
Bacterial and Microeukaryotic Community Compositions and Their Assembly Processes in Lakes on the Eastern Qinghai-Tibet Plateau
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Environmental Variable Monitoring
2.3. DNA Extraction, PCR Amplification, and Sequencing Analysis
2.4. Biodiversity Analysis
2.5. Microbial Community Assembly Processes Analysis
2.6. Co-Occurrence Network Analysis
3. Results
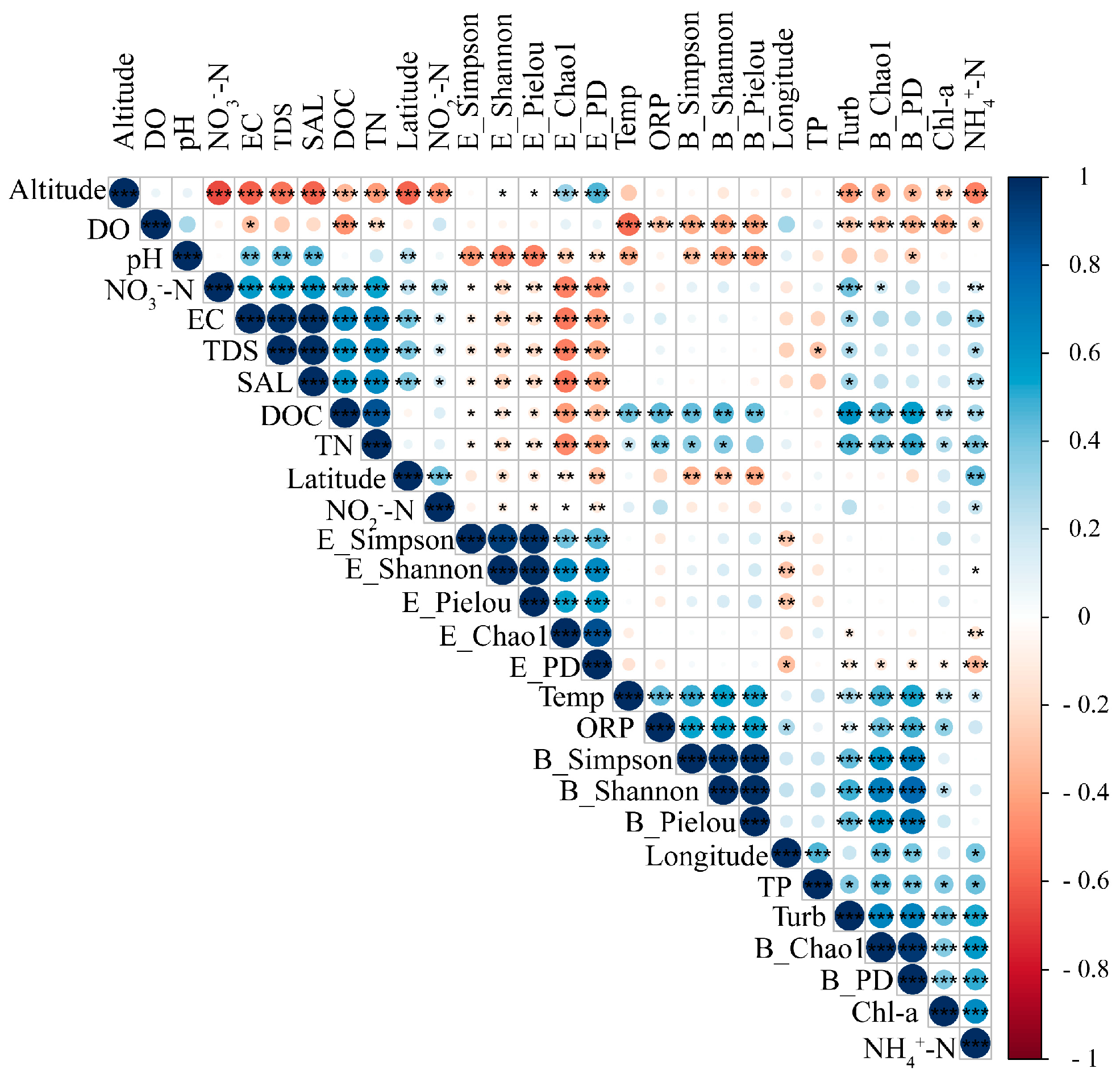
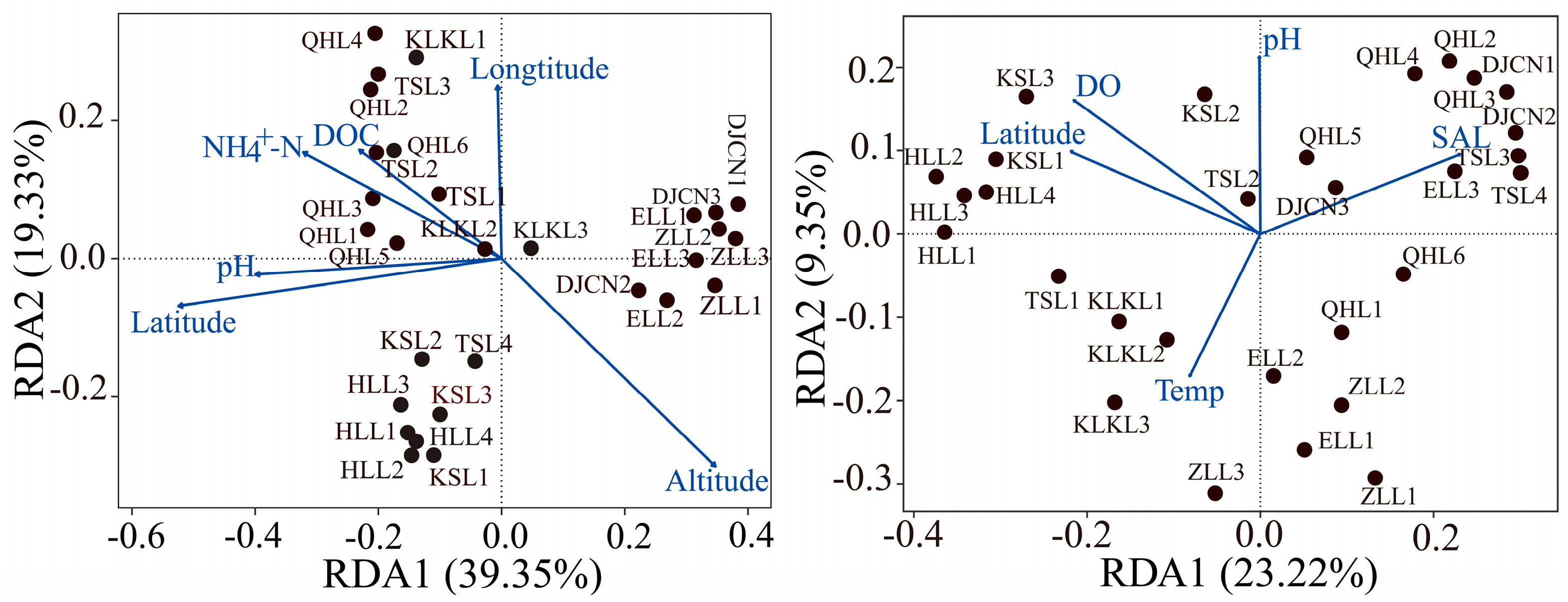
3.1. Variations in Relevant Environmental Factors
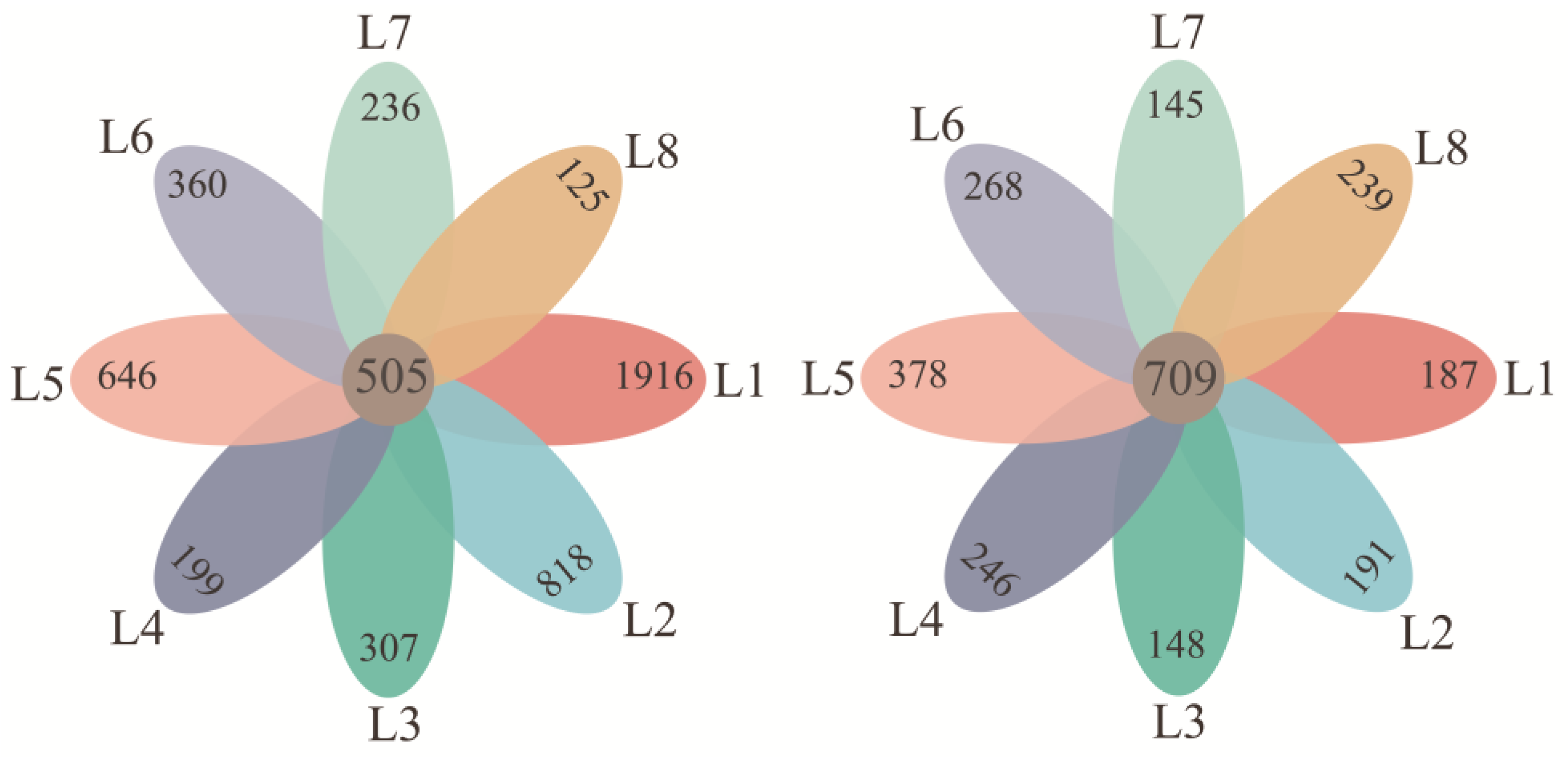
3.2. Microbiological Features
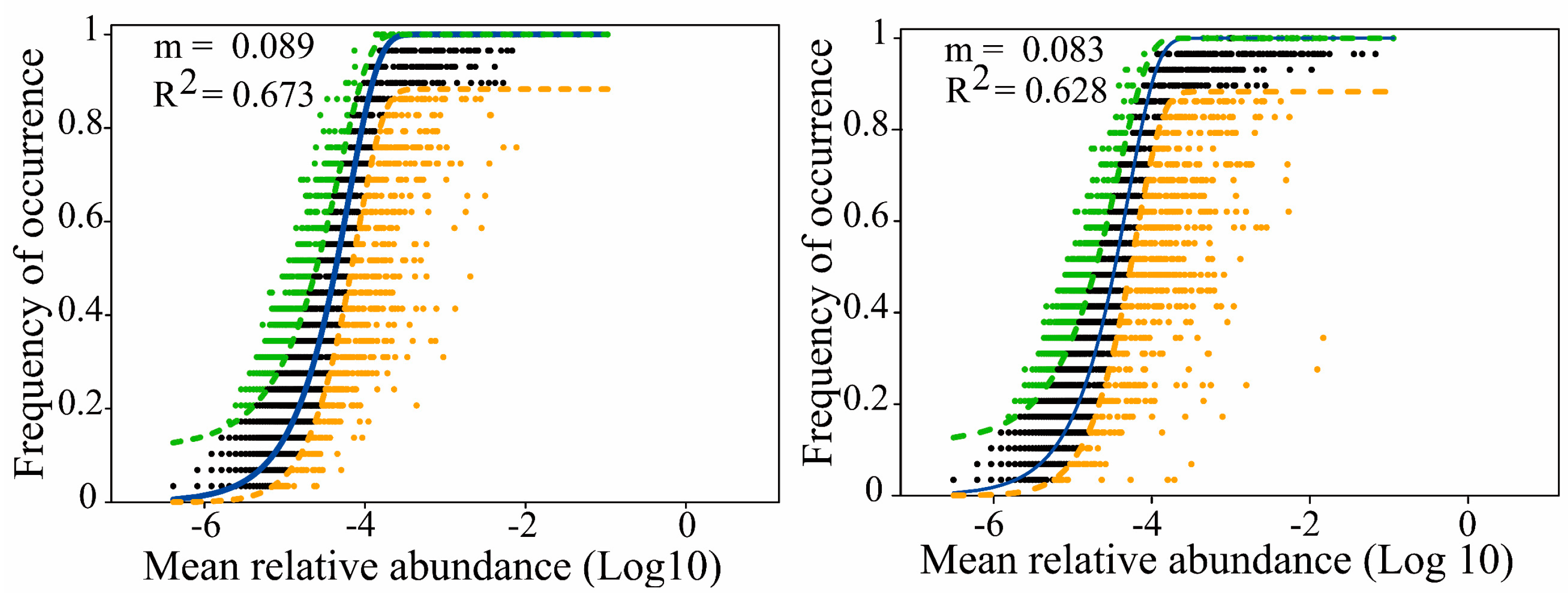
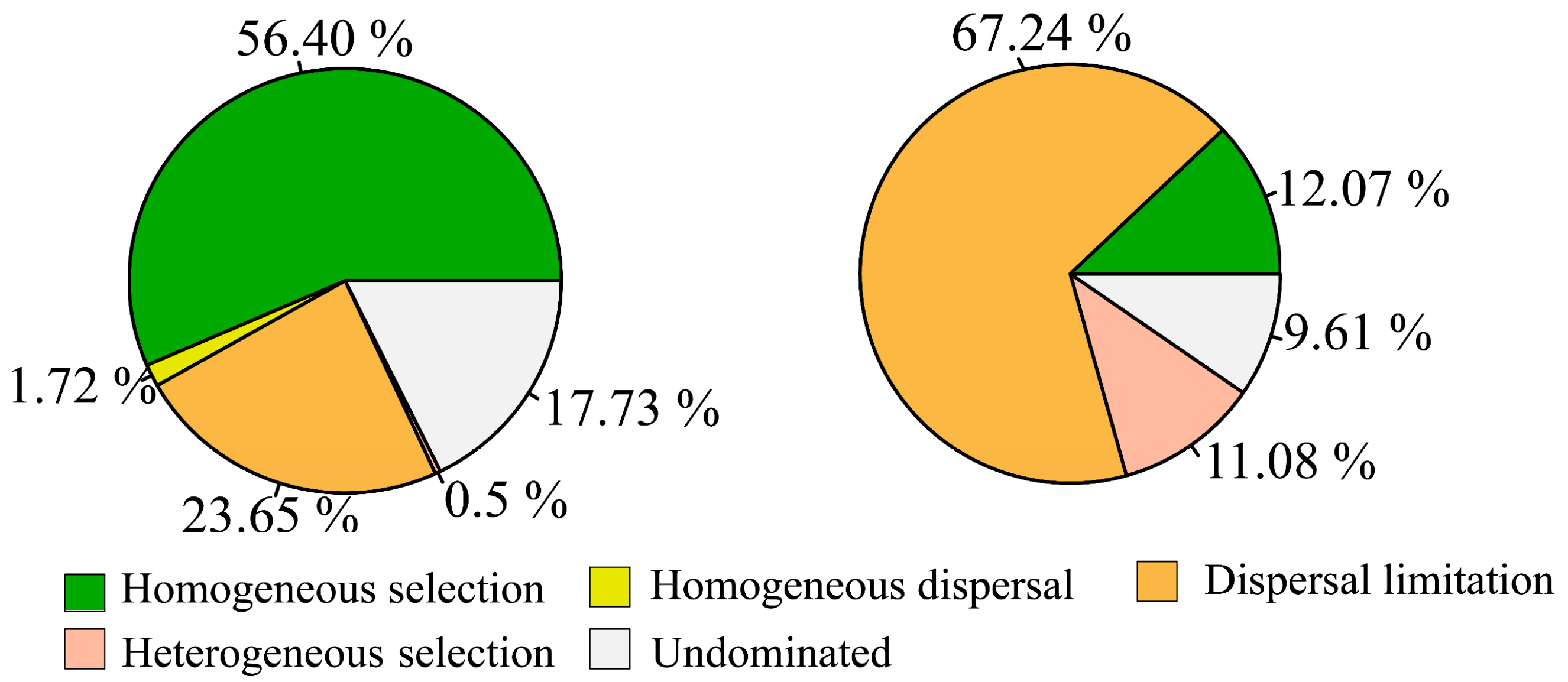
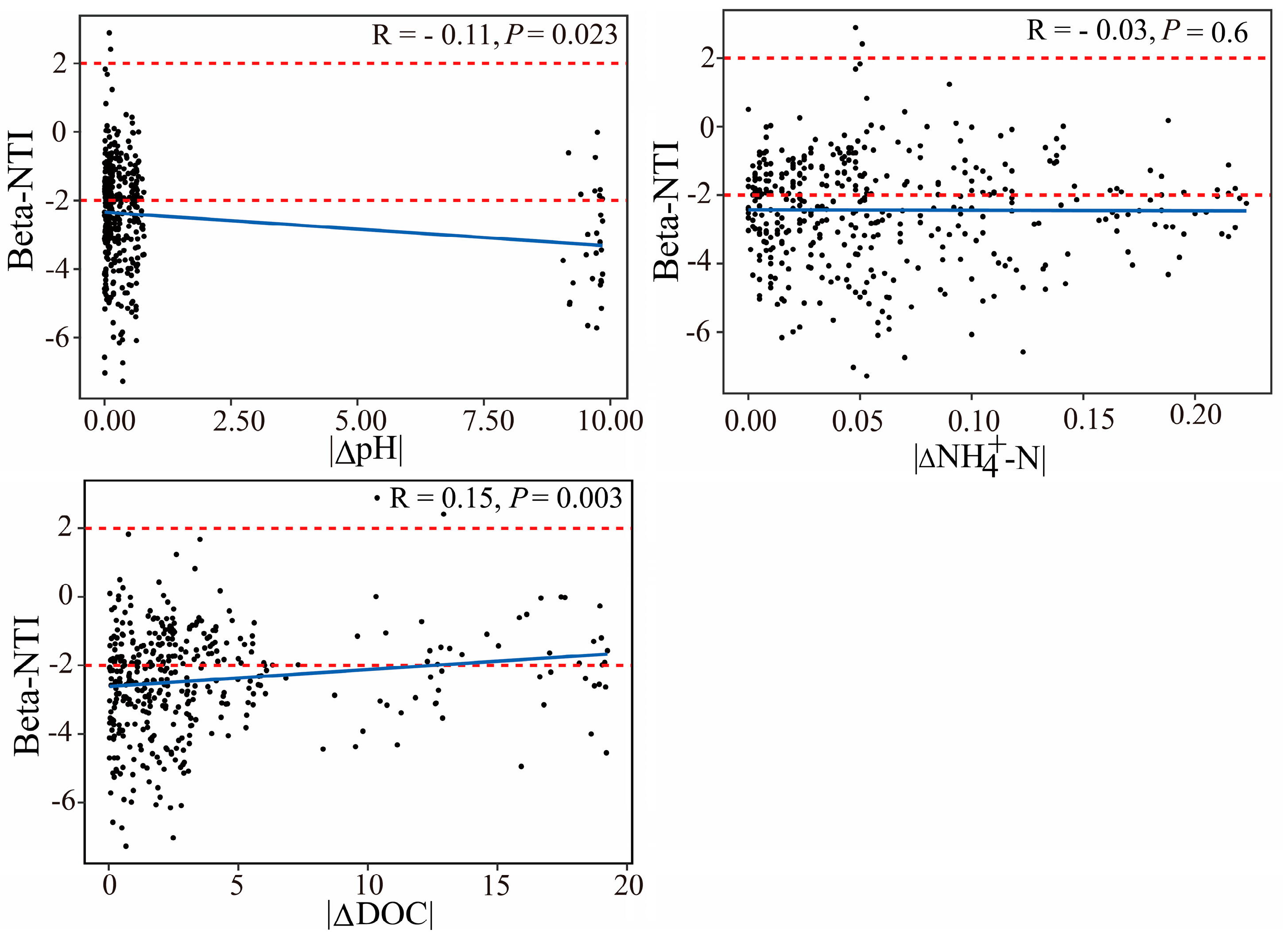
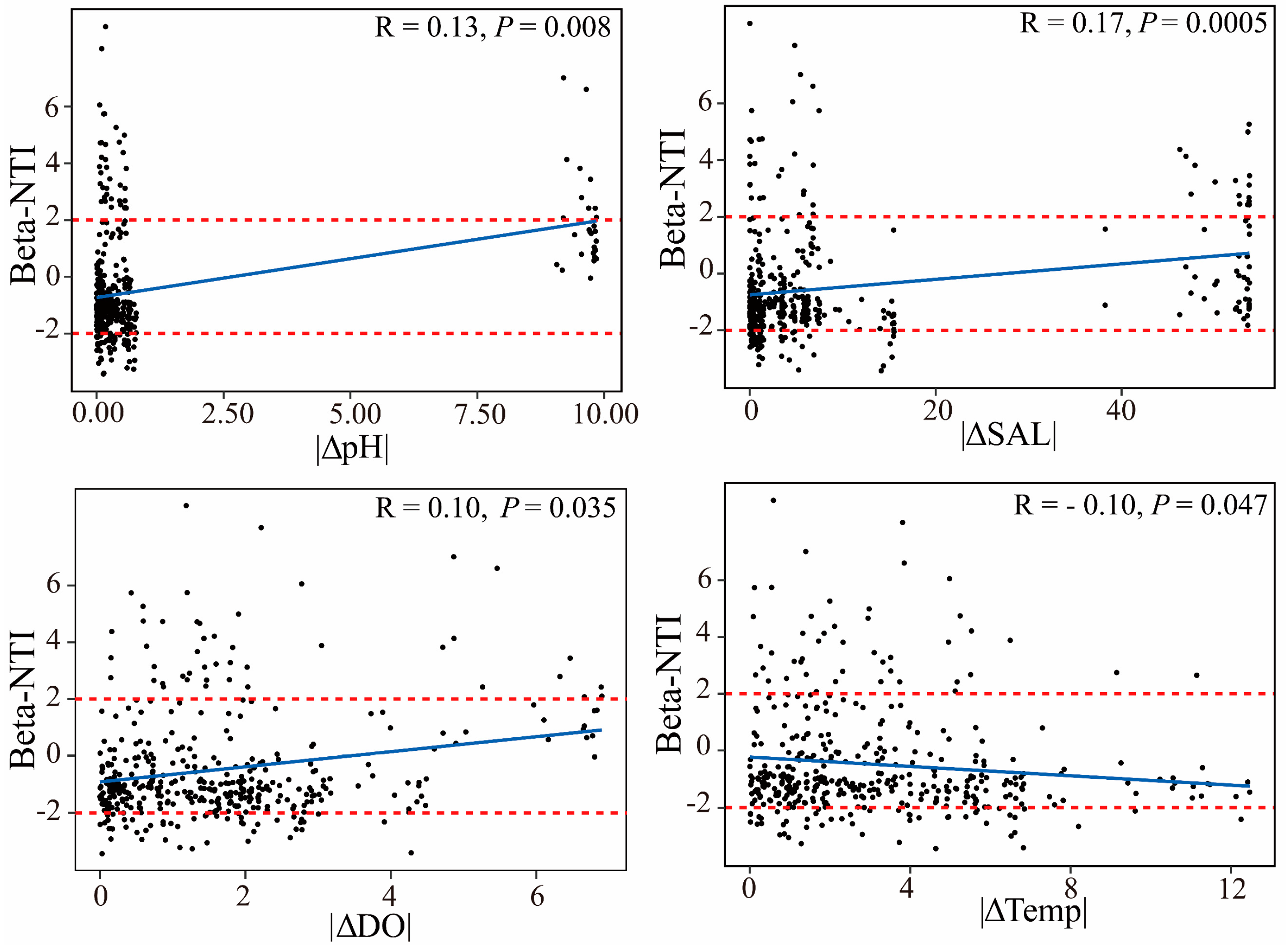
3.3. Disentangling the Microbial Assembly Processes
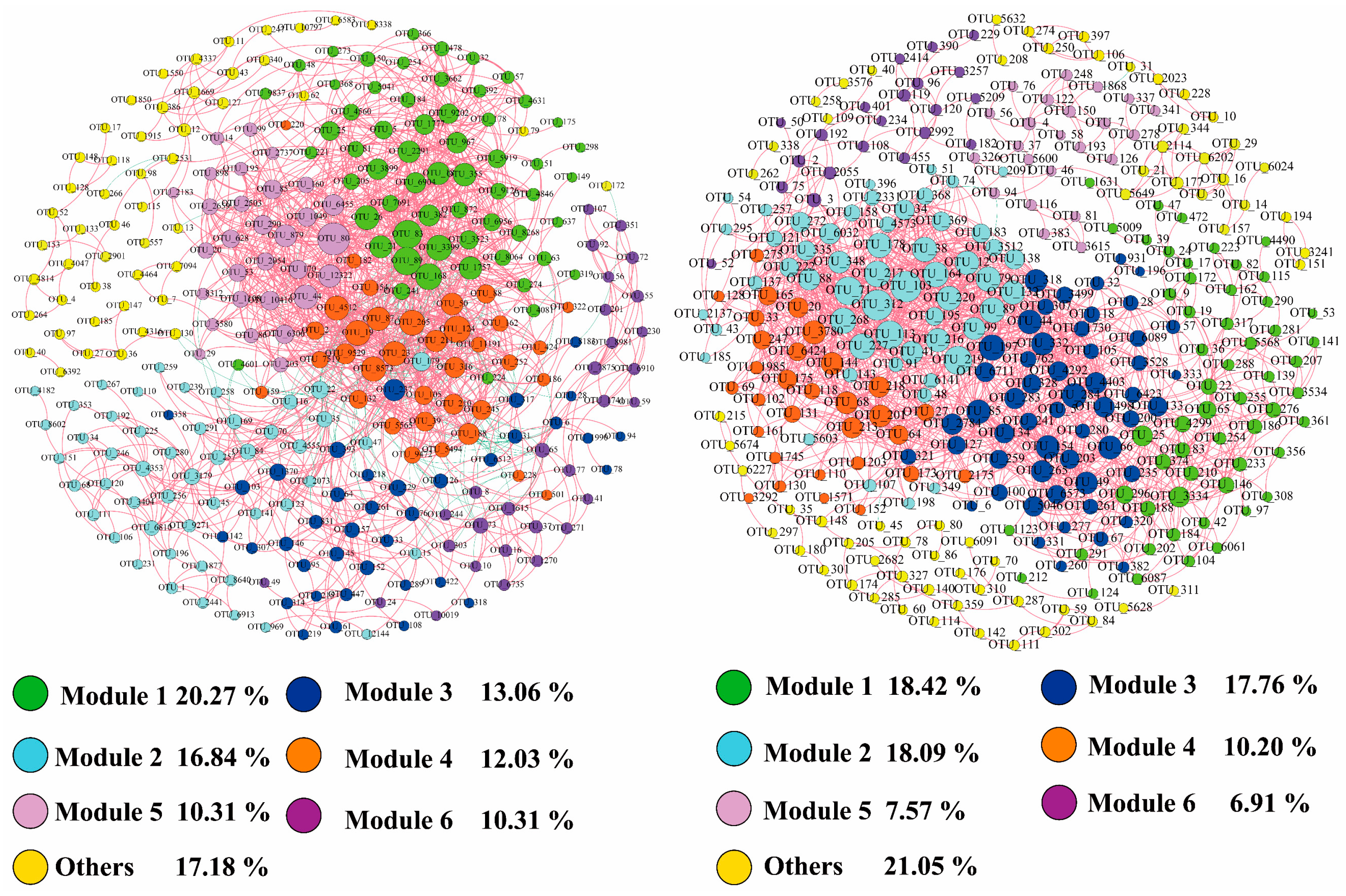
3.4. Network Analysis of Microbial Communities
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sun, J.; Zhou, T.C.; Liu, M.; Chen, Y.C.; Shang, H.; Zhu, L.P.; Ali, S.A.; Yu, H.; Cheng, G.W.; Liu, G.H.; et al. Linkages of the dynamics of glaciers and lakes with the climate elements over the Tibetan Plateau. Earth-Sci. Rev. 2018, 185, 308–324. [Google Scholar] [CrossRef]
- Lei, Y.B.; Yao, T.D.; Yang, K.; Sheng, Y.W.; Kleinherenbrink, M.; Yi, S.; Bird, B.W.; Zhang, X.W.; Zhu, L.; Zhang, G.Q. Lake seasonality across the Tibetan Plateau and their varying relationship with regional mass changes and local hydrology. Geophys. Res. Lett. 2017, 44, 892–900. [Google Scholar] [CrossRef]
- Llorens-Mares, T.; Catalan, J.; Casamayor, E.O. Taxonomy and functional interactions in upper and bottom waters of an oligotrophic high-mountain deep lake (Redon, Pyrenees) unveiled by microbial metagenomics. Sci. Total Environ. 2020, 707, 135929. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.Y.; Chen, J.F.; Tong, T.L.; Xie, S.G.; Liu, Y. Influences of eutrophication on methanogenesis pathways and methanogenic microbial community structures in freshwater lakes. Environ. Pollut. 2020, 260, 114106. [Google Scholar] [CrossRef] [PubMed]
- Ji, M.K.; Kong, W.D.; Yue, L.Y.; Wang, J.B.; Deng, Y.; Zhu, L.P. Salinity reduces bacterial diversity, but increases network complexity in Tibetan Plateau lakes. FEMS Microbiol. Ecol. 2019, 95, fiz190. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Jiang, H.C.; Sun, X.X.; Huang, J.R.; Han, M.X.; Wang, B.C. Distinct co-occurrence patterns of prokaryotic community between the waters and sediments in lakes with different salinity. FEMS Microbiol. Ecol. 2020, 97, fiaa234. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.D.; Ren, K.X.; Isabwe, A.; Chen, H.H.; Liu, M.; Yang, J. Stochastic processes shape microeukaryotic community assembly in a subtropical river across wet and dry seasons. Microbiome 2019, 7, 138. [Google Scholar]
- Wiens, J.J. The niche, biogeography and species interactions. Philos. Trans. R Soc. Lond. B Biol. Sci. 2011, 366, 2336–2350. [Google Scholar] [CrossRef]
- Stegen, J.C.; Lin, X.; Konopka, A.E.; Fredrickson, J.K. Stochastic and deterministic assembly processes in subsurface microbial communities. ISME J. 2012, 6, 1653–1664. [Google Scholar] [CrossRef]
- Stegen, J.C.; Lin, X.; Fredrickson, J.K.; Chen, X.; Kennedy, D.W.; Murray, C.J.; Rockhold, M.L.; Konopka, A. Quantifying community assembly processes and identifying features that impose them. ISME J. 2013, 7, 2069–2079. [Google Scholar] [CrossRef]
- He, Q.; Wang, S.; Hou, W.G.; Feng, K.; Li, F.R.; Hai, W.M.; Zhang, Y.D.; Sun, Y.X.; Deng, Y. Temperature and microbial interactions drive the deterministic assembly processes in sediments of hot springs. Sci. Total Environ. 2021, 772, 145465. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Gao, Y.; Zhang, W.L.; Wang, C.; Wang, P.F.; Niu, L.H.; Wu, H.N. Homogeneous selection dominates the microbial community assembly in the sediment of the Three Gorges Reservoir. Sci. Total Environ. 2019, 690, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Langenheder, S.; Wang, J.; Karjalainen, S.M.; Laamanen, T.M.; Tolonen, K.T.; Vilmi, A.; Heino, J. Bacterial metacommunity organization in a highly connected aquatic system. FEMS Microbiol. Ecol. 2017, 93, fiw225. [Google Scholar] [CrossRef]
- Jiao, C.C.; Zhao, D.Y.; Zeng, J.; Guo, L.; Yu, Z.B. Disentangling the seasonal co-occurrence patterns and ecological stochasticity of planktonic and benthic bacterial communities within multiple lakes. Sci. Total Environ. 2020, 740, 140010. [Google Scholar] [CrossRef] [PubMed]
- Gad, M.; Hou, L.; Li, J.; Wu, Y.; Rashid, A.; Chen, N.; Hu, A. Distinct mechanisms underlying the assembly of microeukaryotic generalists and specialists in an anthropogenically impacted river. Sci. Total Environ. 2020, 748, 141434. [Google Scholar] [CrossRef]
- Mo, Y.Y.; Peng, F.; Gao, X.F.; Xiao, P.; Logares, R.; Jeppesen, E.; Ren, K.X.; Xue, Y.Y.; Yang, J. Low shifts in salinity determined assembly processes and network stability of microeukaryotic plankton communities in a subtropical urban reservoir. Microbiome 2021, 9, 128. [Google Scholar] [CrossRef]
- Zhao, D.Y.; Cao, X.Y.; Huang, R.; Zeng, J.; Shen, F.; Xu, H.M.; Wang, S.C.; He, X.W.; Yu, Z.B. The heterogeneity of composition and assembly processes of the microbial community between different nutrient loading lake zones in Taihu Lake. Appl. Microbiol. Biotechnol. 2017, 101, 5913–5923. [Google Scholar] [CrossRef]
- Gu, Z.Q.; Liu, K.S.; Pedersen, M.W.; Wang, F.; Chen, Y.Y.; Zeng, C.; Liu, Y.Q. Community assembly processes underlying the temporal dynamics of glacial stream and lake bacterial communities. Sci. Total Environ. 2020, 761, 143178. [Google Scholar] [CrossRef]
- Vass, M.; Szekely, A.J.; Lindstrom, E.S.; Langenheder, S. Using null models to compare bacterial and microeukaryotic metacommunity assembly under shifting environmental conditions. Sci. Rep. 2020, 10, 2455. [Google Scholar] [CrossRef]
- Liu, K.S.; Hou, J.Z.; Liu, Y.Q.; Hu, A.Y.; Wang, M.D.; Wang, F.; Chen, Y.Y.; Gu, Z.Q. Biogeography of the free-living and particle-attached bacteria in Tibetan lakes. FEMS Microbiol. Ecol. 2019, 95, fiz088. [Google Scholar] [CrossRef]
- Liu, K.S.; Liu, Y.Q.; Hu, A.Y.; Wang, F.; Chen, Y.Y.; Gu, Z.Q.; Anslan, S.; Hou, J.Z. Different community assembly mechanisms underlie similar biogeography of bacteria and microeukaryotes in Tibetan lakes. FEMS Microbiol. Ecol. 2020, 96, fiaa071. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.D.; Huang, Y.F.; Zhang, S.; Liu, S.F.; Wang, T.; Yang, H.J. Differences in bacterial diversity, composition, and community networks in lake water across three distinct regions on the Qinghai-Tibet Plateau. Front. Environ. Sci. 2022, 10, 1033160. [Google Scholar] [CrossRef]
- Hosam, E.; Taha, S.; Hany, T.; Hiroki, G.; Holger, J.K. Phylogenetic characterization of eukaryotic and prokaryotic gut flora of Nile tilapia, Oreochromis niloticus, along niches of Lake Nasser, Egypt, based on rRNA gene high-throughput sequences. Ecol. Genet. Genom. 2019, 11, 100037. [Google Scholar]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Huttley, G.A. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Zhou, L.; Zhou, Y.Q.; Hu, Y.; Cai, J.; Liu, X.; Bai, C.R.; Tang, X.M.; Zhang, Y.L.; Jang, K.S.; Spencer, R.G.M.; et al. Microbial production and consumption of dissolved organic matter in glacial ecosystems on the Tibetan Plateau. Water Res. 2019, 160, 18–28. [Google Scholar] [CrossRef]
- Faith, D.P. Conservation evaluation and phylogenetic diversity. Biol. Conserv. 1992, 61, 1–10. [Google Scholar] [CrossRef]
- Xu, Z.M.; Li, Y.F.; Lu, Y.H.; Li, Y.D.; Yuan, Z.W.; Dai, M.H.; Liu, H.B. Impacts of the Zhe-Min Coastal Current on the biogeographic pattern of microbial eukaryotic communities. Prog. Oceanogr. 2020, 183, 102309. [Google Scholar] [CrossRef]
- Liu, J.W.; Zhu, S.Q.; Liu, X.Y.; Yao, P.; Ge, T.T.; Zhang, X.H. Spatiotemporal dynamics of the archaeal community in coastal sediments: Assembly process and co-occurrence relationship. ISME J. 2020, 14, 1463–1478. [Google Scholar] [CrossRef]
- Sloan, W.T.; Lunn, M.; Woodcock, S.; Head, I.M.; Nee, S.; Curtis, T.P. Quantifying the roles of immigration and chance in shaping prokaryote community structure. Environ. Microbiol. 2006, 8, 732–740. [Google Scholar] [CrossRef]
- Jiao, S.; Chen, W.M.; Wei, G.H.; Kivlin, S. Linking phylogenetic niche conservatism to soil archaeal biogeography, community assembly and species coexistence. Global Ecol. Biogeogr. 2021, 30, 1488–1501. [Google Scholar] [CrossRef]
- Fine, P.V.A.; Kembel, S.W. Phylogenetic community structure and phylogenetic turnover across space and edaphic gradients in western Amazonian tree communities. Ecography 2011, 34, 552–565. [Google Scholar] [CrossRef]
- Stegen, J.C.; Lin, X.; Fredrickson, J.K.; Konopka, A.E. Estimating and mapping ecological processes influencing microbial community assembly. Front Microbiol. 2015, 6, 370. [Google Scholar] [CrossRef] [PubMed]
- Jiao, S.; Yang, Y.F.; Xu, Y.Q.; Zhang, J.; Lu, Y.H. Balance between community assembly processes mediates species coexistence in agricultural soil microbiomes across eastern China. ISME J. 2020, 14, 202–216. [Google Scholar] [CrossRef] [PubMed]
- Chase, J.M.; Kraft, J.B.; Smith, K.G.; Vellend, M.; Inouye, B.D. Using null models to disentangle variation in community dissimilarity from variation in α-diversity. Ecosphere 2011, 2, 24. [Google Scholar] [CrossRef]
- Wang, B.H.; Zheng, X.F.; Zhang, H.J.; Xiao, F.S.; Gu, H.; Zhang, K.K.; He, Z.L.; Liu, X.; Yan, Q.Y. Bacterial community responses to tourism development in the Xixi National Wetland Park, China. Sci. Total Environ. 2020, 720, 137570. [Google Scholar] [CrossRef] [PubMed]
- Csardi, G.; Nepusz, T. The igraph software. Complex Syst. 2006, 1695, 1–9. [Google Scholar]
- Hou, F.; Zhang, H.; Xie, W.; Zhou, X.; Zhu, X.; Zhang, D. Co-occurrence patterns and assembly processes of microeukaryotic communities in an early-spring diatom bloom. Sci. Total Environ. 2020, 711, 134624. [Google Scholar] [CrossRef]
- Zhang, H.J.; Huang, X.L.; Huang, L.; Bao, F.J.; Xiong, S.L.; Wang, K.; Zhang, D.M. Microeukaryotic biogeography in the typical subtropical coastal waters with multiple environmental gradients. Sci. Total Environ. 2018, 635, 618–628. [Google Scholar] [CrossRef]
- Xiong, J.B.; Liu, Y.Q.; Lin, X.G.; Zhang, H.Y.; Zeng, J.; Hou, J.Z.; Yang, Y.P.; Yao, T.D.; Knight, R.; Chu, H.Y. Geographic distance and pH drive bacterial distribution in alkaline lake sediments across Tibetan Plateau. Environ. Microbiol. 2012, 14, 2457–2466. [Google Scholar] [CrossRef]
- Zhong, Z.P.; Liu, Y.; Miao, L.L.; Wang, F.; Chu, L.M.; Wang, J.L.; Liu, Z.P. Prokaryotic Community Structure Driven by Salinity and Ionic Concentrations in Plateau Lakes of the Tibetan Plateau. Appl. Environ. Microbiol. 2016, 82, 1846–1858. [Google Scholar] [CrossRef]
- Wu, Q.L.; Zwart, G.; Schauer, M.; Kamst-van Agterveld, M.P.; Hahn, M.W. Bacterioplankton community composition along a salinity gradient of sixteen high-mountain lakes located on the Tibetan Plateau, China. Appl. Environ. Microbiol. 2006, 72, 5478–5485. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Jiao, C.C.; Zhao, D.Y.; Xu, H.M.; Huang, R.; Cao, X.Y.; Yu, Z.B.; Wu, Q.L. Patterns and assembly processes of planktonic and sedimentary bacterial community differ along a trophic gradient in freshwater lakes. Ecol. Indic. 2019, 106, 105491. [Google Scholar] [CrossRef]
- Walks, D.J.; Cyr, H. Movement of plankton through lake-stream systems. Freshwat. Biol. 2004, 49, 745–759. [Google Scholar] [CrossRef]
- Robert, E.D.; Michael, D.J.; Chris, K.; Michael, S.; Wilkins, M.J. Microbial Community Cohesion Mediates Community Turnover in Unperturbed Aquifers. mSystems 2018, 3, e00066-00018. [Google Scholar]
- Wu, P.F.; Li, D.X.; Kong, L.F.; Li, Y.Y.; Zhang, H.; Xie, Z.X.; Wang, D.Z. The diversity and biogeography of microeukaryotes in the euphotic zone of the northwestern Pacific Ocean. Sci Total Environ. 2020, 698, 134289. [Google Scholar] [CrossRef]
- Calcagno, V.; Jarne, P.; Loreau, M.; Mouquet, N.; David, P. Diversity spurs diversification in ecological communities. Nat. Commun. 2017, 8, 15810. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Network Properties | Bacteria | Microeukaryotes | |
|---|---|---|---|
| Random network | Average degree (avg K) | 5.064 | 5.382 |
| Average clustering coefficient (avg CC) | 0.05 | 0.07 | |
| Average path distance (GD) | 3.767 | 3.824 | |
| Geodesic efficiency (E) | 0.314 | 0.311 | |
| Harmonic geodesic distance (HD) | 3.181 | 3.217 | |
| Modularity | 0.413 | 0.459 | |
| Empirical network | Nodes | 291 | 304 |
| Edges | 1728 | 1726 | |
| (Positive/negative%) | (92.94%/7.06%) | (99.97%/0.23%) | |
| Average degree | 11.876 | 11.335 | |
| Graph density | 0.041 | 0.037 | |
| Modularity | 0.627 | 0.523 | |
| Average path length | 3.751 | 4.873 | |
| Average clustering coefficient | 0.538 | 0.572 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, D.; Huang, Y.; Jia, H.; Yang, H. Bacterial and Microeukaryotic Community Compositions and Their Assembly Processes in Lakes on the Eastern Qinghai-Tibet Plateau. Microorganisms 2024, 12, 32. https://doi.org/10.3390/microorganisms12010032
Wang D, Huang Y, Jia H, Yang H. Bacterial and Microeukaryotic Community Compositions and Their Assembly Processes in Lakes on the Eastern Qinghai-Tibet Plateau. Microorganisms. 2024; 12(1):32. https://doi.org/10.3390/microorganisms12010032
Chicago/Turabian StyleWang, Dandan, Yuefei Huang, Haichao Jia, and Haijiao Yang. 2024. "Bacterial and Microeukaryotic Community Compositions and Their Assembly Processes in Lakes on the Eastern Qinghai-Tibet Plateau" Microorganisms 12, no. 1: 32. https://doi.org/10.3390/microorganisms12010032