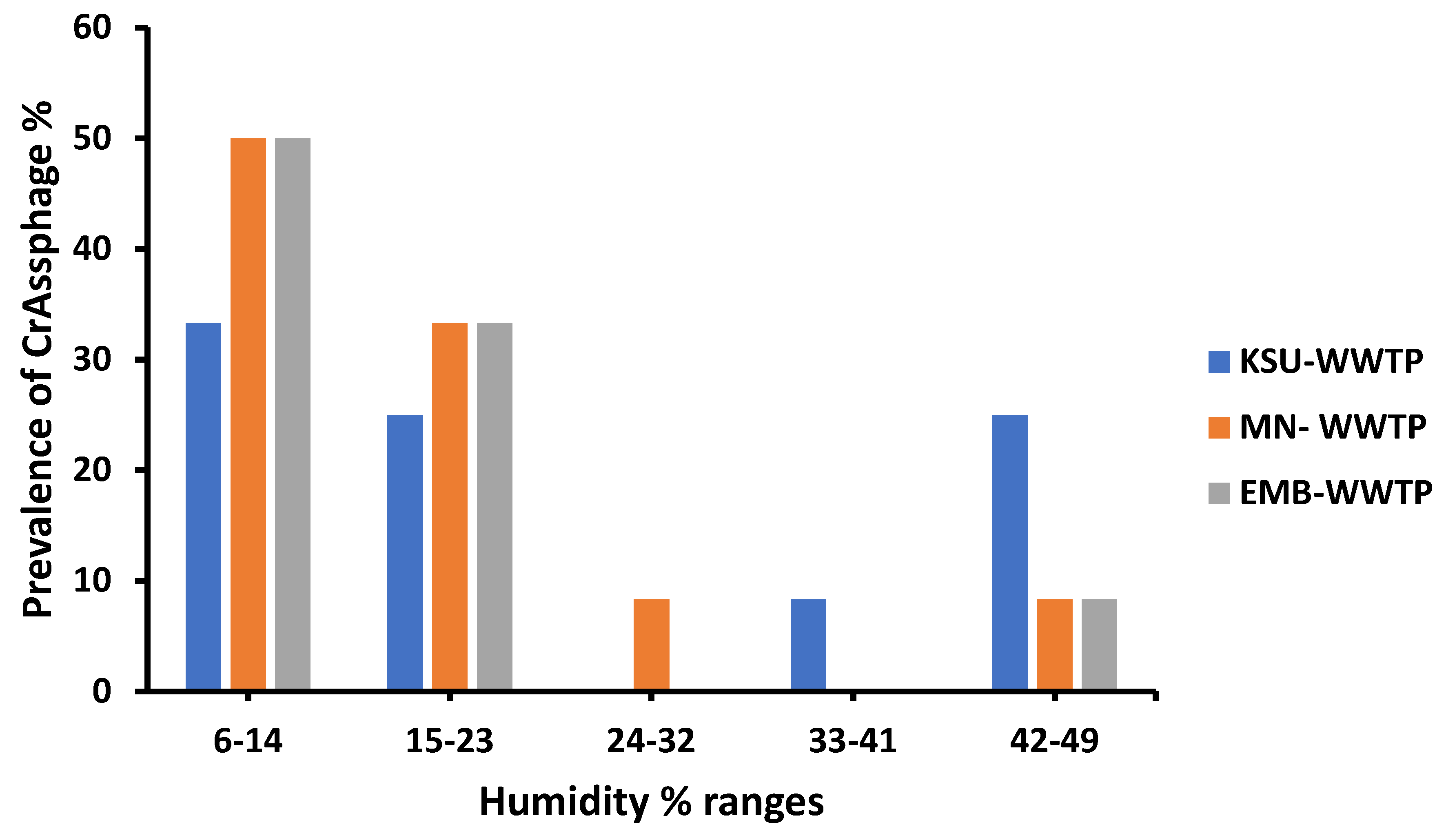
3.6. Humidity Variations Impact on crAssphage Prevalence
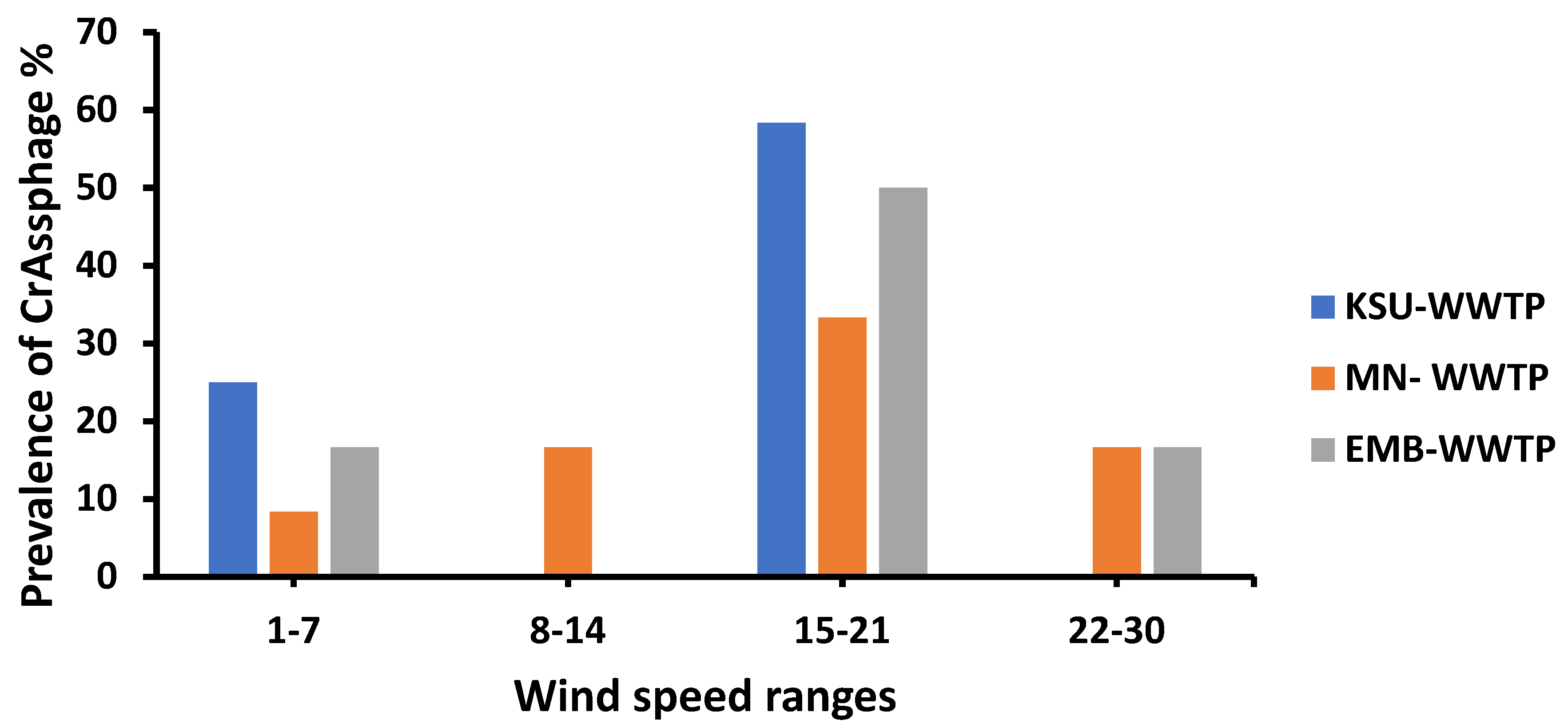
3.7. Wind Speed Influence on the Prevalence of crAssphage
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Strange, J.E.S.; Leekitcharoenphon, P.; Møller, F.D.; Aarestrup, F.M. Metagenomics Analysis of Bacteriophages and Antimicrobial Resistance from Global Urban Sewage. Sci. Rep. 2021, 11, 1600. [Google Scholar] [CrossRef]
- Letarov, A.V. History of Early Bacteriophage Research and Emergence of Key Concepts in Virology. Biochem. Mosc. 2020, 85, 1093–1112. [Google Scholar] [CrossRef] [PubMed]
- Al-Shayeb, B.; Sachdeva, R.; Chen, L.-X.; Ward, F.; Munk, P.; Devoto, A.; Castelle, C.J.; Olm, M.R.; Bouma-Gregson, K.; Amano, Y.; et al. Clades of Huge Phages from across Earth’s Ecosystems. Nature 2020, 578, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Mavrich, T.N.; Hatfull, G.F. Bacteriophage Evolution Differs by Host, Lifestyle and Genome. Nat. Microbiol. 2017, 2, 17112. [Google Scholar] [CrossRef]
- Principi, N.; Silvestri, E.; Esposito, S. Advantages and Limitations of Bacteriophages for the Treatment of Bacterial Infections. Front. Pharmacol. 2019, 10, 513. [Google Scholar] [CrossRef]
- Jassim, S.A.A.; Limoges, R.G. Bacteriophages: Practical Applications for Nature’s Biocontrol; Springer International Publishing: Cham, Switzerland, 2017; ISBN 978-3-319-54050-4. [Google Scholar]
- Simmons, E.L.; Drescher, K.; Nadell, C.D.; Bucci, V. Phage Mobility Is a Core Determinant of Phage–Bacteria Coexistence in Biofilms. ISME J. 2018, 12, 531–543. [Google Scholar] [CrossRef]
- Martinson, J.N.V.; Walk, S.T. Escherichia Coli Residency in the Gut of Healthy Human Adults. EcoSal Plus 2020, 9, 10. [Google Scholar] [CrossRef]
- Yuan, Y.; Gao, M. Jumbo Bacteriophages: An Overview. Front. Microbiol. 2017, 8, 403. [Google Scholar] [CrossRef] [PubMed]
- Ahlgren, N.A.; Perelman, J.N.; Yeh, Y.; Fuhrman, J.A. Multi-year Dynamics of Fine-scale Marine Cyanobacterial Populations Are More Strongly Explained by Phage Interactions than Abiotic, Bottom-up Factors. Environ. Microbiol. 2019, 21, 2948–2963. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Cao, Y.; Young, B.; Yuen, Y.; Jiang, S.; Melendez, D.; Griffith, J.F.; Stewart, J.R. Decay of Coliphages in Sewage-Contaminated Freshwater: Uncertainty and Seasonal Effects. Environ. Sci. Technol. 2016, 50, 11593–11601. [Google Scholar] [CrossRef] [PubMed]
- Dutilh, B.E.; Cassman, N.; McNair, K.; Sanchez, S.E.; Silva, G.G.; Boling, L.; Barr, J.J.; Speth, D.R.; Seguritan, V.; Aziz, R.K. A Highly Abundant Bacteriophage Discovered in the Unknown Sequences of Human Faecal Metagenomes. Nat. Commun. 2014, 5, 1–11. [Google Scholar] [CrossRef]
- Koonin, E.V.; Yutin, N. The CrAss-like Phage Group: How Metagenomics Reshaped the Human Virome. Trends Microbiol. 2020, 28, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Yutin, N.; Makarova, K.S.; Gussow, A.B.; Krupovic, M.; Segall, A.; Edwards, R.A.; Koonin, E.V. Discovery of an Expansive Bacteriophage Family That Includes the Most Abundant Viruses from the Human Gut. Nat. Microbiol. 2018, 3, 38–46. [Google Scholar] [CrossRef]
- Li, Y.; Gordon, E.; Shean, R.C.; Idle, A.; Deng, X.; Greninger, A.L.; Delwart, E. CrAssphage and Its Bacterial Host in Cat Feces. Sci. Rep. 2021, 11, 815. [Google Scholar] [CrossRef]
- Rossi, A.; Treu, L.; Toppo, S.; Zschach, H.; Campanaro, S.; Dutilh, B.E. Evolutionary Study of the Crassphage Virus at Gene Level. Viruses 2020, 12, 1035. [Google Scholar] [CrossRef] [PubMed]
- Papudeshi, B.; Vega, A.A.; Souza, C.; Giles, S.K.; Mallawaarachchi, V.; Roach, M.J.; An, M.; Jacobson, N.; McNair, K.; Mora, M.F.; et al. Novel CrAssphage Isolates Exhibit Conserved Gene Order and Purifying Selection of the Host Specificity Protein. 2023. Available online: https://www.biorxiv.org/content/10.1101/2023.03.05.531146v2 (accessed on 19 August 2023).
- Park, G.W.; Ng, T.F.F.; Freeland, A.L.; Marconi, V.C.; Boom, J.A.; Staat, M.A.; Montmayeur, A.M.; Browne, H.; Narayanan, J.; Payne, D.C. CrAssphage as a Novel Tool to Detect Human Fecal Contamination on Environmental Surfaces and Hands. Emerg. Infect. Dis. 2020, 26, 1731. [Google Scholar] [CrossRef]
- Liang, Y.Y.; Zhang, W.; Tong, Y.G.; Chen, S.P. CrAssphage Is Not Associated with Diarrhoea and Has High Genetic Diversity. Epidemiol. Infect. 2016, 144, 3549–3553. [Google Scholar] [CrossRef] [PubMed]
- Stachler, E.; Kelty, C.; Sivaganesan, M.; Li, X.; Bibby, K.; Shanks, O.C. Quantitative CrAssphage PCR Assays for Human Fecal Pollution Measurement. Environ. Sci. Technol. 2017, 51, 9146–9154. [Google Scholar] [CrossRef]
- Cinek, O.; Mazankova, K.; Kramna, L.; Odeh, R.; Alassaf, A.; Ibekwe, M.U.; Ahmadov, G.; Mekki, H.; Abdullah, M.A.; Elmahi, B.M.E.; et al. Quantitative CrAssphage Real-Time PCR Assay Derived from Data of Multiple Geographically Distant Populations. J. Med. Virol. 2018, 90, 767–771. [Google Scholar] [CrossRef]
- Ahmed, W.; Lobos, A.; Senkbeil, J.; Peraud, J.; Gallard, J.; Harwood, V.J. Evaluation of the Novel CrAssphage Marker for Sewage Pollution Tracking in Storm Drain Outfalls in Tampa, Florida. Water Res. 2018, 131, 142–150. [Google Scholar] [CrossRef]
- Holm, R.H.; Nagarkar, M.; Yeager, R.A.; Talley, D.; Chaney, A.C.; Rai, J.P.; Mukherjee, A.; Rai, S.N.; Bhatnagar, A.; Smith, T. Surveillance of RNase P, PMMoV, and CrAssphage in Wastewater as Indicators of Human Fecal Concentration across Urban Sewer Neighborhoods, Kentucky. FEMS Microbes 2022, 3, xtac003. [Google Scholar] [CrossRef] [PubMed]
- Tandukar, S.; Sherchan, S.P.; Haramoto, E. Reduction of Human Enteric and Indicator Viruses at a Wastewater Treatment Plant in Southern Louisiana, USA. Food Environ. Virol. 2020, 12, 260–263. [Google Scholar] [CrossRef] [PubMed]
- García-Aljaro, C.; Ballesté, E.; Muniesa, M.; Jofre, J. Determination of CrAssphage in Water Samples and Applicability for Tracking Human Faecal Pollution. Microb. Biotechnol. 2017, 10, 1775–1780. [Google Scholar] [CrossRef]
- Ahmed, W.; Payyappat, S.; Cassidy, M.; Besley, C.; Power, K. Novel CrAssphage Marker Genes Ascertain Sewage Pollution in a Recreational Lake Receiving Urban Stormwater Runoff. Water Res. 2018, 145, 769–778. [Google Scholar] [CrossRef]
- Farkas, K.; Adriaenssens, E.M.; Walker, D.I.; McDonald, J.E.; Malham, S.K.; Jones, D.L. Critical Evaluation of CrAssphage as a Molecular Marker for Human-Derived Wastewater Contamination in the Aquatic Environment. Food Environ. Virol. 2019, 11, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Crank, K.; Li, X.; North, D.; Ferraro, G.B.; Iaconelli, M.; Mancini, P.; La Rosa, G.; Bibby, K. CrAssphage Abundance and Correlation with Molecular Viral Markers in Italian Wastewater. Water Res. 2020, 184, 116161. [Google Scholar] [CrossRef]
- Kitajima, M.; Iker, B.C.; Pepper, I.L.; Gerba, C.P. Relative Abundance and Treatment Reduction of Viruses during Wastewater Treatment Processes—Identification of Potential Viral Indicators. Sci. Total Environ. 2014, 488–489, 290–296. [Google Scholar] [CrossRef]
- Qiu, Y.; Lee, B.E.; Neumann, N.; Ashbolt, N.; Craik, S.; Maal-Bared, R.; Pang, X.L. Assessment of Human Virus Removal during Municipal Wastewater Treatment in Edmonton, Canada. J. Appl. Microbiol. 2015, 119, 1729–1739. [Google Scholar] [CrossRef]
- Sidhu, J.P.S.; Sena, K.; Hodgers, L.; Palmer, A.; Toze, S. Comparative Enteric Viruses and Coliphage Removal during Wastewater Treatment Processes in a Sub-Tropical Environment. Sci. Total Environ. 2018, 616–617, 669–677. [Google Scholar] [CrossRef]
- Stachler, E.; Bibby, K. Metagenomic Evaluation of the Highly Abundant Human Gut Bacteriophage CrAssphage for Source Tracking of Human Fecal Pollution. Environ. Sci. Technol. Lett. 2014, 1, 405–409. [Google Scholar] [CrossRef]
- Ballesté, E.; Pascual-Benito, M.; Martín-Díaz, J.; Blanch, A.R.; Lucena, F.; Muniesa, M.; Jofre, J.; García-Aljaro, C. Dynamics of CrAssphage as a Human Source Tracking Marker in Potentially Faecally Polluted Environments. Water Res. 2019, 155, 233–244. [Google Scholar] [CrossRef]
- Malla, B.; Makise, K.; Nakaya, K.; Mochizuki, T.; Yamada, T.; Haramoto, E. Evaluation of Human-and Animal-Specific Viral Markers and Application of CrAssphage, Pepper Mild Mottle Virus, and Tobacco Mosaic Virus as Potential Fecal Pollution Markers to River Water in Japan. Food Environ. Virol. 2019, 11, 446–452. [Google Scholar] [CrossRef]
- Bivins, A.; Crank, K.; Greaves, J.; North, D.; Wu, Z.; Bibby, K. Cross-Assembly Phage and Pepper Mild Mottle Virus as Viral Water Quality Monitoring Tools—Potential, Research Gaps, and Way Forward. Curr. Opin. Environ. Sci. Health 2020, 16, 54–61. [Google Scholar] [CrossRef]
- Nour, I.; Hanif, A.; Alanazi, F.; Zakri, A.M.; Al-Ashkar, I.; Alhetheel, A.; Eifan, S. Evaluation of Three Different Concentration and Extraction Methods for Recovery Efficiency of Human Adenovirus and Human Rotavirus Virus A. J. Virol. Methods 2021, 295, 114212. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547. [Google Scholar] [CrossRef] [PubMed]
- Cervantes-Echeverría, M.; Equihua-Medina, E.; Cornejo-Granados, F.; Hernández-Reyna, A.; Sánchez, F.; López-Contreras, B.E.; Canizales-Quinteros, S.; Ochoa-Leyva, A. Whole-Genome of Mexican-CrAssphage Isolated from the Human Gut Microbiome. BMC Res. Notes 2018, 11, 902. [Google Scholar] [CrossRef] [PubMed]
- Kongprajug, A.; Mongkolsuk, S.; Sirikanchana, K. CrAssphage as a Potential Human Sewage Marker for Microbial Source Tracking in Southeast Asia. Environ. Sci. Technol. Lett. 2019, 6, 159–164. [Google Scholar] [CrossRef]
- Wilder, M.L.; Middleton, F.; Larsen, D.A.; Du, Q.; Fenty, A.; Zeng, T.; Insaf, T.; Kilaru, P.; Collins, M.; Kmush, B.; et al. Co-Quantification of CrAssphage Increases Confidence in Wastewater-Based Epidemiology for SARS-CoV-2 in Low Prevalence Areas. Water Res. X 2021, 11, 100100. [Google Scholar] [CrossRef]
- Wu, Z.; Greaves, J.; Arp, L.; Stone, D.; Bibby, K. Comparative Fate of CrAssphage with Culturable and Molecular Fecal Pollution Indicators during Activated Sludge Wastewater Treatment. Environ. Int. 2020, 136, 105452. [Google Scholar] [CrossRef]
- Tandukar, S.; Sherchan, S.P.; Haramoto, E. Applicability of CrAssphage, Pepper Mild Mottle Virus, and Tobacco Mosaic Virus as Indicators of Reduction of Enteric Viruses during Wastewater Treatment. Sci. Rep. 2020, 10, 3616. [Google Scholar] [CrossRef]
- Farkas, K.; Walker, D.I.; Adriaenssens, E.M.; McDonald, J.E.; Hillary, L.S.; Malham, S.K.; Jones, D.L. Viral Indicators for Tracking Domestic Wastewater Contamination in the Aquatic Environment. Water Res. 2020, 181, 115926. [Google Scholar] [CrossRef]
- Hamza, I.A.; Bibby, K. Critical Issues in Application of Molecular Methods to Environmental Virology. J. Virol. Methods 2019, 266, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Edwards, R.A.; Vega, A.A.; Norman, H.M.; Ohaeri, M.; Levi, K.; Dinsdale, E.A.; Cinek, O.; Aziz, R.K.; McNair, K.; Barr, J.J.; et al. Global Phylogeography and Ancient Evolution of the Widespread Human Gut Virus CrAssphage. Nat. Microbiol. 2019, 4, 1727–1736. [Google Scholar] [CrossRef] [PubMed]
- Mafumo, N.; Bezuidt, O.K.I.; le Roux, W.; Makhalanyane, T.P. CrAssphage May Be Viable Markers of Contamination in Pristine and Contaminated River Water. mSystems 2023, 8, e01282-22. [Google Scholar] [CrossRef]
- Gulyaeva, A.; Garmaeva, S.; Ruigrok, R.A.A.A.; Wang, D.; Riksen, N.P.; Netea, M.G.; Wijmenga, C.; Weersma, R.K.; Fu, J.; Vila, A.V.; et al. Discovery, Diversity, and Functional Associations of CrAss-like Phages in Human Gut Metagenomes from Four Dutch Cohorts. Cell Rep. 2022, 38, 110204. [Google Scholar] [CrossRef]
- Stachler, E.; Akyon, B.; de Carvalho, N.A.; Ference, C.; Bibby, K. Correlation of CrAssphage QPCR Markers with Culturable and Molecular Indicators of Human Fecal Pollution in an Impacted Urban Watershed. Environ. Sci. Technol. 2018, 52, 7505–7512. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Genes | Primers | Amplicon Size | Reference |
|---|---|---|---|
| MCP | F: 5′-TGACCGTGATACTCAAGATG-3′ R: 5′-GACGTACTATACGAACATTCTG-3′ | 371 bp | Current study |
| MP-PBD | F: 5′-CCTGTTACTRATTCTACTRC-3′ R: 5′-ATTCWTRAAGAGTTCTACGAATCC-3′ | 223 bp |
| Sampling Location | CrAssphage +ve | CrAssphage Prevalence % |
|---|---|---|
| KSU-WWTP | 10 | 83.3% |
| MN-WWTP | 9 | 75% |
| EMB-WWTP | 10 | 83.3% |
| Environmental Factor | R2 | RMSE | Equation |
|---|---|---|---|
| High temperature (TH) | 0.9526 * | 1.8982 | %PrevCrAss = −10.1247 + 0.992 × TH |
| Low temperature (TL) | 0.1629 | 6.94868 | %PrevCrAss = 12.211 + 0.417 × TL |
| Relative humidity (RH%) | 0.62089 ** | 9.8919 | %PrevCrAss = 38.333 − 0.9259 × RH% |
| Wind speed (WS) | 0.03 | 17.404 | %PrevCrAss = 16.0317 + 0.357 × WS |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alotaibi, R.; Eifan, S.; Hanif, A.; Nour, I.; Alkathiri, A. Prevalence and Genetic Diversity of Cross-Assembly Phages in Wastewater Treatment Plants in Riyadh, Saudi Arabia. Microorganisms 2023, 11, 2167. https://doi.org/10.3390/microorganisms11092167
Alotaibi R, Eifan S, Hanif A, Nour I, Alkathiri A. Prevalence and Genetic Diversity of Cross-Assembly Phages in Wastewater Treatment Plants in Riyadh, Saudi Arabia. Microorganisms. 2023; 11(9):2167. https://doi.org/10.3390/microorganisms11092167
Chicago/Turabian StyleAlotaibi, Riyadh, Saleh Eifan, Atif Hanif, Islam Nour, and Abdulrahman Alkathiri. 2023. "Prevalence and Genetic Diversity of Cross-Assembly Phages in Wastewater Treatment Plants in Riyadh, Saudi Arabia" Microorganisms 11, no. 9: 2167. https://doi.org/10.3390/microorganisms11092167
APA StyleAlotaibi, R., Eifan, S., Hanif, A., Nour, I., & Alkathiri, A. (2023). Prevalence and Genetic Diversity of Cross-Assembly Phages in Wastewater Treatment Plants in Riyadh, Saudi Arabia. Microorganisms, 11(9), 2167. https://doi.org/10.3390/microorganisms11092167