

Competitive Analysis of Rumen and Hindgut Microbiota Composition and Fermentation Function in Diarrheic and Non-Diarrheic Postpartum Dairy Cows
 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Management and Sample Collection
2.2. Sample Collection
2.3. Milk Yield, Composition, and Feed Digestibility Measurements
2.4. Ruminal and Fecal Fermentation Profiles and LPS Measurements
2.5. Rumen and Hindgut Microbiota Composition and Network Analysis
2.5.1. DNA Extraction and Amplicon Sequencing
2.5.2. Sequence Data Analysis
2.5.3. Microbiota Network Construction
2.6. Statistics
3. Results
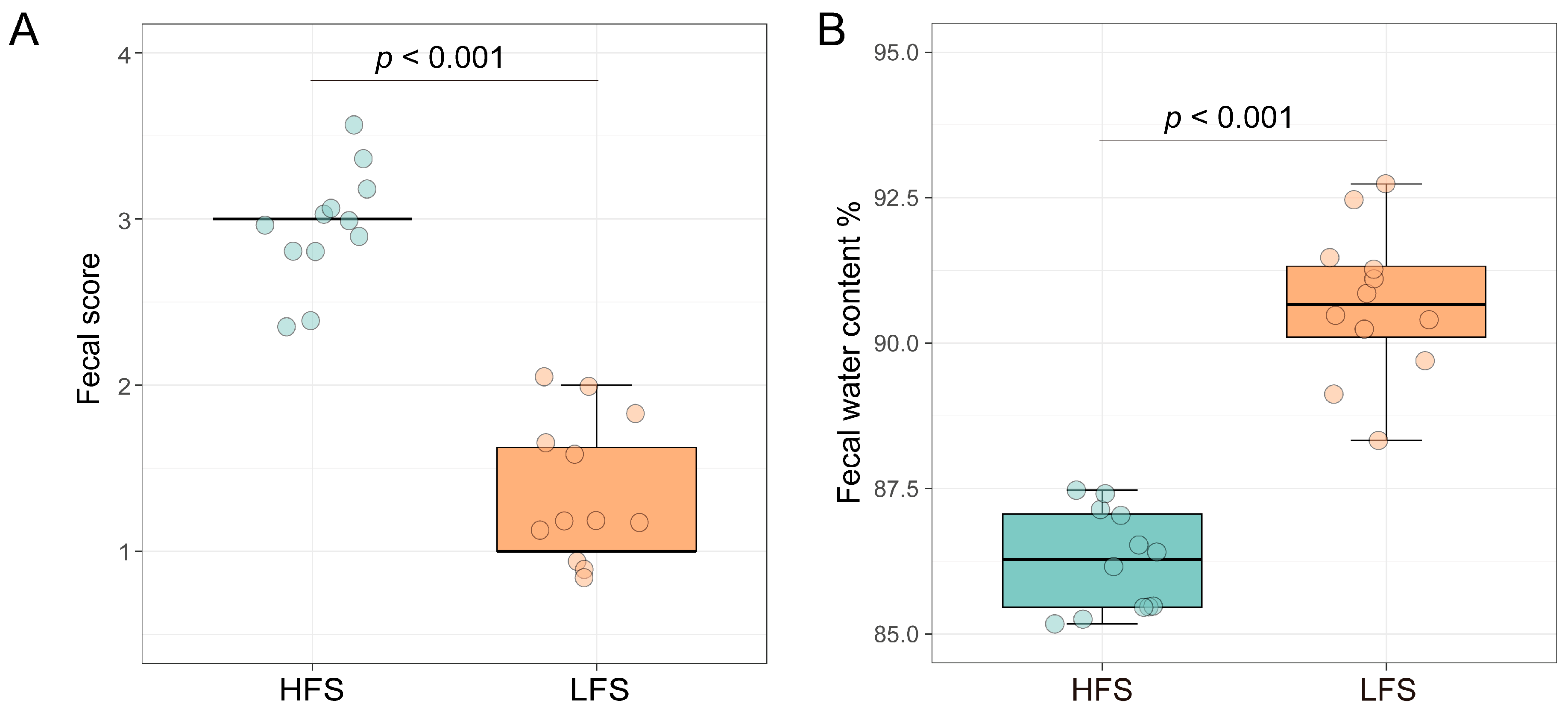
3.1. Rumen and Hindgut Fermentation Profiles and LPS Concentrations
3.2. Ruminal and Fecal Bacterial Composition and Diversity
3.3. Relationship between Rumen and Fecal Bacteria and Fecal Water Content and Fermentation Profiles
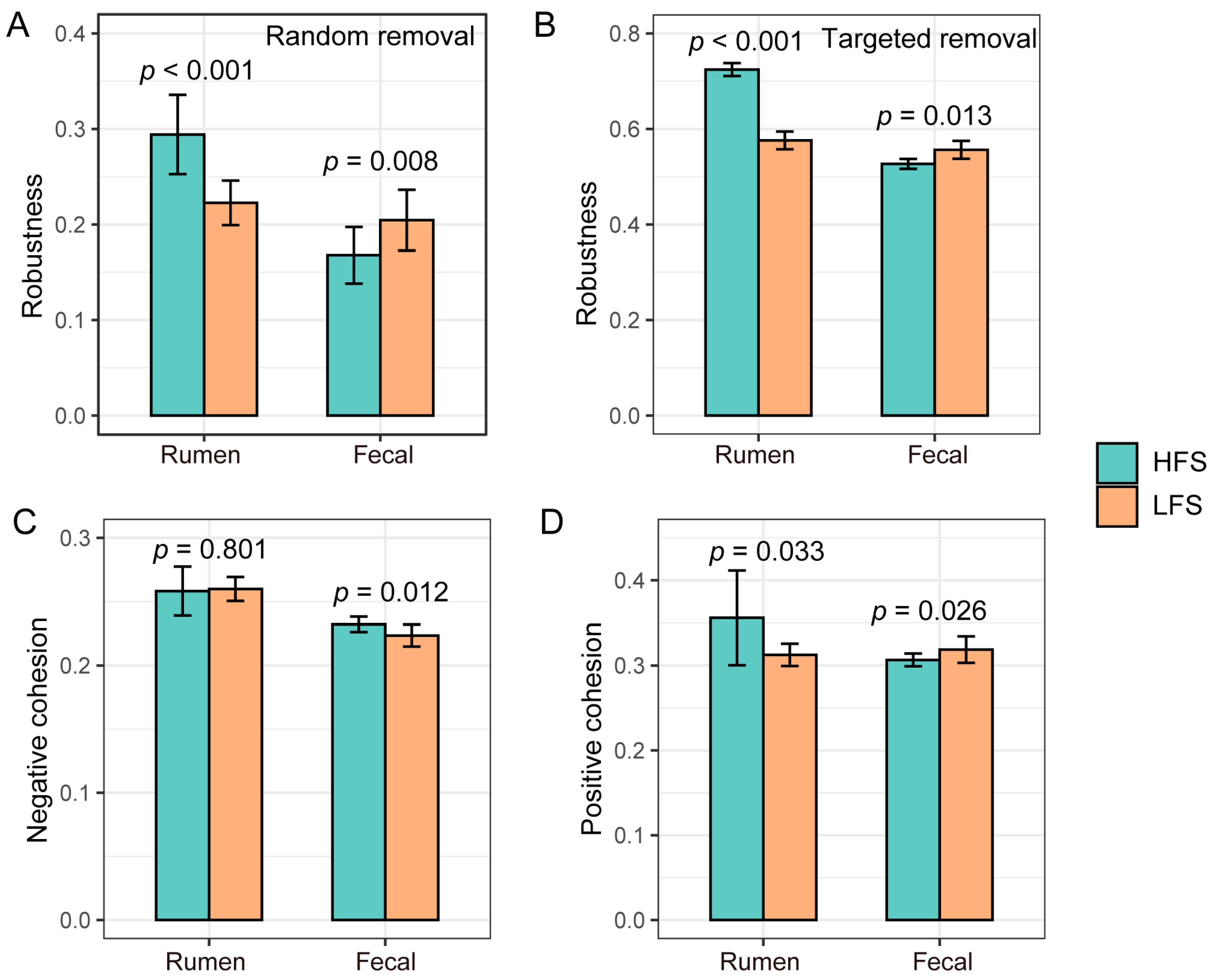
3.4. Difference in Rumen and Fecal Bacterial Community Networks and Topologies between Two Groups of Cows
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Steele, M.A.; Penner, G.B.; Chaucheyras-Durand, F.; Guan, L.L. Development and physiology of the rumen and the lower gut: Targets for improving gut health. J. Dairy Sci. 2016, 99, 4955–4966. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, P.C. Diagnostics of dairy and beef cattle diarrhea. Vet. Clin. N. Am. Food Anim. Pract. 2012, 28, 443–464. [Google Scholar] [CrossRef] [PubMed]
- Chigerwe, M.; Heller, M.C. Diagnosis and Treatment of Infectious Enteritis in Adult Ruminants. Vet. Clin. N. Am. Food Anim. Pract. 2017, 34, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Yin, J.; Xu, K.; Li, T.; Yin, Y. What Is the Impact of Diet on Nutritional Diarrhea Associated with Gut Microbiota in Weaning Piglets: A System Review. BioMed Res. Int. 2019, 2019, 6916189. [Google Scholar] [CrossRef]
- Rist, V.T.S.; Weiss, E.; Eklund, M.; Mosenthin, R. Impact of dietary protein on microbiota composition and activity in the gastrointestinal tract of piglets in relation to gut health: A review. Animal 2013, 7, 1067–1078. [Google Scholar] [CrossRef]
- Zhang, K.; Xu, Y.; Yang, Y.; Guo, M.; Zhang, T.; Zong, B.; Huang, S.; Suo, L.; Ma, B.; Wang, X.; et al. Gut microbiota-derived metabolites contribute negatively to hindgut barrier function development at the early weaning goat model. Anim. Nutr. 2022, 10, 111–123. [Google Scholar] [CrossRef]
- Han, X.; Lei, X.; Yang, X.; Shen, J.; Zheng, L.; Jin, C.; Cao, Y.; Yao, J. A Metagenomic Insight into the Hindgut Microbiota and Their Metabolites for Dairy Goats Fed Different Rumen Degradable Starch. Front. Microbiol. 2021, 12, 651631. [Google Scholar] [CrossRef]
- Penner, G.; Beauchemin, K.; Mutsvangwa, T. Severity of Ruminal Acidosis in Primiparous Holstein Cows During the Periparturient Period. J. Dairy Sci. 2007, 90, 365–375. [Google Scholar] [CrossRef]
- Li, S.; Khafipour, E.; Krause, D.; Kroeker, A.; Rodriguez-Lecompte, J.; Gozho, G.; Plaizier, J. Effects of subacute ruminal acidosis challenges on fermentation and endotoxins in the rumen and hindgut of dairy cows. J. Dairy Sci. 2012, 95, 294–303. [Google Scholar] [CrossRef]
- Liang, Y.-C.; Liu, H.-J.; Chen, S.-H.; Chen, C.-C.; Chou, L.-S.; Tsai, L.H. Effect of lipopolysaccharide on diarrhea and gastrointestinal transit in mice: Roles of nitric oxide and prostaglandin E2. World J. Gastroenterol. 2005, 11, 357–361. [Google Scholar] [CrossRef]
- NRC. Nutrient Requirements of Dairy Cattle, 7th ed.; Subcommittee on Dairy Cattle Nutrition, Committee on Animal Nutrition, Board on Agriculture and Natural Resources, National Research Council; National Academy Press: Washington, DC, USA, 2001.
- Hulsen, J. Cow Signals, A Practical Guide for Dairy Farm Management; Roodbont Publishers: Zutphen, The Netherlands, 2006. [Google Scholar]
- Liu, J.; Li, H.; Zhu, W.; Mao, S. Dynamic changes in rumen fermentation and bacterial community following rumen fluid transplantation in a sheep model of rumen acidosis: Implications for rumen health in ruminants. FASEB J. 2019, 33, 8453–8467. [Google Scholar] [CrossRef] [PubMed]
- Rogers, M.J.; Moore, R.; Cohen, J. The relationship between faecal endotoxin and faecal microflora of the C57BL mouse. Epidemiol. Infect. 1985, 95, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Van Soest, P.J.; Robertson, J.B.; Lewis, B.A. Methods for dietary fiber, neutral detergent fiber, and nonstarch polysaccharides in relation to animal nutrition. J. Dairy Sci. 1991, 74, 3583–3597. [Google Scholar] [CrossRef] [PubMed]
- AOAC International. Official Methods of Analysis, 17th ed.; AOAC International: Rockville, MD, USA, 2000; Volume 1–2. [Google Scholar]
- Van Keulen, J.; Young, B.A. Evaluation of Acid-Insoluble Ash as a Natural Marker in Ruminant Digestibility Studies. J. Anim. Sci. 1977, 44, 282–287. [Google Scholar] [CrossRef]
- Schneider, B.H.; Flatt, W.P. The Evaluation of Feeds through Digestibility Experiments. Can. Vet. J. 1977, 18, 141. [Google Scholar] [CrossRef]
- Broderick, G.A.; Kang, J.H. Automated Simultaneous Determination of Ammonia and Total Amino Acids in Ruminal Fluid and In Vitro Media. J. Dairy Sci. 1980, 63, 64–75. [Google Scholar] [CrossRef]
- Cao, Z.J.; Li, S.L.; Xing, J.J.; Ma, M.; Wang, L.L. Effects of maize grain and lucerne particle size on ruminal fermentation, digestibility and performance of cows in midlactation. J. Anim. Physiol. Anim. Nutr. 2007, 92, 157–167. [Google Scholar] [CrossRef]
- Liu, J.-H.; Xu, T.-T.; Liu, Y.-J.; Zhu, W.-Y.; Mao, S.-Y. A high-grain diet causes massive disruption of ruminal epithelial tight junctions in goats. Am. J. Physiol. Integr. Comp. Physiol. 2013, 305, R232–R241. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; Mcmurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Jiang, Y.-H.; Yang, Y.; He, Z.; Luo, F.; Zhou, J. Molecular ecological network analyses. BMC Bioinform. 2012, 13, 113. [Google Scholar] [CrossRef] [PubMed]
- Montesinos-Navarro, A.; Hiraldo, F.; Tella, J.L.; Blanco, G. Network structure embracing mutualism–antagonism continuums increases community robustness. Nat. Ecol. Evol. 2017, 1, 1661–1669. [Google Scholar] [CrossRef] [PubMed]
- Dunne, J.A.; Williams, R.J.; Martinez, N.D. Food-web structure and network theory: The role of connectance and size. Proc. Natl. Acad. Sci. USA 2002, 99, 12917–12922. [Google Scholar] [CrossRef] [PubMed]
- Herren, C.M.; McMahon, K.D. Cohesion: A method for quantifying the connectivity of microbial communities. ISME J. 2017, 11, 2426–2438. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Kleen, J.L.; Hooijer, G.A.; Rehage, J.; Noordhuizen, J.P.T.M. Subacute ruminal acidosis (SARA): A Review. J. Vet. Med. Ser. A 2003, 50, 406–414. [Google Scholar] [CrossRef]
- Plaizier, J.C.; Krause, D.O.; Gozho, G.N.; McBride, B.W. Subacute ruminal acidosis in dairy cows: The physiological causes, incidence and consequences. Vet. J. 2008, 176, 21–31. [Google Scholar] [CrossRef]
- Zhang, R.; Liu, J.; Jiang, L.; Wang, X.; Mao, S. The Remodeling Effects of High-Concentrate Diets on Microbial Composition and Function in the Hindgut of Dairy Cows. Front. Nutr. 2022, 8, 809406. [Google Scholar] [CrossRef]
- Ricci, S.; Pacífico, C.; Castillo-Lopez, E.; Rivera-Chacon, R.; Schwartz-Zimmermann, H.E.; Reisinger, N.; Berthiller, F.; Zebeli, Q.; Petri, R.M. Progressive microbial adaptation of the bovine rumen and hindgut in response to a step-wise increase in dietary starch and the influence of phytogenic supplementation. Front. Microbiol. 2022, 13, 920427. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-S.; Liang, X.; Wei, X.-H.; Jin, Z.; Chen, F.-L.; Tang, Q.-F.; Tan, X.-M. Gegen Qinlian Decoction Treats Diarrhea in Piglets by Modulating Gut Microbiota and Short-Chain Fatty Acids. Front. Microbiol. 2019, 10, 825. [Google Scholar] [CrossRef]
- Morrison, D.J.; Preston, T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes 2016, 7, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Wu, W.; Liu, Z.; Cong, Y. Microbiota metabolite short chain fatty acids, GPCR, and inflammatory bowel diseases. J. Gastroenterol. 2016, 52, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, S.; Toh, H.; Hase, K.; Oshima, K.; Nakanishi, Y.; Yoshimura, K.; Tobe, T.; Clarke, J.M.; Topping, D.L.; Suzuki, T.; et al. Bifidobacteria can protect from enteropathogenic infection through production of acetate. Nature 2011, 469, 543–547. [Google Scholar] [CrossRef] [PubMed]
- Falony, G.; Vlachou, A.; Verbrugghe, K.; De Vuyst, L. Cross-feeding between Bifidobacterium longum BB536 and acetate-converting, butyrate-producing colon bacteria during growth on oligofructose. Appl. Environ. Microbiol. 2006, 72, 7835–7841. [Google Scholar] [CrossRef]
- Benno, Y.; Mitsuoka, T. Impact of Bifidobacterium longum on human fecal microflora. Microbiol. Immunol. 1992, 36, 683–694. [Google Scholar] [CrossRef]
- Louis, P.; Flint, H.J. Formation of propionate and butyrate by the human colonic microbiota. Environ. Microbiol. 2017, 19, 29–41. [Google Scholar] [CrossRef]
- Cremin, J.D.; Fitch, M.D.; Fleming, S.E. Glucose alleviates ammonia-induced inhibition of short-chain fatty acid metabolism in rat colonic epithelial cells. Am. J. Physiol. Liver Physiol. 2003, 285, G105–G114. [Google Scholar] [CrossRef]
- Dias, A.L.G.; Freitas, J.A.; Micai, B.; Azevedo, R.A.; Greco, L.F.; Santos, J.E.P. Effect of supplemental yeast culture and dietary starch content on rumen fermentation and digestion in dairy cows. J. Dairy Sci. 2018, 101, 201–221. [Google Scholar] [CrossRef]
- Kulkarni, N.A.; Chethan, H.S.; Srivastava, R.; Gabbur, A.B. Role of probiotics in ruminant nutrition as natural modulators of health and productivity of animals in tropical countries: An overview. Trop. Anim. Health Prod. 2022, 54, 110. [Google Scholar] [CrossRef]
- Chen, H.; Liu, Y.; Huang, K.; Yang, B.; Zhang, Y.; Yu, Z.; Wang, J. Fecal microbiota dynamics and its relationship to diarrhea and health in dairy calves. J. Anim. Sci. Biotechnol. 2022, 13, 132. [Google Scholar] [CrossRef] [PubMed]
- Machado, D.; Maistrenko, O.M.; Andrejev, S.; Kim, Y.; Bork, P.; Patil, K.R.; Patil, K.R. Polarization of microbial communities between competitive and cooperative metabolism. Nat. Ecol. Evol. 2021, 5, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.M.; Guo, X.; Wu, L.; Zhang, Y.; Xiao, N.; Ning, D.; Shi, Z.; Zhou, X.; Wu, L.; Yang, Y.; et al. Climate warming enhances microbial network complexity and stability. Nat. Clim. Chang. 2021, 11, 343–348. [Google Scholar] [CrossRef]
- Shade, A.; Peter, H.; Allison, S.D.; Baho, D.L.; Berga, M.; Bürgmann, H.; Huber, D.H.; Langenheder, S.; Lennon, J.T.; Martiny, J.B.H.; et al. Fundamentals of microbial community resistance and resilience. Front. Microbiol. 2012, 3, 417. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Items 1 | Groups | SEM | p-Value | |
|---|---|---|---|---|
| LFS (n = 12) | HFS (n = 12) | |||
| pH | 6.54 | 6.59 | 0.05 | 0.668 |
| NH3-N mg/dL | 43.56 | 44.16 | 5.40 | 0.913 |
| TVFA mM | 83.37 | 77.90 | 4.77 | 0.266 |
| VFA (mol/100 mol) | ||||
| Acetate | 58.82 | 61.83 | 1.43 | 0.048 |
| Propionate | 26.13 | 22.07 | 1.38 | 0.008 |
| Isobutyrate | 0.45 | 0.53 | 0.08 | 0.359 |
| Butyrate | 12.16 | 13.06 | 0.61 | 0.155 |
| Isovalerate | 1.04 | 1.09 | 0.14 | 0.732 |
| Valerate | 1.41 | 1.43 | 0.14 | 0.868 |
| A/P | 2.32 | 2.85 | 0.20 | 0.016 |
| Fecal fermentation profile | ||||
| NH3-N mg/g of feces | 4.04 | 2.61 | 0.31 | 0.020 |
| TVFA µmol/g of feces | 63.14 | 81.06 | 5.23 | 0.002 |
| VFA (mol/100 mol) | ||||
| Acetate | 75.19 | 77.02 | 1.25 | 0.158 |
| Propionate | 13.33 | 12.11 | 0.64 | 0.069 |
| Isobutyrate | 1.33 | 0.82 | 0.15 | 0.003 |
| Butyrate | 7.76 | 8.40 | 0.98 | 0.525 |
| Isovalerate | 0.81 | 0.42 | 0.17 | 0.034 |
| Valerate | 1.57 | 1.24 | 0.16 | 0.054 |
| A/P | 5.71 | 6.49 | 0.37 | 0.046 |
| Items 1 | Groups | SEM | p-Value | |
|---|---|---|---|---|
| LFS (n = 12) | HFS (n = 12) | |||
| Fecal LPS (EU/g of wet feces) | 13,746.20 | 16,185.49 | 1049.20 | 0.260 |
| Rumen fluid LPS (EU/mL) | 14,255.22 | 12,208.48 | 1645.38 | 0.550 |
| Blood LPS (EU/mL) | 1.63 | 1.41 | 0.16 | 0.519 |
| Blood LBP (mg/L) | 2.01 | 2.51 | 0.16 | 0.117 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hao, Y.; Ouyang, T.; Wang, W.; Wang, Y.; Cao, Z.; Yang, H.; Guan, L.L.; Li, S. Competitive Analysis of Rumen and Hindgut Microbiota Composition and Fermentation Function in Diarrheic and Non-Diarrheic Postpartum Dairy Cows. Microorganisms 2024, 12, 23. https://doi.org/10.3390/microorganisms12010023
Hao Y, Ouyang T, Wang W, Wang Y, Cao Z, Yang H, Guan LL, Li S. Competitive Analysis of Rumen and Hindgut Microbiota Composition and Fermentation Function in Diarrheic and Non-Diarrheic Postpartum Dairy Cows. Microorganisms. 2024; 12(1):23. https://doi.org/10.3390/microorganisms12010023
Chicago/Turabian StyleHao, Yangyi, Tong Ouyang, Wei Wang, Yajing Wang, Zhijun Cao, Hongjian Yang, Le Luo Guan, and Shengli Li. 2024. "Competitive Analysis of Rumen and Hindgut Microbiota Composition and Fermentation Function in Diarrheic and Non-Diarrheic Postpartum Dairy Cows" Microorganisms 12, no. 1: 23. https://doi.org/10.3390/microorganisms12010023
APA StyleHao, Y., Ouyang, T., Wang, W., Wang, Y., Cao, Z., Yang, H., Guan, L. L., & Li, S. (2024). Competitive Analysis of Rumen and Hindgut Microbiota Composition and Fermentation Function in Diarrheic and Non-Diarrheic Postpartum Dairy Cows. Microorganisms, 12(1), 23. https://doi.org/10.3390/microorganisms12010023