
Diverse Small Circular DNA Viruses Identified in an American Wigeon Fecal Sample
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Fecal Sampling and High-Throughput Sequencing
2.2. Sequence Assembly and Identification of Viral Contigs
2.3. Analyses of Cressdnaviruses
2.4. Analyses of Microviruses
3. Results and Discussion
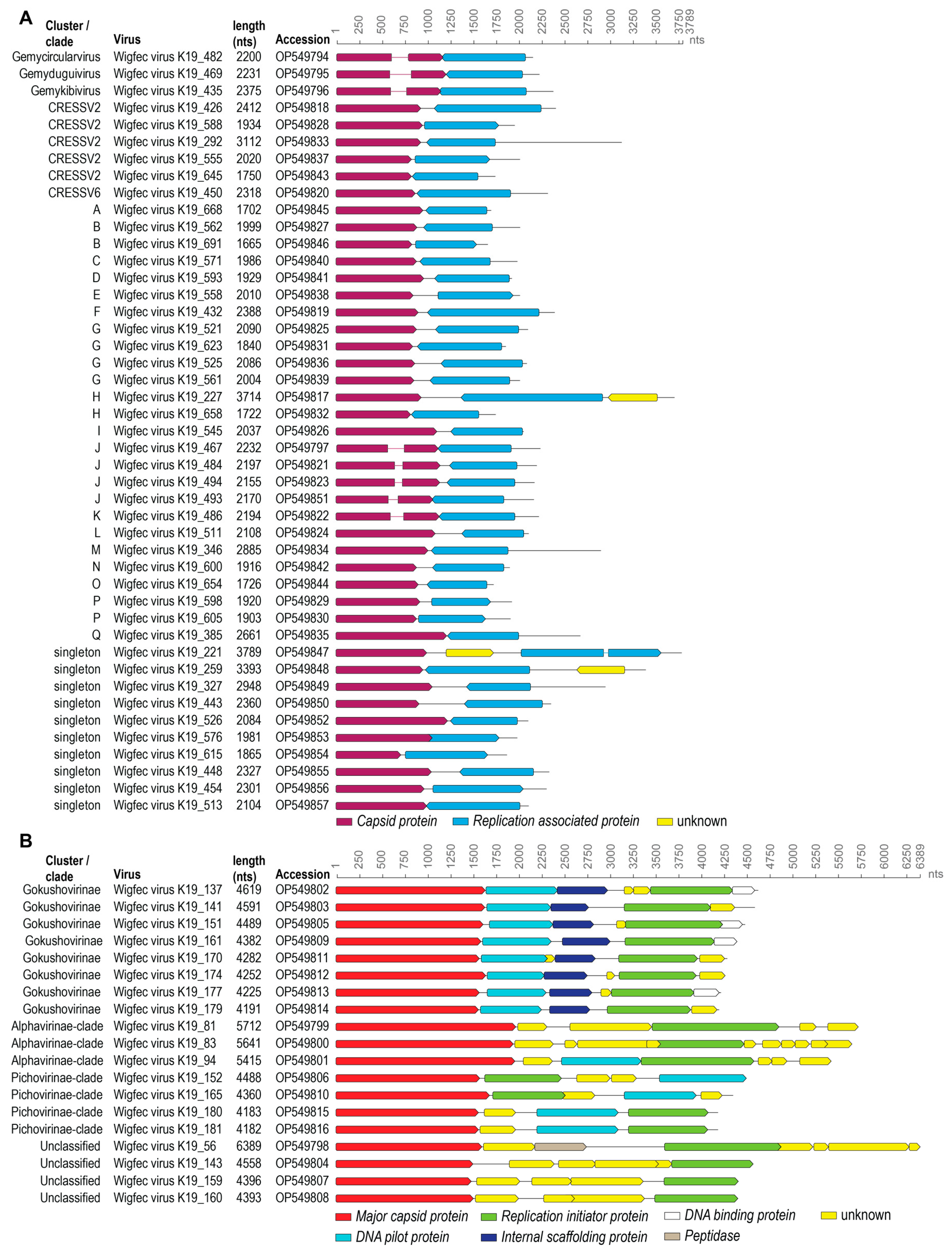
3.1. Identification of Viral Genomes
3.2. Genomoviruses
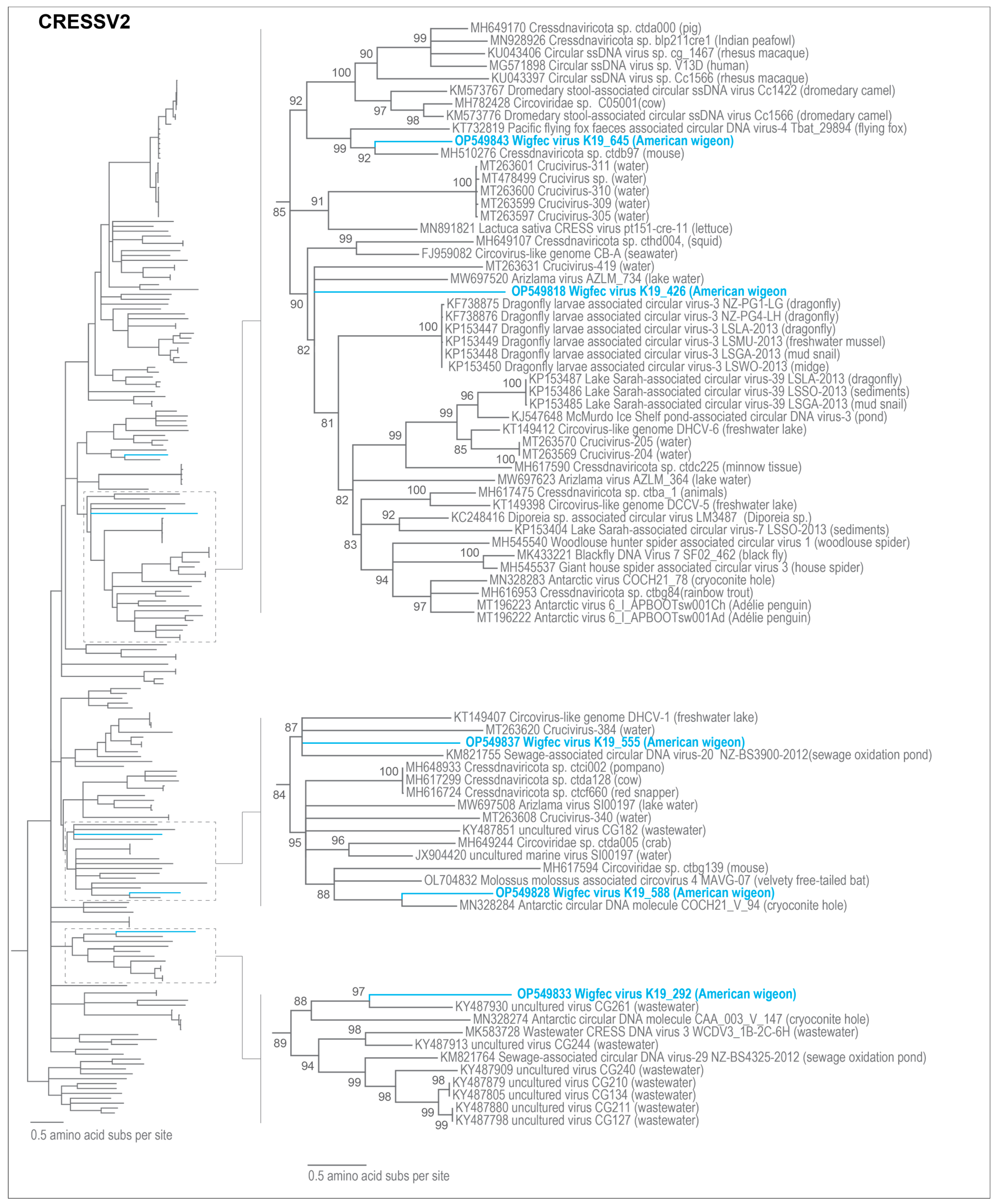
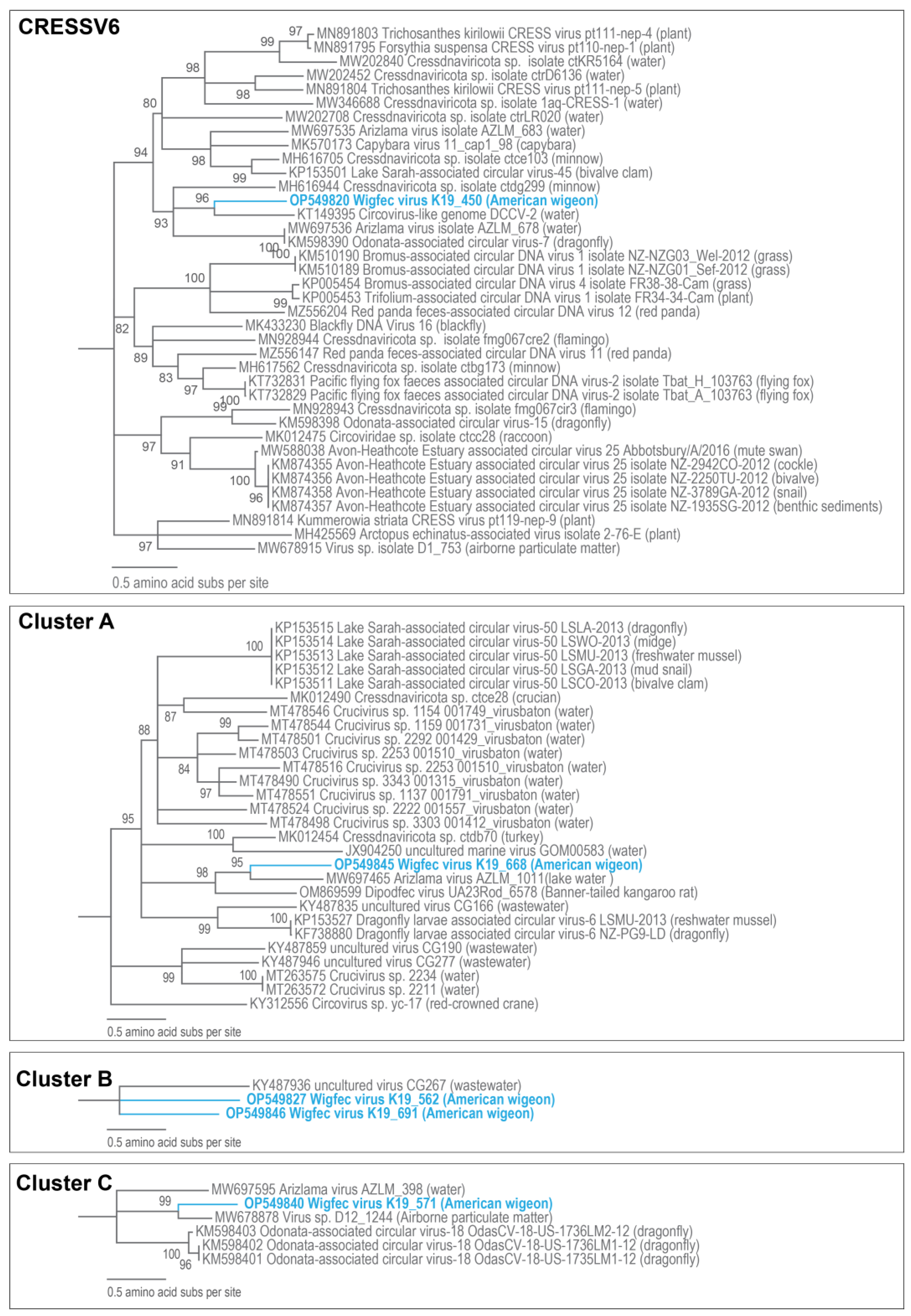
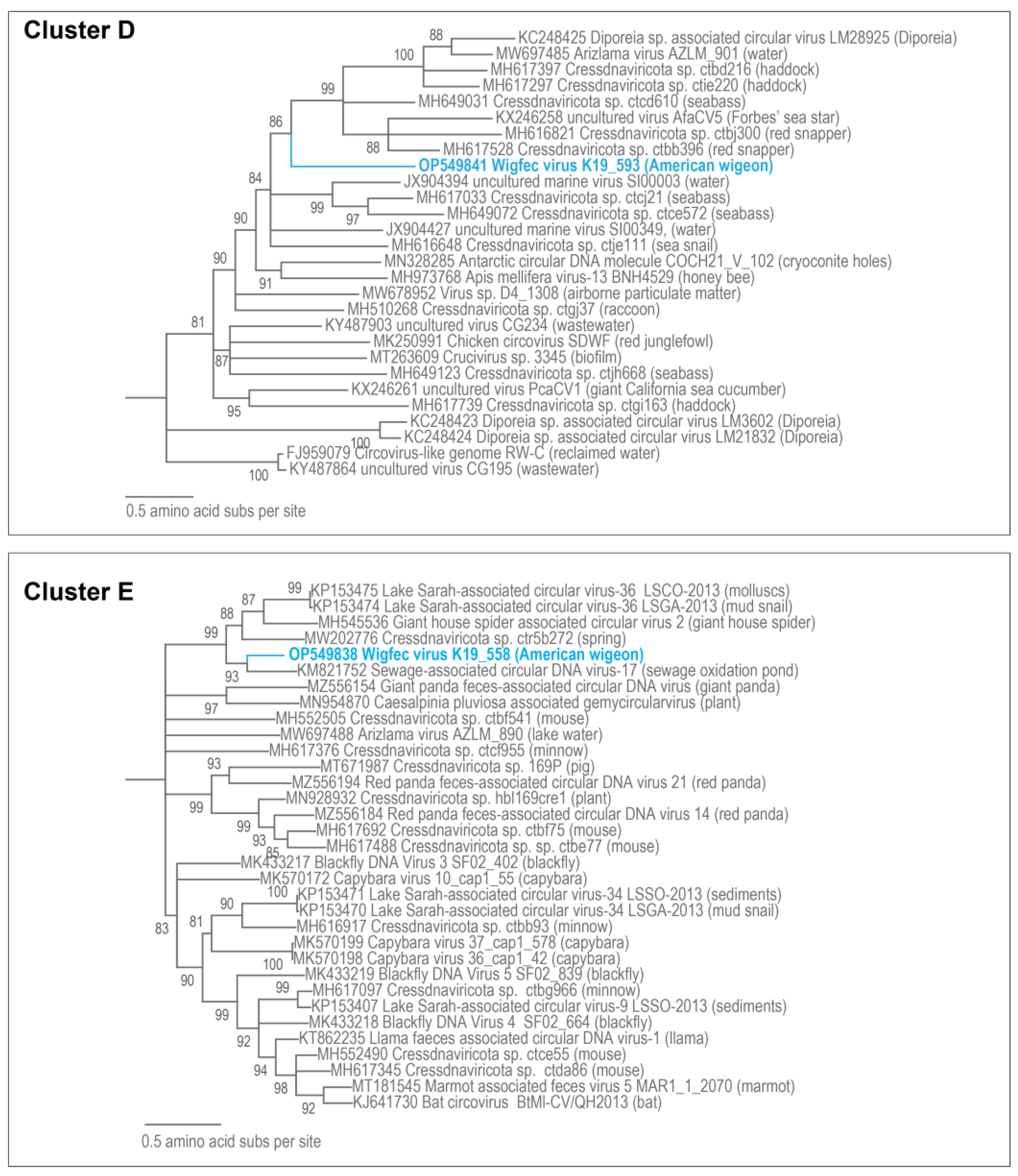
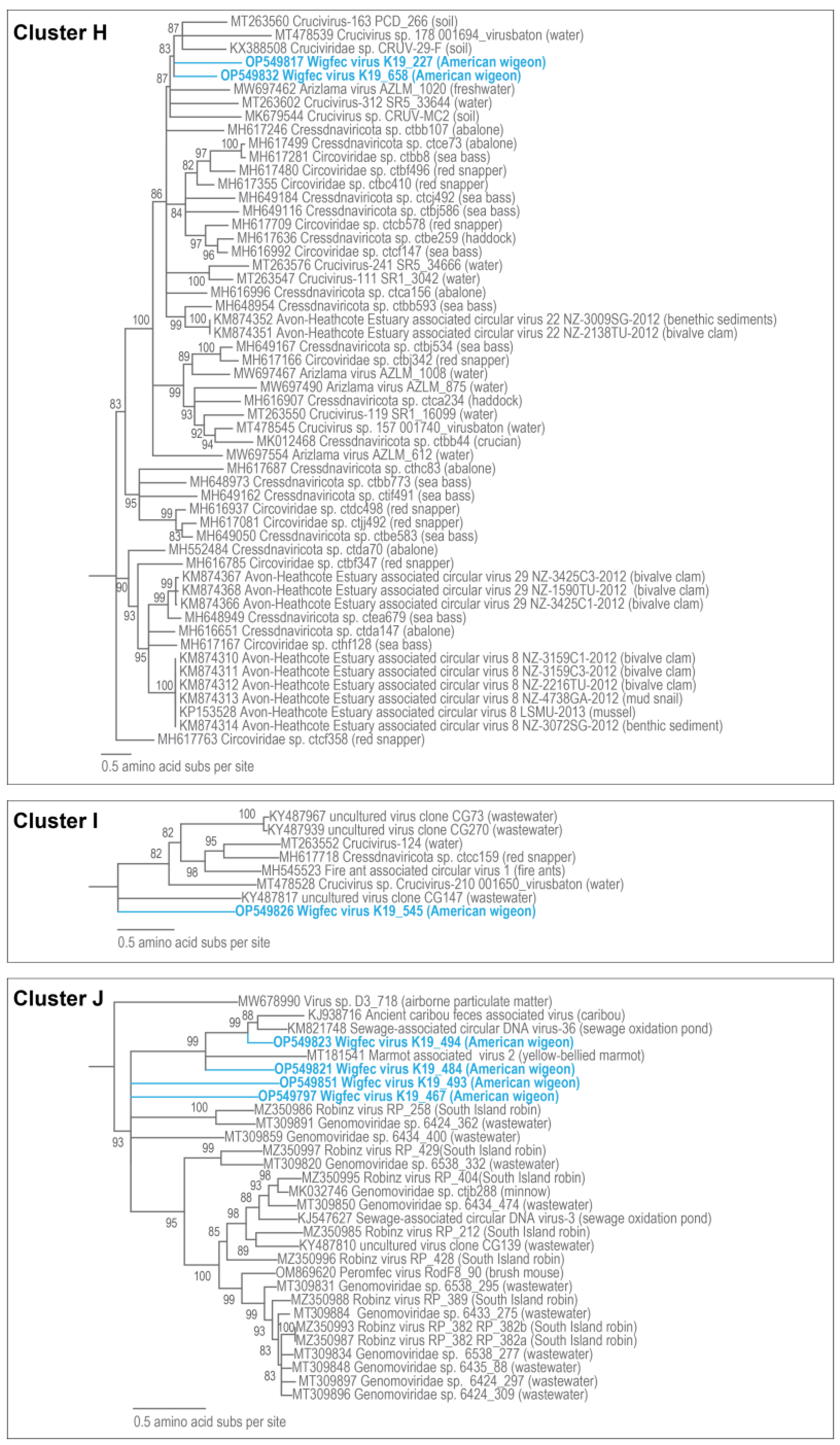
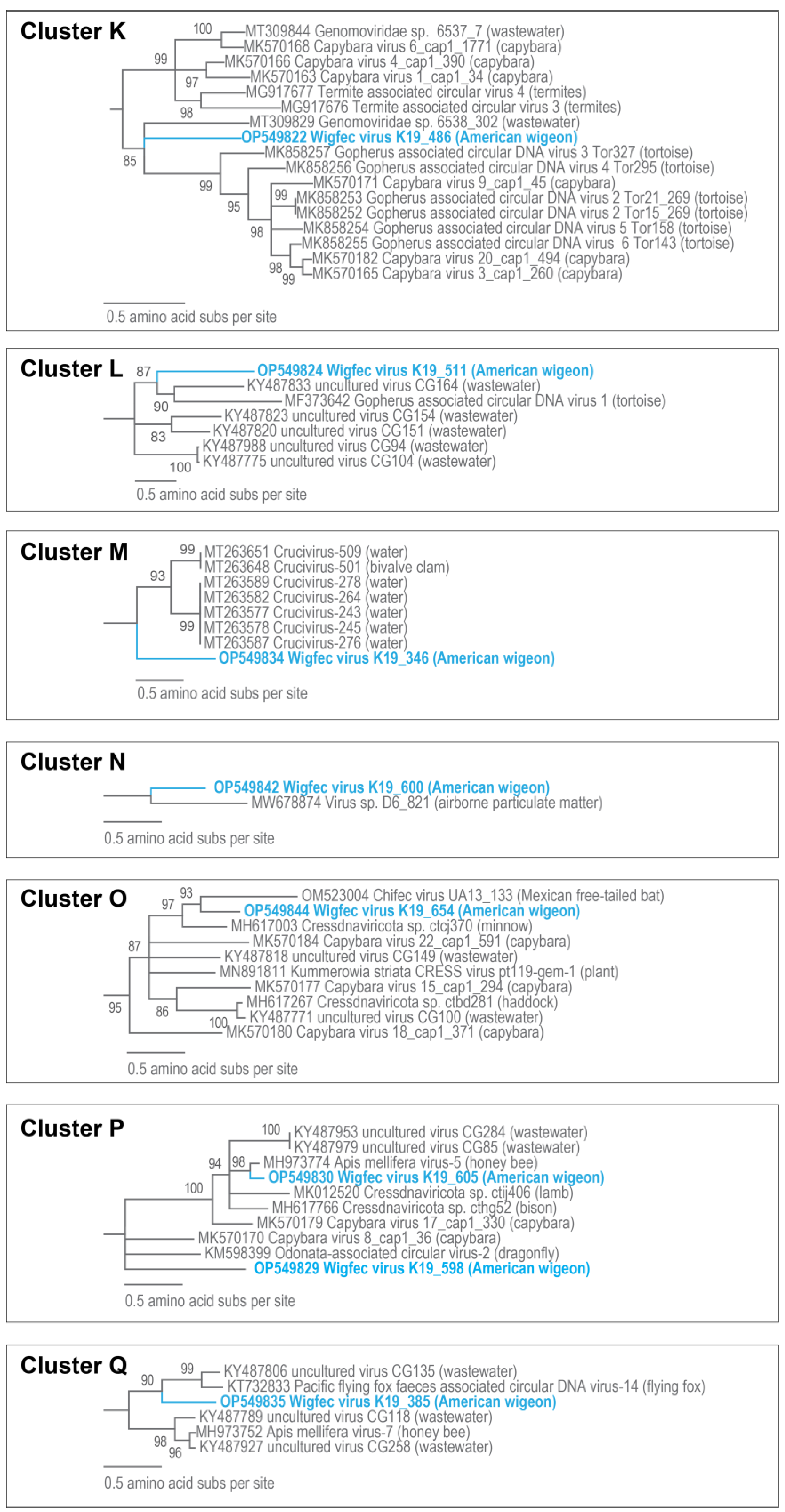
3.3. Unclassified Cressdnaviruses
3.4. Microviruses
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Johnsgard, P.A.; Johnsgard, P.A. Waterfowl of North America; Indiana University Press: Bloomington, Indiana, 1975; p. 575. [Google Scholar]
- Johnson, D.H.; Grier, J.W. Determinants of Breeding Distributions of Ducks. Wildl. Monogr. 1988, 100, 3–37. [Google Scholar]
- de Sobrino, C.N.; Feldheim, C.F.; Arnold, T.W. Distribution and derivation of dabbling duck harvests in the Pacific Flyway. Calif. Fish Game 2017, 103, 118–137. [Google Scholar]
- Dessborn, L.; Brochet, A.L.; Elmberg, J.; Legagneux, P.; Gauthier-Clerc, M.; Guillemain, M. Geographical and temporal patterns in the diet of pintail Anas acuta, wigeon Anas penelope, mallard Anas platyrhynchos and teal Anas crecca in the Western Palearctic. Eur. J. Wildl. Res. 2011, 57, 1119–1129. [Google Scholar] [CrossRef]
- Ip, H.S.; Torchetti, M.K.; Crespo, R.; Kohrs, P.; DeBruyn, P.; Mansfield, K.G.; Baszler, T.; Badcoe, L.; Bodenstein, B.; Shearn-Bochsler, V.; et al. Novel Eurasian highly pathogenic avian influenza A H5 viruses in wild birds, Washington, USA, 2014. Emerg. Infect. Dis. 2015, 21, 886–890. [Google Scholar] [CrossRef] [PubMed]
- Bevins, S.N.; Dusek, R.J.; White, C.L.; Gidlewski, T.; Bodenstein, B.; Mansfield, K.G.; DeBruyn, P.; Kraege, D.; Rowan, E.; Gillin, C.; et al. Widespread detection of highly pathogenic H5 influenza viruses in wild birds from the Pacific Flyway of the United States. Sci. Rep. 2016, 6, 28980. [Google Scholar] [CrossRef] [PubMed]
- Hopken, M.W.; Piaggio, A.J.; Pabilonia, K.L.; Pierce, J.; Anderson, T.; Abdo, Z. Predicting whole genome sequencing success for archived avian influenza virus (Orthomyxoviridae) samples using real-time and droplet PCRs. J. Virol. Methods 2020, 276, 113777. [Google Scholar] [CrossRef] [PubMed]
- Chu, D.K.; Leung, C.Y.; Gilbert, M.; Joyner, P.H.; Ng, E.M.; Tse, T.M.; Guan, Y.; Peiris, J.S.; Poon, L.L. Avian coronavirus in wild aquatic birds. J. Virol. 2011, 85, 12815–12820. [Google Scholar] [CrossRef]
- Khalifeh, A.; Custer, J.M.; Kraberger, S.; Varsani, A. Novel viruses belonging to the family Circoviridae identified in wild American wigeon samples. Arch. Virol. 2021, 166, 3437–3441. [Google Scholar] [CrossRef]
- Krupovic, M.; Varsani, A.; Kazlauskas, D.; Breitbart, M.; Delwart, E.; Rosario, K.; Yutin, N.; Wolf, Y.I.; Harrach, B.; Zerbini, F.M.; et al. Cressdnaviricota: A Virus Phylum Unifying Seven Families of Rep-Encoding Viruses with Single-Stranded, Circular DNA Genomes. J. Virol. 2020, 94, 10–1128. [Google Scholar] [CrossRef]
- Krupovic, M.; Varsani, A. Naryaviridae, Nenyaviridae, and Vilyaviridae: Three new families of single-stranded DNA viruses in the phylum Cressdnaviricota. Arch. Virol. 2022, 167, 2907–2921. [Google Scholar] [CrossRef]
- Walker, P.J.; Siddell, S.G.; Lefkowitz, E.J.; Mushegian, A.R.; Adriaenssens, E.M.; Alfenas-Zerbini, P.; Dempsey, D.M.; Dutilh, B.E.; Garcia, M.L.; Curtis Hendrickson, R.; et al. Recent changes to virus taxonomy ratified by the International Committee on Taxonomy of Viruses (2022). Arch. Virol. 2022, 167, 2429–2440. [Google Scholar] [CrossRef]
- Custer, J.M.; White, R.; Taylor, H.; Schmidlin, K.; Fontenele, R.S.; Stainton, D.; Kraberger, S.; Briskie, J.V.; Varsani, A. Diverse single-stranded DNA viruses identified in New Zealand (Aotearoa) South Island robin (Petroica australis) fecal samples. Virology 2022, 565, 38–51. [Google Scholar] [CrossRef]
- Dayaram, A.; Goldstien, S.; Arguello-Astorga, G.R.; Zawar-Reza, P.; Gomez, C.; Harding, J.S.; Varsani, A. Diverse small circular DNA viruses circulating amongst estuarine molluscs. Infect. Genet. Evol. 2015, 31, 284–295. [Google Scholar] [CrossRef]
- de la Higuera, I.; Kasun, G.W.; Torrance, E.L.; Pratt, A.A.; Maluenda, A.; Colombet, J.; Bisseux, M.; Ravet, V.; Dayaram, A.; Stainton, D.; et al. Unveiling Crucivirus Diversity by Mining Metagenomic Data. mBio 2020, 11, e01410-20. [Google Scholar] [CrossRef]
- Kraberger, S.; Cook, C.N.; Schmidlin, K.; Fontenele, R.S.; Bautista, J.; Smith, B.; Varsani, A. Diverse single-stranded DNA viruses associated with honey bees (Apis mellifera). Infect. Genet. Evol. 2019, 71, 179–188. [Google Scholar] [CrossRef]
- Lund, M.C.; Larsen, B.B.; Rowsey, D.M.; Otto, H.W.; Gryseels, S.; Kraberger, S.; Custer, J.M.; Steger, L.; Yule, K.M.; Harris, R.E.; et al. Using archived and biocollection samples towards deciphering the DNA virus diversity associated with rodent species in the families cricetidae and heteromyidae. Virology 2023, 585, 42–60. [Google Scholar] [CrossRef]
- Nayfach, S.; Paez-Espino, D.; Call, L.; Low, S.J.; Sberro, H.; Ivanova, N.N.; Proal, A.D.; Fischbach, M.A.; Bhatt, A.S.; Hugenholtz, P.; et al. Metagenomic compendium of 189,680 DNA viruses from the human gut microbiome. Nat. Microbiol. 2021, 6, 960–970. [Google Scholar] [CrossRef]
- Orton, J.P.; Morales, M.; Fontenele, R.S.; Schmidlin, K.; Kraberger, S.; Leavitt, D.J.; Webster, T.H.; Wilson, M.A.; Kusumi, K.; Dolby, G.A.; et al. Virus Discovery in Desert Tortoise Fecal Samples: Novel Circular Single-Stranded DNA Viruses. Viruses 2020, 12, 143. [Google Scholar] [CrossRef]
- Tisza, M.J.; Pastrana, D.V.; Welch, N.L.; Stewart, B.; Peretti, A.; Starrett, G.J.; Pang, Y.S.; Krishnamurthy, S.R.; Pesavento, P.A.; McDermott, D.H.; et al. Discovery of several thousand highly diverse circular DNA viruses. Elife 2020, 9, e51971. [Google Scholar] [CrossRef] [PubMed]
- Kinsella, C.M.; Bart, A.; Deijs, M.; Broekhuizen, P.; Kaczorowska, J.; Jebbink, M.F.; van Gool, T.; Cotten, M.; van der Hoek, L. Entamoeba and Giardia parasites implicated as hosts of CRESS viruses. Nat. Commun. 2020, 11, 4620. [Google Scholar] [CrossRef] [PubMed]
- Kinsella, C.M.; van der Hoek, L. Vertebrate-tropism of a cressdnavirus lineage implicated by poxvirus gene capture. Proc. Natl. Acad. Sci. USA 2023, 120, e2303844120. [Google Scholar] [CrossRef] [PubMed]
- Varsani, A.; Krupovic, M. Sequence-based taxonomic framework for the classification of uncultured single-stranded DNA viruses of the family Genomoviridae. Virus Evol. 2017, 3, vew037. [Google Scholar] [CrossRef]
- Varsani, A.; Krupovic, M. Family Genomoviridae: 2021 taxonomy update. Arch. Virol. 2021, 166, 2911–2926. [Google Scholar] [CrossRef] [PubMed]
- Rosario, K.; Duffy, S.; Breitbart, M. A field guide to eukaryotic circular single-stranded DNA viruses: Insights gained from metagenomics. Arch. Virol. 2012, 157, 1851–1871. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Li, B.; Fu, Y.; Jiang, D.; Ghabrial, S.A.; Li, G.; Peng, Y.; Xie, J.; Cheng, J.; Huang, J.; et al. A geminivirus-related DNA mycovirus that confers hypovirulence to a plant pathogenic fungus. Proc. Natl. Acad. Sci. USA 2010, 107, 8387–8392. [Google Scholar] [CrossRef]
- Li, P.; Wang, S.; Zhang, L.; Qiu, D.; Zhou, X.; Guo, L. A tripartite ssDNA mycovirus from a plant pathogenic fungus is infectious as cloned DNA and purified virions. Sci. Adv. 2020, 6, eaay9634. [Google Scholar] [CrossRef]
- Breitbart, M.; Fane, B.A. Microviridae. eLS 2021, 2, 1–14. [Google Scholar] [CrossRef]
- Cherwa, J.E.; Fane, B.A. Microviridae: Microviruses and Gokushoviruses. In eLS; John Wiley & Sons, Ltd.: Chichester, UK, 2011. [Google Scholar] [CrossRef]
- Olo Ndela, E.; Roux, S.; Henke, C.; Sczyrba, A.; Sime Ngando, T.; Varsani, A.; Enault, F. Reekeekee- and roodoodooviruses, two different Microviridae clades constituted by the smallest DNA phages. Virus Evol. 2023, 9, veac123. [Google Scholar] [CrossRef]
- Kirchberger, P.C.; Martinez, Z.A.; Ochman, H. Organizing the Global Diversity of Microviruses. mBio 2022, 13, e00588-22. [Google Scholar] [CrossRef]
- Krupovic, M.; Forterre, P. Microviridae goes temperate: Microvirus-related proviruses reside in the genomes of Bacteroidetes. PLoS ONE 2011, 6, e19893. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Li, D.; Luo, R.; Liu, C.M.; Leung, C.M.; Ting, H.F.; Sadakane, K.; Yamashita, H.; Lam, T.W. MEGAHIT v1.0: A fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods 2016, 102, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Kieft, K.; Zhou, Z.; Anantharaman, K. VIBRANT: Automated recovery, annotation and curation of microbial viruses, and evaluation of viral community function from genomic sequences. Microbiome 2020, 8, 90. [Google Scholar] [CrossRef]
- Asplund, M.; Kjartansdottir, K.R.; Mollerup, S.; Vinner, L.; Fridholm, H.; Herrera, J.A.R.; Friis-Nielsen, J.; Hansen, T.A.; Jensen, R.H.; Nielsen, I.B.; et al. Contaminating viral sequences in high-throughput sequencing viromics: A linkage study of 700 sequencing libraries. Clin. Microbiol. Infect. 2019, 25, 1277–1285. [Google Scholar] [CrossRef] [PubMed]
- Holmes, E.C. Reagent contamination in viromics: All that glitters is not gold. Clin. Microbiol. Infect. 2019, 25, 1167–1168. [Google Scholar] [CrossRef]
- Naccache, S.N.; Greninger, A.L.; Lee, D.; Coffey, L.L.; Phan, T.; Rein-Weston, A.; Aronsohn, A.; Hackett, J., Jr.; Delwart, E.L.; Chiu, C.Y. The perils of pathogen discovery: Origin of a novel parvovirus-like hybrid genome traced to nucleic acid extraction spin columns. J. Virol. 2013, 87, 11966–11977. [Google Scholar] [CrossRef]
- Porter, A.F.; Cobbin, J.; Li, C.; Eden, J.-S.; Holmes, E.C. Metagenomic identification of viral sequences in laboratory reagents. Viruses 2021, 13, 2122. [Google Scholar] [CrossRef]
- Bushnell, B. BBMap: A Fast, Accurate, Splice-Aware Aligner; Lawrence Berkeley National Lab. (LBNL): Berkeley, CA, USA, 2014. [Google Scholar]
- Kazlauskas, D.; Varsani, A.; Koonin, E.V.; Krupovic, M. Multiple origins of prokaryotic and eukaryotic single-stranded DNA viruses from bacterial and archaeal plasmids. Nat. Commun. 2019, 10, 3425. [Google Scholar] [CrossRef]
- Kazlauskas, D.; Varsani, A.; Krupovic, M. Pervasive Chimerism in the Replication-Associated Proteins of Uncultured Single-Stranded DNA Viruses. Viruses 2018, 10, 187. [Google Scholar] [CrossRef]
- Zallot, R.; Oberg, N.; Gerlt, J.A. The EFI Web Resource for Genomic Enzymology Tools: Leveraging Protein, Genome, and Metagenome Databases to Discover Novel Enzymes and Metabolic Pathways. Biochemistry 2019, 58, 4169–4182. [Google Scholar] [CrossRef]
- Fontenele, R.S.; Lacorte, C.; Lamas, N.S.; Schmidlin, K.; Varsani, A.; Ribeiro, S.G. Single Stranded DNA Viruses Associated with Capybara Faeces Sampled in Brazil. Viruses 2019, 11, 710. [Google Scholar] [CrossRef] [PubMed]
- Harding, C.; Larsen, B.B.; Otto, H.W.; Potticary, A.L.; Kraberger, K.; Custer, J.M.; Suazo, C.; Upham, N.S.; Worobey, M.; van Doorslaer, K.; et al. Diverse DNA virus genomes identified in fecal samples of Mexican free-tailed bats (Tadarida brasiliensis) captured in Chiricahua Mountains of southeast Arizona (USA). Virology 2022, 580, 8–111. [Google Scholar] [CrossRef] [PubMed]
- Kraberger, S.; Schmidlin, K.; Fontenele, R.S.; Walters, M.; Varsani, A. Unravelling the Single-Stranded DNA Virome of the New Zealand Blackfly. Viruses 2019, 11, 532. [Google Scholar] [CrossRef] [PubMed]
- Levy, H.; Fontenele, R.S.; Harding, C.; Suazo, C.; Kraberger, S.; Schmidlin, K.; Djurhuus, A.; Black, C.E.; Hart, T.; Smith, A.L.; et al. Identification and Distribution of Novel Cressdnaviruses and Circular molecules in Four Penguin Species in South Georgia and the Antarctic Peninsula. Viruses 2020, 12, 1029. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Capella-Gutierrez, S.; Silla-Martinez, J.M.; Gabaldon, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Anisimova, M.; Gascuel, O. Approximate likelihood-ratio test for branches: A fast, accurate, and powerful alternative. Syst. Biol. 2006, 55, 539–552. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. ProtTest 3: Fast selection of best-fit models of protein evolution. Bioinformatics 2011, 27, 1164–1165. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Stover, B.C.; Muller, K.F. TreeGraph 2: Combining and visualizing evidence from different phylogenetic analyses. BMC Bioinform. 2010, 11, 7. [Google Scholar] [CrossRef] [PubMed]
- Muhire, B.M.; Varsani, A.; Martin, D.P. SDT: A virus classification tool based on pairwise sequence alignment and identity calculation. PLoS ONE 2014, 9, e108277. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [PubMed]
- Finn, D.R.; Maldonado, J.; de Martini, F.; Yu, J.; Penton, C.R.; Fontenele, R.S.; Schmidlin, K.; Kraberger, S.; Varsani, A.; Gile, G.H.; et al. Agricultural practices drive biological loads, seasonal patterns and potential pathogens in the aerobiome of a mixed-land-use dryland. Sci. Total Environ. 2021, 798, 149239. [Google Scholar] [CrossRef]
- Rosario, K.; Mettel, K.A.; Benner, B.E.; Johnson, R.; Scott, C.; Yusseff-Vanegas, S.Z.; Baker, C.C.M.; Cassill, D.L.; Storer, C.; Varsani, A.; et al. Virus discovery in all three major lineages of terrestrial arthropods highlights the diversity of single-stranded DNA viruses associated with invertebrates. PeerJ 2018, 6, e5761. [Google Scholar] [CrossRef]
- Li, W.; Gu, Y.; Shen, Q.; Yang, S.; Wang, X.; Wan, Y.; Zhang, W. A novel gemycircularvirus from experimental rats. Virus Genes 2015, 51, 302–305. [Google Scholar] [CrossRef]
- Hewson, I.; Eaglesham, J.B.; Höök, T.O.; LaBarre, B.A.; Sepúlveda, M.S.; Thompson, P.D.; Watkins, J.M.; Rudstam, L.G. Investigation of viruses in Diporeia spp. from the Laurentian Great Lakes and Owasco Lake as potential stressors of declining populations. J. Great Lakes Res. 2013, 39, 499–506. [Google Scholar] [CrossRef]
- Sommers, P.; Fontenele, R.S.; Kringen, T.; Kraberger, S.; Porazinska, D.L.; Darcy, J.L.; Schmidt, S.K.; Varsani, A. Single-Stranded DNA Viruses in Antarctic Cryoconite Holes. Viruses 2019, 11, 1022. [Google Scholar] [CrossRef] [PubMed]
- Pearson, V.M.; Caudle, S.B.; Rokyta, D.R. Viral recombination blurs taxonomic lines: Examination of single-stranded DNA viruses in a wastewater treatment plant. PeerJ 2016, 4, e2585. [Google Scholar] [CrossRef] [PubMed]
- Kraberger, S.; Argüello-Astorga, G.R.; Greenfield, L.G.; Galilee, C.; Law, D.; Martin, D.P.; Varsani, A. Characterisation of a diverse range of circular replication-associated protein encoding DNA viruses recovered from a sewage treatment oxidation pond. Infect. Genet. Evol. 2015, 31, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Yuan, S.; Yan, T.; Shan, H.; Cheng, Z. Identification and characterization of chicken circovirus from commercial broiler chickens in China. Transbound. Emerg. Dis. 2020, 67, 6–10. [Google Scholar] [CrossRef]
- Ng, T.F.; Chen, L.F.; Zhou, Y.; Shapiro, B.; Stiller, M.; Heintzman, P.D.; Varsani, A.; Kondov, N.O.; Wong, W.; Deng, X.; et al. Preservation of viral genomes in 700-y-old caribou feces from a subarctic ice patch. Proc. Natl. Acad. Sci. USA 2014, 111, 16842–16847. [Google Scholar] [CrossRef]
- Roux, S.; Krupovic, M.; Poulet, A.; Debroas, D.; Enault, F. Evolution and diversity of the Microviridae viral family through a collection of 81 new complete genomes assembled from virome reads. PLoS ONE 2012, 7, e40418. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Top BLASTn Hit | ||||||
|---|---|---|---|---|---|---|
| Cluster | Accession | Virus | Hit Coverage | E Value | % Identity | Hit Accession |
| Genomovirus | OP549794 | Genomoviridae sp. D1_734 | 38% | 1 × 10−83 | 75.51% | MW678940 |
| Genomovirus | OP549795 | Genomoviridae sp. D2_1183 | 100% | 0 | 97.94% | MW678959 |
| Genomovirus | OP549796 | Plant-associated genomovirus 2 GnOP3_BA764 | 27% | 6 × 10−44 | 73.26% | MH939416 |
| CRESSV2 | OP549818 | Circoviridae sp. CN13_L19_1119 | 22% | 1 × 10−20 | 65.49% | MT208232 |
| CRESSV2 | OP549828 | Cressdnaviricota sp. Miresoil virus 170 | 12% | 2 × 10−42 | 77.54% | OM154655 |
| CRESSV2 | OP549833 | Circoviridae sp. CN13_L15_2544 | 13% | 2 × 10−38 | 69.90% | MT203404 |
| CRESSV2 | OP549837 | Circoviridae sp. CN13_L18_554 | 5% | 2 × 10−17 | 79.49% | MT207945 |
| CRESSV2 | OP549843 | Circoviridae sp. CN13_L03_572 | 45% | 2 × 10−138 | 75.24% | MT201433 |
| CRESSV6 | OP549820 | CRESS virus sp. ctWTK558 | 64% | 7 × 10−88 | 65.79% | MW202429 |
| A | OP549845 | Banfec virus 6 V16_S08b | 15% | 1 × 10−37 | 73.98% | OQ599930 |
| B | OP549827 | Cressdnaviricota sp. Miresoil virus 407 | 5% | 4 × 10−08 | 74.55% | OM154420 |
| B | OP549846 | Cressdnaviricota sp Miresoil virus 476 | 14% | 2 × 10−29 | 73.53% | OM154360 |
| C | OP549840 | Virus sp. D12_1244 | 53% | 1 × 10−45 | 65.68% | MW678878 |
| D | OP549841 | Cressdnaviricota sp. ctje111 | 14% | 2 × 10−17 | 68.53% | MH616648 |
| E | OP549838 | Sewage-associated circular DNA virus-17 NZ-BS4236-2012 | 37% | 5 × 10−120 | 73.39% | KM821752 |
| F | OP549819 | Avon-Heathcote Estuary-associated circular virus 3 NZ-3887G-2012 | 8% | 1 × 10−15 | 71.35% | KM874297 |
| G | OP549825 | Chicken circovirus 2 CCV-2 | 37% | 7 × 10−81 | 69.69% | MN420497 |
| G | OP549831 | Chicken circovirus 4 CCV-4 | 38% | 1 × 10−38 | 68.44% | MN428454 |
| G | OP549836 | Chicken circovirus 4 CCV-4 | 30% | 4 × 10−46 | 67.50% | MN428454 |
| G | OP549839 | Chicken circovirus 4 CCV-4 | 100% | 0 | 90.87% | MN428454 |
| H | OP549817 | Crucivirus-93 GP1_93001 | 9% | 3 × 10−19 | 67.64% | MT263542 |
| H | OP549832 | Cruciviridae sp. CRUV-29-F | 31% | 7 × 10−29 | 67.35% | KX388508 |
| I | OP549826 | Cressdnaviricota sp. Miresoil virus 407 | 5% | 4 × 10−08 | 74.55% | OM154420 |
| J | OP549797 | Capybara virus 5_cap1_460 | 10% | 8 × 10−17 | 69.80% | MK570167 |
| J | OP549821 | Egret CRESS-DNA virus egret03 | 49% | 9 × 10−99 | 68.37% | MT797255 |
| J | OP549823 | Sewage-associated circular DNA virus-36 NZ-BS3974-2012 | 48% | 9 × 10−156 | 72.43% | KM821748 |
| J | OP549851 | Trichosanthes kirilowii geminiviridae pt111-gem-5 | 6% | 9 × 10−10 | 72.79% | MN823663 |
| K | OP549822 | Gopherus-associated circular DNA virus 3 Tor327 | 14% | 2 × 10−18 | 68.63% | MK858257 |
| L | OP549824 | - | - | - | - | - |
| M | OP549834 | Circoviridae sp. CN55_L18_367 | 24% | 6 × 10−33 | 66.39% | MT207798 |
| N | OP549842 | Cressdnaviricota sp. Miresoil virus 418 | 45% | 4 × 10−90 | 69.28% | OM154411 |
| O | OP549844 | Capybara virus 15_cap1_294 | 10% | 7 × 10−10 | 69.66% | MK570177 |
| P | OP549829 | Cressdnaviricota sp. Miresoil virus 170 | 12% | 2 × 10−42 | 77.54% | OM154655 |
| P | OP549830 | Apis mellifera virus-5 BNH861 | 54% | 0 | 79.92% | MH973774 |
| Q | OP549835 | Uncultured virus clone CG135 | 33% | 2 × 10−44 | 66.06% | KY487806 |
| Singleton | OP549847 | Crucivirus sp. Crucivirus-391 001074_virusbaton | 7% | 1 × 10−17 | 68.86% | MT478484 |
| Singleton | OP549848 | - | - | - | - | - |
| Singleton | OP549849 | Circoviridae sp. CN3_L17_554 | 1% | 0.004 | 92.31% | MT206222 |
| Singleton | OP549850 | Cressdnaviricota sp. Miresoil virus 557 | 16% | 9 × 10−23 | 68.20% | OM154279 |
| Singleton | OP549852 | - | - | - | - | - |
| Singleton | OP549853 | - | - | - | - | - |
| Singleton | OP549854 | - | - | - | - | - |
| Singleton | OP549855 | Cressdnaviricota sp. Miresoil virus 386 | 8% | 0.039 | 68.02% | OM154441 |
| Singleton | OP549856 | Cressdnaviricota sp. Miresoil virus 388 | 3% | 0.039 | 75.32% | OM154439 |
| Singleton | OP549857 | - | - | - | - | - |
| Microvirus | OP549798 | Tortoise microvirus 72_SP_41 | 44% | 1 × 10−115 | 72.38% | MK765625 |
| Microvirus | OP549799 | Microviridae sp. ctdg534 | 15% | 1 × 10−38 | 67.76% | MH616940 |
| Microvirus | OP549800 | Microviridae sp. ctdg534 | 20% | 6 × 10−61 | 69.09% | MH616940 |
| Microvirus | OP549801 | Microviridae sp. ctci549 | 54% | 7 × 10−162 | 71.14% | MH617187 |
| Microvirus | OP549802 | Apis mellifera-associated microvirus 29 INH_SP_235 | 13% | 1 × 10−38 | 68.86% | MH992203 |
| Microvirus | OP549803 | Microviridae sp. CN7_L19_255 | 81% | 0 | 70.73% | MT208143 |
| Microvirus | OP549804 | Microviridae sp. Dog06 | 34% | 3 × 10−172 | 69.41% | MG883726 |
| Microvirus | OP549805 | Arizlama microvirus AZLM_329 | 52% | 6 × 10−136 | 74.36% | MW697640 |
| Microvirus | OP549806 | Microviridae sp. ctba71 | 33% | 5 × 10−68 | 69.33% | MH616766 |
| Microvirus | OP549807 | Arizlama microvirus AZLM_380 | 31% | 7 × 10−104 | 72.45% | MW697604 |
| Microvirus | OP549808 | Microvirus sp. 6433_74 | 31% | 1 × 10−106 | 72.40% | MT310103 |
| Microvirus | OP549809 | Arizlama microvirus AZLM_274 | 35% | 4 × 10−170 | 69.40% | MW697684 |
| Microvirus | OP549810 | Microviridae sp. SD_MC_24 | 12% | 2 × 10−33 | 67.44% | MH572460 |
| Microvirus | OP549811 | Microvirus sp. BS1_385 | 65% | 3 × 10−146 | 69.91% | MT309971 |
| Microvirus | OP549812 | Robinz microvirus RP_38 | 3% | 1 × 10−11 | 73.91% | MZ364230 |
| Microvirus | OP549813 | Microviridae sp. ctbi780 | 25% | 6 × 10−98 | 70.78% | MH622931 |
| Microvirus | OP549814 | Microvirus sp. BS1_385 | 62% | 5 × 10−124 | 72.73% | MT309971 |
| Microvirus | OP549815 | Tortoise microvirus 93_SP_131 | 20% | 9 × 10−45 | 66.97% | MK765646 |
| Microvirus | OP549816 | Tortoise microvirus 93_SP_131 | 22% | 7 × 10−40 | 66.06% | MK765646 |
| Cluster | Accession | Motif I | Motif II | Motif III | GRS Domain | Walker A | Walker B | Motif C |
|---|---|---|---|---|---|---|---|---|
| Genomovirus | OP549794 | LLTYAQ | IHLHV | KAYDYAIK | DVFDVGGYHPNIERVG | GESQLGKTLWAR | AIFDDI | WLAN |
| Genomovirus | OP549795 | LLTYAQ | THYHA | KMFDYATK | RAFDVDGYHPNILRGI | GPTRTGKTSWAR | AVFDDI | WCNN |
| Genomovirus | OP549796 | LVTYAQ | THLHV | KGWEYATK | HIFDVDGYHPNVVPGY | GPTRLGKTVWAR | AVFDDM | WLSN |
| CRESSV2 | OP549818 | VFTWNN | EHLQG | QAHTYCKK | GPSGTGKSHFIS | IWLDDV | VTSN | |
| CRESSV2 | OP549828 | CFTINN | PHLQG | QNHKYCTK | GETGSGKSKSVR | VCIDDF | VTSQ | |
| CRESSV2 | OP549833 | CFTLNG | KHLQC | QNRRYCIK | GNTGAGKSYLAR | ILLDDF | VTSN | |
| CRESSV2 | OP549837 | VFTLNN | PHLQG | QAKAYCQK | GDPKAGKSEGAR | IIIEDM | VTSN | |
| CRESSV2 | OP549843 | TFTINT | PHFQG | QNYDYCSK | GPPRTGKSHKAR | VLIDEL | VTSN | |
| CRESSV6 | OP549820 | ALTYSN | DHFHA | AWLTYIKK | GESGIGKTNWAK | IIFDDV | FTAN | |
| A | OP549845 | FLTINN | PHIQG | ACIIYCTK | GPAGCRKTRTAV | IIIDDF | ITCE | |
| B | OP549827 | IYTLNN | PHLQG | DAANYCMK | GPTGVGKTRSVV | TLFDDY | ITCP | |
| B | OP549846 | TFTLNN | PHLQG | AAINYCMK | GETGAGKTRYVF | ALLDDF | ITAP | |
| C | OP549840 | VFTINN | PHLQG | QAIIYCEK | GPTGSGKSRYAW | AIIDDF | VTCP | |
| D | OP549841 | VVTFWC | HHWQA | SNAKYCSK | GSTGRGKSHRTF | VIINEF | VNSS | |
| E | OP549838 | CFTLNN | PHLQG | SNREYCSK | GLPGVGKSRRAH | VIIDDF | VTSN | |
| F | OP549819 | ILTIPV | HHWQI | AADKYVHK | GGSGLGKTRRAW | VIIDEF | ITSN | |
| G | OP549825 | AFTLNN | PHLQG | DSCYYCVK | SRGGAGKSYFAR | IIIIDI | IFAN | |
| G | OP549831 | CFTLNN | PHLQG | DNDAYCGG | TEGNVGKSAFTK | LIIWDM | IFSN | |
| G | OP549836 | CFTLNN | PHLQG | INLKYCSK | SIGNIGKSAFIK | CIMFDI | IFAN | |
| G | OP549839 | IMVLNN | PHLQG | QNDIYCSK | RPGHFGKSQFVK | CVMFDI | CFAN | |
| H | OP549817 | DFTIWA | LHYQG | GEPFYVLK | PEGASGKSTLRN | LYVLDL | VFTN | |
| H | OP549832 | DFTIKE | LHYQG | DNNFYVMK | TQGNNGKSTLKA | LYILDI | VFMN | |
| I | OP549826 | LLTYGK | EHMHV | NAKNYLAK | PVGGSGKTQFAK | GLIFNL | VFAN | |
| J | OP549797 | FLTYSQ | FHLHG | RVLHYTQK | RLFDVGIYHPNIGALR | GASRTGKTTWAR | IILDDI | HLCN |
| J | OP549821 | FLTYSQ | IHYHV | RCRHYLRK | DIFDFGGCHAIQPIKN | GPTRLGKTDWAR | LVIDDF | WLCN |
| J | OP549823 | FLTYSQ | IHFHV | NRRHYIRK | NIFDCAGYHPNILPIR | GPTECGKSVWAR | LVLDDM | WICN |
| J | OP549851 | LLTYAQ | QHFHC | NCWEYCTK | RAFDIGQNHPNIKRVG | GPTRTGKTIWAR | LIMDDF | YICN |
| K | OP549822 | FLTYSR | FHLHG | KTLNYIYK | RFLDITGP[DGTVY]HPKLEPVK | GPTKTGKSAWAR | VVFDDI | ILCN |
| L | OP549824 | FLTYPQ | LHLHV | AVMRYCTK | GPTNSGKTALAK | IIYDEA | FTSN | |
| M | OP549834 | AYTDFQ | KHIQG | QNFVYCSK | GPTNIGKTQFAI | IVFDDM | FTYN | |
| N | OP549842 | FLTYSQ | FHLHC | KCLAYVIK | GESGVGKSTIAT | VVIEDI | ITSN | |
| O | OP549844 | FLTYPQ | HHIHA | NVIKYCTK | TKPNLGKTFLFG | VVLDEY | VLSN | |
| P | OP549829 | FLTYAQ | PHMHV | AIKKYCMK | GPSNTGKSFWLR | LWADEY | ICSN | |
| P | OP549830 | LLTYPQ | PHIHA | RSHKYCQK | GPSNSGKSYWLT | LFSDEY | IVSN | |
| Q | OP549835 | FITFPQ | QHLHI | AAIAYITK | GVANVGKTTIIS | AYIDEF | ILSN | |
| S | OP549847 | KFTPQK | LHYQC | ALQAYSMK | PSGQVGKTWFGN | CYIIDL | VFAN | |
| S | OP549848 | CFTLNN | PHVQG | QARDYCCK | GNTGTGKSTYAR | VVLDEF | AISN | |
| S | OP549849 | CFTLNN | HHLQG | QARDYCRK | GPTGCGKTSTAY | VLIDDF | ITTN | |
| S | OP549850 | LFTLWL | RHFQS | QNIDYCTK | GSSGTGKTRFAY | VLFDDY | ITSN | |
| S | OP549852 | FLTYQS | PHRHV | KCCFYMCK | - | YLVVSL | VVAN | |
| S | OP549853 | AMTINN | RHIHV | GWIKYCMK | - | VNYEEL | LYFN | |
| S | OP549854 | WLTVNP | FHLHA | DSGQYVFE | - | - | LYTL | |
| S | OP549855 | FLTYAQ | PHRHC | NCAKYVLK | GDTGSGKSKYAR | VLLEDV | ITSN | |
| S | OP549856 | VFTINY | HHIQG | EARDYALK | GETGRGKTRRAA | VLFDDF | ITSN | |
| S | OP549857 | CLTHYG | EHQQA | EARKYCMK | GDTGTGKSHLAH | MIINEF | LTSP |
| Top Rep Hit | ||||
|---|---|---|---|---|
| Cluster | Accession | Virus | Rep % Identity | Accession |
| Genomovirus | OP549794 | Gemycircularvirus gemy-ch-rat1 | 59.7 | KR912221 |
| Genomovirus | OP549796 | Cybaeus spider-associated circular virus 2 BC_I1644B_C3 | 60.3 | MH545507 |
| Genomovirus | OP549795 | Genomoviridae sp. D2_1183 | 100 | MW678959 |
| CRESSV2 | OP549818 | Diporeia sp.-associated circular virus LM3487 | 45.8 | KC248416 |
| CRESSV2 | OP549837 | Sewage-associated circular DNA virus-20 NZ-BS3900-2012 | 48.4 | KM821755 |
| CRESSV2 | OP549833 | Uncultured virus clone CG261 | 48.2 | KY487930 |
| CRESSV2 | OP549843 | Cressdnaviricota sp. ctdb97 | 56.3 | MH510276 |
| CRESSV2 | OP549828 | Antarctic circular DNA molecule COCH21_V_94 | 57.2 | MN328284 |
| CRESSV6 | OP549820 | Circovirus-like genome DCCV-2 | 47.0 | KT149395 |
| A | OP549845 | Arizlama virus AZLM_1011 | 51.3 | MW697465 |
| B | OP549827 | Uncultured virus clone CG267 | 42.6 | KY487936 |
| B | OP549846 | Uncultured virus clone CG267 | 44.0 | KY487936 |
| C | OP549840 | Virus sp. D12_1244 | 63.8 | MW678878 |
| D | OP549841 | Cressdnaviricota sp. ctcd610 | 45.4 | MH649031 |
| E | OP549838 | Sewage-associated circular DNA virus-17 SaCV-17_NZ-BS4236-2012 | 72.1 | KM821752 |
| F | OP549819 | Avon-Heathcote Estuary-associated circular virus 26 NZ-2311TU-2012 | 48.4 | KM874359 |
| G | OP549831 | Cressdnaviricota sp. ctcd828 | 54.0 | MH649233 |
| G | OP549825 | Chicken circovirus 2 strain CCV-2 | 63.2 | MN420497 |
| G | OP549836 | Chicken circovirus 4 CCV-4 | 58.8 | MN428454 |
| G | OP549839 | Chicken circovirus 4 CCV-4 | 98.6 | MN428454 |
| H | OP549832 | Cressdnaviricota sp. ctca156 | 53.3 | MH616996 |
| H | OP549817 | Cressdnaviricota sp. ctbb593 | 48.4 | MH648954 |
| I | OP549826 | Crucivirus-124 BS_313 | 42.6 | MT263552 |
| J | OP549821 | Ancient caribou feces associated virus | 53.3 | KJ938716 |
| J | OP549823 | Sewage-associated circular DNA virus-36 NZ-BS3974-2012 | 64.9 | KM821748 |
| J | OP549851 | Genomoviridae sp. 6538_332 | 36.9 | MT309820 |
| J | OP549797 | Genomoviridae sp. 6434_400 | 45.8 | MT309859 |
| K | OP549822 | Genomoviridae sp. 6538_302 | 47.6 | MT309829 |
| L | OP549824 | Uncultured virus clone CG104 | 40.9 | KY487775 |
| M | OP549834 | Crucivirus-243 SR3_42497 | 47.4 | MT263577 |
| N | OP549842 | Virus sp. D6_821 | 47.4 | MW678874 |
| O | OP549844 | Cressdnaviricota sp. ctcj370 | 58.6 | MH617003 |
| P | OP549830 | Apis mellifera virus-5 BNH861 | 85.7 | MH973774 |
| P | OP549829 | Capybara virus 8_cap1_36 | 46.2 | MK570170 |
| Q | OP549835 | Uncultured virus clone CG135 | 52.0 | KY487806 |
| Singleton | OP549847 | McMurdo Ice Shelf pond-associated circular DNA virus-8 alg49-57 | 35.4 | KJ547653 |
| Singleton | OP549848 | Red panda feces-associated circular DNA virus 7 Rpf101envir01-8 | 39.1 | MZ556225 |
| Singleton | OP549849 | Dipodfec virus UA04Rod_4537 | 41.4 | OM869597 |
| Singleton | OP549850 | False black widow spider-associated circular virus 1 BC_I1659B_H2 | 41.4 | MH545542 |
| Singleton | OP549852 | Sewage-associated circular DNA virus-13 NZ-BS4044-2012 | 34.6 | KJ547624 |
| Singleton | OP549853 | Cressdnaviricota sp. Miresoil virus 251 | 26.4 | OM154576 |
| Singleton | OP549854 | Cressdnaviricota sp. ctcc233 | 34.2 | MH617714 |
| Singleton | OP549855 | Cressdnaviricota sp. Miresoil virus 418 | 33.9 | OM154411 |
| Singleton | OP549856 | Circovirus sp. panda384 | 37.5 | MZ556112 |
| Singleton | OP549857 | Avon-Heathcote Estuary-associated circular virus 19 NZ-4942GA-2012 | 29.9 | KM874347 |
| Top MCP Hit | ||||
|---|---|---|---|---|
| Subfamily/Clade | Accession | Virus | MCP % Identity | Accession |
| Unclassified | OP549798 | Tortoise microvirus 72 | 65.4% | MK765625 |
| Alphavirinae-clade | OP549799 | Microviridae sp. ctcf586 | 38.1% | MH617122 |
| Alphavirinae-clade | OP549800 | Microvirus sp. BS1_235, | 37.3% | MT310011 |
| Alphavirinae-clade | OP549801 | Microviridae sp. Flamingo05 | 58.2% | MG883728 |
| Gokushovirinae | OP549802 | Microviridae sp. ctcf650 | 48.0% | MH62292 |
| Gokushovirinae | OP549803 | Apis mellifera associated microvirus 28 INH_SP_214 | 71.2% | MH992195 |
| Unclassified | OP549804 | Microvirus sp. 1712115_732 | 71.4% | MT310269 |
| Gokushovirinae | OP549805 | Microvirus sp. 6433_52 | 70.0% | MT310113 |
| Pichovirinae-clade | OP549806 | Microviridae sp. ctba71 | 59.9% | MH616766 |
| Unclassified | OP549807 | Apis mellifera associated microvirus 42 INH_SP_292 | 67.4% | MH992217 |
| Unclassified | OP549808 | Microvirus sp. 1712115_732 | 71.9% | MT310269 |
| Gokushovirinae | OP549809 | Arizlama microvirus AZLM_259 | 72.3% | MW697690 |
| Pichovirinae-clade | OP549810 | Microviridae sp. SD_MC_53 | 51.9% | MH572461 |
| Gokushovirinae | OP549811 | Microviridae sp. ctsGZ299 | 76.4% | MW202558 |
| Gokushovirinae | OP549812 | Microviridae sp. ctgc091 | 43.9% | MH617728 |
| Gokushovirinae | OP549813 | Arizlama microvirus AZLM_329 | 65.6% | MW697640 |
| Gokushovirinae | OP549814 | Microvirus sp. BS1_385 | 77.1% | MT309971 |
| Pichovirinae-clade | OP549815 | Microviridae sp. ctcf880 | 56.40% | MH617497 |
| Pichovirinae-clade | OP549816 | Microviridae sp. ctcf880 | 56.6% | MH617497 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olivo, D.; Khalifeh, A.; Custer, J.M.; Kraberger, S.; Varsani, A. Diverse Small Circular DNA Viruses Identified in an American Wigeon Fecal Sample. Microorganisms 2024, 12, 196. https://doi.org/10.3390/microorganisms12010196
Olivo D, Khalifeh A, Custer JM, Kraberger S, Varsani A. Diverse Small Circular DNA Viruses Identified in an American Wigeon Fecal Sample. Microorganisms. 2024; 12(1):196. https://doi.org/10.3390/microorganisms12010196
Chicago/Turabian StyleOlivo, Diego, Anthony Khalifeh, Joy M. Custer, Simona Kraberger, and Arvind Varsani. 2024. "Diverse Small Circular DNA Viruses Identified in an American Wigeon Fecal Sample" Microorganisms 12, no. 1: 196. https://doi.org/10.3390/microorganisms12010196