
Detection of Nitrate-Reducing/Denitrifying Bacteria from Contaminated and Uncontaminated Tallgrass Prairie Soil: Limitations of PCR Primers


Abstract
:1. Introduction
2. Materials and Methods
2.1. Summary of Sampling Sites and Sampling Protocol
2.2. Selection and Isolation of Nitrate- and Nitrite-Reducing Bacteria
2.3. Obtaining Pure Cultures from Microtiter Plate Wells Positive for DN or NR
2.4. DNA Extraction from Strains
2.5. Molecular Detection of 16S rRNA Gene Sequences and Nitrate Reduction/Denitrification Functional Genes
2.6. DNA Sequencing and Analysis
3. Results
3.1. Distribution of Strains by Origin and Capacity to Reduce Nitrate/Nitrite
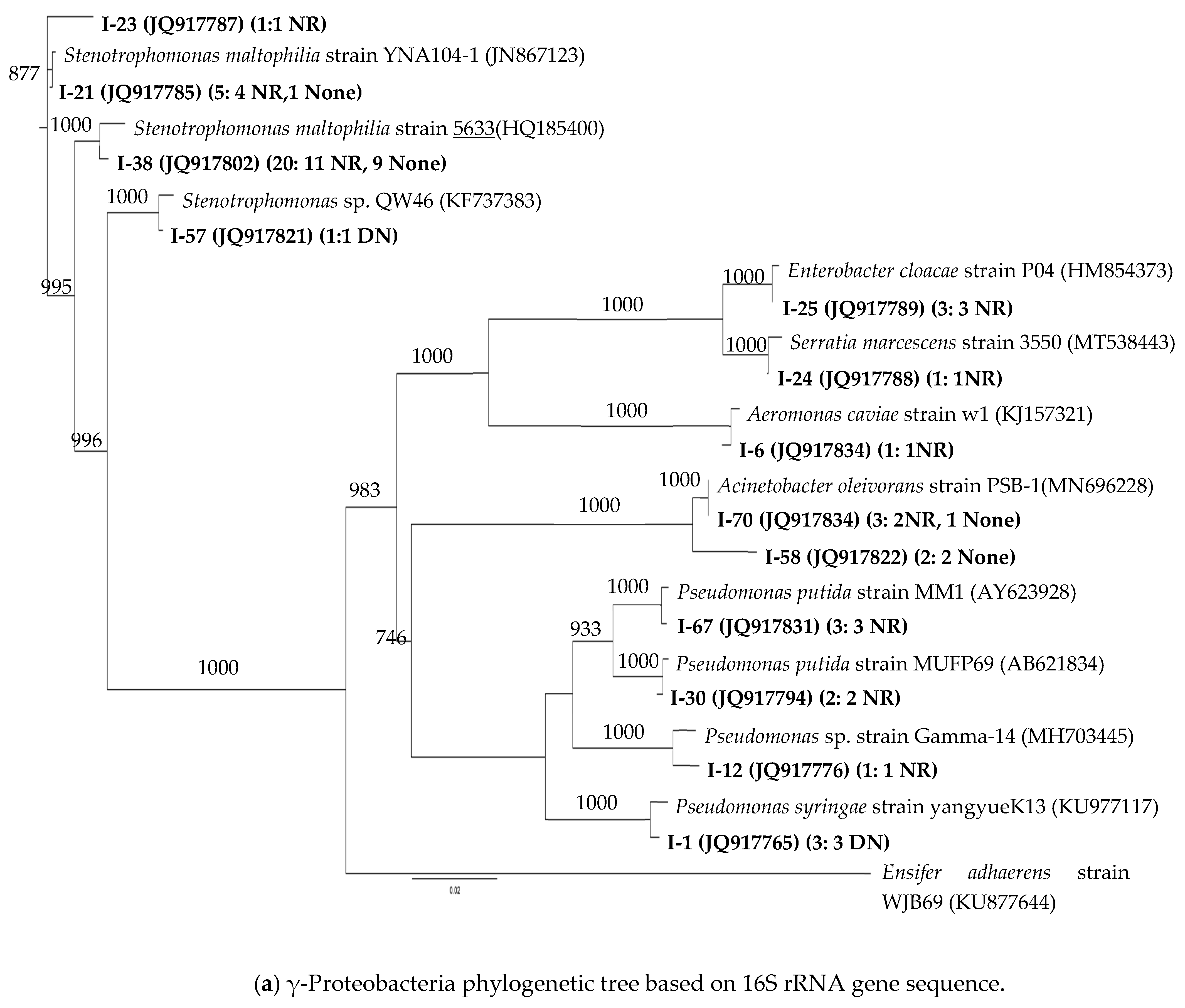
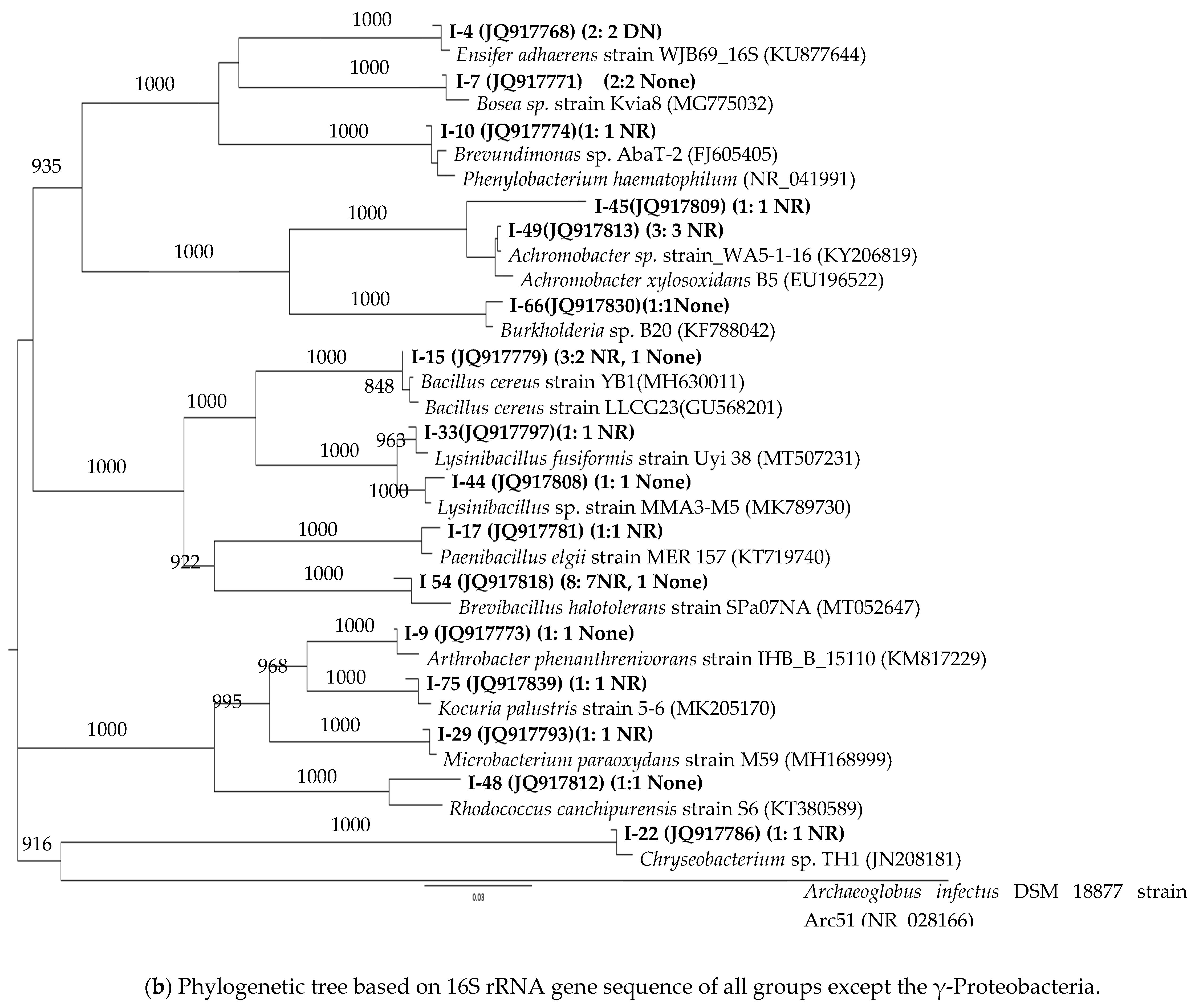
3.2. 16S rRNA Phylogenetic Affiliations of the Strains
3.3. Molecular Detection of Nitrate-Reducing Functional Genes napA and narG
3.4. Molecular Detection of Denitrifying Functional Genes nirS, nirK, cnorB, qnorB, and nosZ
4. Discussion
4.1. 16S rRNA Phylogeny and the Reduction of Nitrate/Nitrite
4.2. Considerations When Estimating Numbers of NR and DN Bacteria by PCR
4.3. Long-Term Impact of Brine/Oil Contamination on Nitrogen Cycling in Tallgrass Prairie Soil
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fierer, N.; Jackson, R.B. The diversity and biogeography of soil bacterial communities. Proc. Natl. Acad. Sci. USA 2006, 103, 626–631. [Google Scholar] [CrossRef] [PubMed]
- Bragg, J.R.; Prince, R.C.; Harner, E.J.; Atlas, R.M. Effectiveness of bioremediation for the Exxon Valdez oil spill. Nature 1994, 368, 413–418. [Google Scholar] [CrossRef]
- Harayama, S.; Kasai, Y.; Hara, A. Microbial communities in oil-contaminated seawater. Curr. Opin. Biotechnol. 2004, 15, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Head, I.M.; Jones, D.M.; Roling, W.F. Marine microorganisms make a meal of oil. Nat Rev. Microbiol. 2006, 4, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Boch, J.; Nau-Wagner, G.; Kneip, S.; Bremer, E. Glycine betaine aldehyde dehydrogenase from Bacillus subtilis: Characterization of an enzyme required for the synthesis of the osmoprotectant glycine betaine. Arch. Microbiol. 1997, 168, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Egamberdiyeva, D. Characterization of Pseudomonas species isolated from the rhizosphere of plants grown in serozem soil, semi arid region of Uzbekistan. Sci. World J. 2005, 5, 501–509. [Google Scholar] [CrossRef]
- Nie, M.; Wang, Y.; Yu, J.; Xiao, M.; Jiang, L.; Yang, J.; Fang, C.; Chen, J.; Li, B. Understanding plant-microbe interactions for phytoremediation of petroleum-polluted soil. PLoS ONE 2011, 6, e17961. [Google Scholar] [CrossRef] [PubMed]
- Song, B.; Palleroni, N.J.; Häggblom, M.M. Isolation and characterization of diverse halobenzoate-degrading denitrifying bacteria from soils and sediments. Appl. Environ. Microbiol. 2000, 66, 3446–3453. [Google Scholar] [CrossRef]
- Vargas, C.; Song, B.; Camps, M.; Häggblom, M.M. Anaerobic degradation of fluorinated aromatic compounds. Appl. Microbiol. Biotechnol. 2000, 53, 342–347. [Google Scholar] [CrossRef]
- An, Y.J.; Joo, Y.H.; Hong, I.Y.; Ryu, H.W.; Cho, K.S. Microbial characterization of toluene-degrading denitrifying consortia obtained from terrestrial and marine ecosystems. Appl. Microbiol. Biotechnol. 2004, 65, 611–619. [Google Scholar] [CrossRef]
- AbuBakr, S.M. Nitrate reducing and denitrifying bacteria in oil/brine contaminated soils. Atlas J. Biol. 2020, 1, 699–705. [Google Scholar]
- Duncan, K.E.; Kolhatkar, R.; Subramaniam, G.; Narasimhan, R.; Jennings, E.; Hettenbach, S.; Brown, A.; McComas, C.; Potter, W.; Sublett, K. Microbial dynamics in oil-impacted prairie soil. Appl. Biochem. Biotechnol. 1999, 77–79, 421–434. [Google Scholar] [CrossRef] [PubMed]
- AbuBakr, S.M.; Duncan, K.E.; Thoma, G.; Sublette, K. The Effect of Environmental Factors on the Abundance of Culturable Nitrate Reducing/Denitrifying Bacteria from Contaminated and Uncontaminated Tallgrass Prairie Soil. Atlas J. Biol. 2019, 1, 592–602. [Google Scholar] [CrossRef]
- Zumft, W.G. Cell biology and molecular basis of denitrification. Microbiol. Mol. Biol. Rev. 1997, 61, 533–536. [Google Scholar]
- Philippot, L.; Piutti, S.; Martin-Laurent, F.; Hallet, S.; Germon, J.C. Molecular analysis of the nitrate-reducing community from unplanted and maize-planted soils. Appl. Environ. Microbiol. 2002, 68, 6121–6128. [Google Scholar] [CrossRef]
- He, W.; Liu, S.; Jiang, Z.; Zheng, J.; Li, X.; Zhang, D. The Diversity and Nitrogen Metabolism of Culturable Nitrate-Utilizing Bacteria Within the Oxygen Minimum Zone of the Changjiang (Yangtze River). Estuary. Front. Mar. Sci. 2021, 8, 720413. [Google Scholar] [CrossRef]
- Philippot, L.; Hojberg, O. Dissimilatory nitrate reductases in bacteria. Biochim. Biophys. Acta 1999, 1446, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Philippot, L. Denitrifying genes in bacterial and archaeal genomes. Biochim. Biophys. Acta 2002, 1577, 355–376. [Google Scholar] [CrossRef]
- Roussel-Delif, L.; Tarnawski, S.; Hamelin, J.; Philippot, L.; Aragno, M.; Fromin, N. Frequency and diversity of nitrate reductase genes among nitrate-dissimilating Pseudomonas in the rhizosphere of perennial grasses grown in field conditions. Microb. Ecol. 2005, 49, 63–72. [Google Scholar] [CrossRef]
- Kelly, C.N.; Schwaner, G.W.; Cumming, J.R.; Driscoll, T.P. Metagenomic reconstruction of nitrogen and carbon cycling pathways in forest soil: Influence of different hardwood tree species. Soil Biol. Biochem. 2021, 156, 108226. [Google Scholar] [CrossRef]
- Ye, R.W.; Averill, B.A.; Tiedje, J.M. Denitrification: Production and consumption of nitric oxide. Appl. Environ. Microbiol. 1994, 60, 1053–1058. [Google Scholar] [CrossRef] [PubMed]
- Casciotti, K.L.; Ward, B.B. Dissimilatory nitrite reductase genes from autotrophic ammonia-oxidizing bacteria. Appl. Environ. Microbiol. 2001, 67, 2213–2221. [Google Scholar] [CrossRef]
- Braker, G.; Zhou, J.; Wu, L.; Devol, A.H.; Tiedje, J.M. Nitrite reductase genes (nirK and nirS) as functional markers to investigate diversity of denitrifying bacteria in Pacific Northwest marine sediment communities. Appl. Environ. Microbiol. 2000, 66, 2096–2104. [Google Scholar] [CrossRef] [PubMed]
- Gruntzig, V.; Nold, S.C.; Zhou, J.; Tiedje, J.M. Pseudomonas stutzeri nitrite reductase gene abundance in environmental samples measured by real-time PCR. Appl. Environ. Microbiol. 2001, 67, 760–768. [Google Scholar] [CrossRef]
- Avrahami, S.; Conrad, R.; Braker, G. Effect of soil ammonium concentration on N2O release and on the community structure of ammonia oxidizers and denitrifiers. Appl. Environ. Microbiol. 2002, 68, 5685–5692. [Google Scholar] [CrossRef]
- Braker, G.; Tiedje, J.M. Nitric oxide reductase (norB) genes from pure cultures and environmental samples. Appl. Environ. Microbiol. 2003, 69, 3476–3483. [Google Scholar] [CrossRef] [PubMed]
- Zumft, W.G. The denitrifying prokaryotes. In The Prokaryotes; Balows, A., Truper, H.G., Dworkin, M., Harder, W., Schleifer, K.-H., Eds.; Springer: New York, NY, USA, 1992; pp. 554–582. [Google Scholar]
- Braker, G.; Fesefeldt, A.; Witzel, K.P. Development of PCR primer systems for amplification of nitrite reductase genes (nirK and nirS) to detect denitrifying bacteria in environmental samples. Appl. Environ. Microbiol. 1998, 64, 3769–3775. [Google Scholar] [CrossRef]
- Scala, D.J.; Kerkhof, L.J. Nitrous oxide reductase (nosZ) gene-specific PCR primers for detection of denitrifiers and three nosZ genes from marine sediments. FEMS Microbiol. Lett. 1998, 162, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Hallin, S.; Lindgren, P.E. PCR detection of genes encoding nitrite reductases in denitrifying bacteria. Appl. Environ. Microbiol. 1999, 65, 1652–1657. [Google Scholar] [CrossRef]
- Scala, D.J.; Kerkhof, L.J. Diversity of nitrous oxide reductase (nosZ) genes in continental shelf sediments. Appl. Environ. Microbiol. 1999, 65, 1681–1687. [Google Scholar] [CrossRef]
- Qiu, X.Y.; Hurt, R.A.; Wu, L.Y.; Chen, C.H.; Tiedje, J.M.; Zhou, J.Z. Detection and quantification of copper-denitrifying bacteria by quantitative competitive PCR. J. Microbiol. Methods 2004, 59, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Sublette, K.; Jennings, E.; Mehta, C.; Duncan, K.; Brokaw, J.; Todd, T.; Thoma, G. Monitoring soil ecosystem recovery following bioremediation of a terrestrial crude oil spill with and without a fertilizer amendment. J. Soils Sediments 2007, 16, 181–208. [Google Scholar] [CrossRef]
- Duncan, K.; Jennings, E.; Hettenbach, S.; Potter, W.; Sublette, K.; Subramaniam, G.; Narasimhan, R. Nitrogen Cycling and Nitric Oxide Emissions in Oil-Impacted Prairie Soils’. Bioremediation J. 1998, 1, 195–208. [Google Scholar] [CrossRef]
- Harris, T.M.; Tapp, J.B.; Sublette, K.L. Remediation of oil-field brine-impacted soil using a subsurface drainage system and hay. Environ. Geosci. 2005, 12, 101–113. [Google Scholar] [CrossRef]
- Sublette, K.L.; Moralwar, A.; Ford, L.P.; Duncan, K.; Thoma, G.; Brokaw, J. Remediation of a spill of crude oil and brine without gypsum. Environ. Geosci. 2005, 12, 115–125. [Google Scholar] [CrossRef]
- Sublette, K.L.; Tapp, J.B.; Fisher, J.B.; Jennings, E.; Duncan, K.; Thoma, G.; Brokaw, J.; Todd, T. Lessons learned in remediation and restoration in the Oklahoma prairie: A review. J. Appl. Geochem. 2007, 22, 2225–2239. [Google Scholar] [CrossRef]
- Shim, H.; Yang, S.T. Biodegradation of benzene, toluene, ethylbenzene, and o-xylene by a coculture of Pseudomonas putida and Pseudomonas fluorescens immobilized in a fibrous-bed bioreactor. J. Biotechnol. 1999, 67, 99–112. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.Y.; Jun, Y.S.; Cho, K.S.; Ryu, H.W. Degradation characteristics of toluene, benzene, ethylbenzene, and xylene by Stenotrophomonas maltophilia T3-c. J. Air Waste Manag. 2002, 52, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Van Hamme, J.D.; Singh, A.; Ward, O.P. Recent advances in petroleum microbiology. Microbiol. Mol. Biol. Rev. 2003, 67, 503–549. [Google Scholar] [CrossRef] [PubMed]
- Dandie, C.E.; Miller, M.N.; Burton, D.L.; Zebarth, B.J.; Trevors, J.T.; Goyer, C. Nitric oxide reductase-targeted real-time PCR quantification of denitrifier populations in soil. Appl. Environ. Microbiol. 2007, 73, 4250–4258. [Google Scholar] [CrossRef]
- Atlas, R.M. Microbial degradation of petroleum hydrocarbons: An environmental perspective. Microbiol Rev. 1981, 45, 180–209. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Chen, Q.; Gong, Z. Microbial remediation of petroleum-contaminated soil focused on the mechanism and microbial response: A review. Environ. Sci. Pollut. Res. Int. 2024, 31, 33325–33346. [Google Scholar] [CrossRef]
- Soussi, M.; Santamaria, M.; Ocãna, A.; Lluch, C. Effects of salinity on protein and lipolysaccharide pattern in a salt-tolerant strain of Mesorhizobium ciceri. J. Appl. Microbiol. 2001, 90, 476–481. [Google Scholar] [CrossRef]
- Hung, M.H.; Bhagwath, A.A.; Shen, F.T.; Devasya, R.P.; Young, C.C. Indigenous rhizobia associated with native shrubby legumes in Taiwan. Pedobiologia 2005, 49, 577–584. [Google Scholar] [CrossRef]
- Alcántara-Hernández, R.J.; Valenzuela-Encinas, C.; Marsch, R.; Dendooven, L. Respiratory and dissimilatory nitrate-reducing communities from an extreme saline alkaline soil of the former lake Texcoco (Mexico). Extremophiles 2009, 13, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Andersen, S.M.; Johnsen, K.; Sørensen, J.; Nielsen, P.; Jacobsen, C.S. Pseudomonas frederiksbergensis sp. nov., isolated from soil at a coal gasification site. Int. J. Syst. Evol. Microbiol. 2000, 6, 1957–1964. [Google Scholar] [CrossRef] [PubMed]
- Atlas, R.M. Handbook of Microbiological Media; CRC Press, Inc.: Boca Raton, FL, USA, 1993. [Google Scholar]
- Jones, R.A.; Broder, M.W.; Stotzky, G. Effects of genetically engineered microorganisms on nitrogen transformations and nitrogen-transforming microbial populations in soil. Appl. Environ. Microbiol. 1991, 57, 3212–3219. [Google Scholar] [CrossRef] [PubMed]
- Baron, E.J.; Finegold, S.M. Bailey and Scott’s Diagnostic Microbiology, 8th ed.; CV Mosby Co.: St. Louis, MO, USA, 1990; pp. 286–402; 435–438. [Google Scholar]
- Smibert, R.M.; Krieg, N.R. Phenotypic characterization. In Methods for General and Molecular Bacteriology; Gerhardt, P., Murray, R.G.E., Wood, W.A., Krieg, N.R., Eds.; American Society for Microbiology: Washington, DC, USA, 1994; p. 649. [Google Scholar]
- Wilson, K.H.; Blitchington, R.B.; Greene, R.C. Amplification of bacterial 16S ribosomal DNA with polymerase chain reaction. J. Clin. Microbiol. 1990, 28, 1942–1946. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed]
- Pruesse, E.; Quast, C.; Knittel, K.; Fuchs, B.M.; Ludwig, W.; Peplies, J.; Glöckner, F.O. SILVA: A comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007, 35, 7188–7196. [Google Scholar] [CrossRef] [PubMed]
- DeSantis, T.Z., Jr.; Hugenholtz, P.; Keller, K.; Brodie, E.L.; Larsen, N.; Piceno, Y.M.; Phan, R.; Andersen, G.L. NAST: A multiple sequence alignment server for comparative analysis of 16S rRNA genes. Nucleic Acids Res. 2006, 34, 394–399. [Google Scholar] [CrossRef]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmongin, F.; Higgins, D.G. The ClustalX windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25, 4876–4882. [Google Scholar] [CrossRef]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [PubMed]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 666–670. [Google Scholar] [CrossRef]
- Sun, L.; Sheng, Q.; Ge, Y.; He, L.; Sheng, X. The quorum sensing SinI/R system contributes to cadmium immobilization in Ensifer adhaerens NER9 in the cadmium-contaminated solution. J. Hazard. Mater. 2024, 15, 470:134300. [Google Scholar] [CrossRef] [PubMed]
- Nijburg, J.; Coolen, M.J.L.; Gerards, S.; Gunnewiek, P.J.A.K.; Lannbroek, H.J. Effects of nitrate availability and the presence of Glyceria maxima on the composition and activity of the dissimilatory nitrate-reducing bacteria community. Appl. Environ. Microbiol. 1997, 63, 931–937. [Google Scholar] [CrossRef] [PubMed]
- Shirey, J.J.; Sextone, A.J. Denitrification and nitrate-reducing bacterial populations. FEMS Microbiol. Ecol. 1989, 62, 59–70. [Google Scholar] [CrossRef]
- Verbaendert, I.; Boon, N.; De Vos, P.; Heylen, K. Denitrification is a common feature among members of the genus Bacillus. Syst. Appl. Microbiol. 2011, 34, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Rich, J.J.; Heichen, R.S.; Bottomley, P.J.; Cromack Jr, K.; Myrold, D.D. Community composition and functioning of denitrifying bacteria from adjacent meadow and forest soils. Appl. Environ. Microbiol. 2003, 69, 5974–5982. [Google Scholar] [CrossRef] [PubMed]
- Dandie, C.E.; Burton, D.L.; Zebarth, B.J.; Trevors, J.T.; Goyer, C. Analysis of denitrification genes and comparison of nosZ, cnorB and 16S rDNA from culturable denitrifying bacteria in potato cropping systems. Syst. Appl. Microbiol. 2007, 30, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.M.; Stres, B.; Rosenquist, M.; Hallin, S. Phylogenetic analysis of nitrite, nitric oxide, and nitrous oxide respiratory enzymes reveal a complex evolutionary history for denitrification. Mol. Biol. Evol. 2008, 25, 1955–1966. [Google Scholar] [CrossRef] [PubMed]
- Philippot, L.; Andert, J.; Jones, C.M.; Bru, D.; Hallin, S. Importance of denitrifiers lacking the genes encoding the nitrous oxide reductase for N2O emissions from soil. Glob. Chang. Biol. 2011, 17, 1497–1504. [Google Scholar] [CrossRef]
- Shrewsbury, L.H.; Smith, J.L.; Huggins, D.R.; Carpenter-Boggs, L.; Reardon, C.L. Denitrifier abundance has a greater influence on denitrification rates at larger landscape scales but is a lesser driver than environmental variables. Soil Biol. Bioch. 2016, 103, 221–231. [Google Scholar] [CrossRef]
- Ma, Y.; Zilles, J.L.; Kent, A.D. An evaluation of primers for detecting denitrifiers via their functional genes. Environ. Microbiol. 2019, 4, 1196–1210. [Google Scholar] [CrossRef] [PubMed]
- Klatte, T.; Evans, L.; Whitehead, R.N.; Cole, J.A. Four PCR primers necessary for the detection of periplasmic nitrate reductase genes in all groups of Proteobacteria and in environmental DNA. Biochem. Soc. Trans. 2011, 39, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Sanford, R.A.; Wagner, D.D.; Wu, Q.; Chee-Sanford, J.C.; Thomas, S.H.; Cruz-García, C.; Rodríguez, G.; Massol-Deyá, A.; Krishnani, K.K.; Ritalahti, K.M.; et al. Unexpected nondenitrifier nitrous oxide reductase gene diversity and abundance in soils. Proc. Natl. Acad. Sci. USA 2012, 109, 19709–19714. [Google Scholar] [CrossRef]
- Mefferd, C.C.; Zhou, E.; Seymour, C.O.; Bernardo, N.A.; Srivastava, S.; Bengtson, A.J.; Jiao, J.Y.; Dong, H.; Li, W.J.; Hedlund, B.P. Incomplete denitrification phenotypes in diverse Thermus species from diverse geothermal spring sediments and adjacent soils in southwest China. Extremophiles 2022, 26, 23. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, S.O.; Bharathi, P.A.; Bonin, P.C.; Michotey, V.D. Denitrification: An important pathway for nitrous oxide production in tropical mangrove sediments (Goa, India). J. Environ. Qual. 2010, 39, 1507–1516. [Google Scholar] [CrossRef] [PubMed]
- Mackelprang, R.; Grube, A.M.; Lamendella, R.; Jesus, E.d.C.; Copeland, A.; Liang, C.; Jackson, R.D.; Rice, C.W.; Kapucija, S.; Parsa, B.; et al. Microbial Community Structure and Functional Potential in Cultivated and Native Tallgrass Prairie Soils of the Midwestern United States. Front. Microbiol. 2018, 9, 1775. [Google Scholar] [CrossRef] [PubMed]
- Chu, S.; Zhang, D.; Wang, D.; Zhi, Y.; Zhou, P. Heterologous expression and biochemical characterization of assimilatory nitrate and nitrite reductase reveals adaption and potential of Bacillus megaterium NCT-2 in secondary salinization soil. Int. J. Biol. Macromol. 2017, 101, 1019–1028. [Google Scholar] [CrossRef] [PubMed]
- Spini, G.; Spina, F.; Poli, A.; Blieux, A.-L.; Regnier, T.; Gramellini, C.; Varese, G.C.; Puglisi, E. Molecular and microbiological Insights on the enrichment procedures for the isolation of petroleum degrading bacteria and fungi. Front. Microbiol. 2018, 9, 2543. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Taxonomic Group | Genus * (Total # of Strains) | Brine/Oil | Oil | Prairie |
|---|---|---|---|---|
| γ-Proteobacteria | Stenotrophomonas (27) | 3NR ** | 2NR, 3“None” | 11NR, 1DN 7 “None” |
| Pseudomonas (9) | 2NR, 2DN | 1NR | 3NR, 1DN | |
| Aeromonas (1) | 1NR | 0 | 0 | |
| Serratia (1) | 0 | 1NR | 0 | |
| Enterobacter (3) | 0 | 3NR | 0 | |
| Acinetobacter (5) | 0 | 0 | 2NR, 3 “None” | |
| Total # γ Proteobacteria (46) | 8 | 10 | 28 | |
| α-Proteobacteria | Ensifer (2) | 1 DN | 0 | 1 DN |
| Bosea (2) | 2 “None” | 0 | 0 | |
| Phenylobacterium (1) | 1NR | 0 | 0 | |
| Total # α- Proteobacteria (5) | 4 | 0 | 1 | |
| β-Proteobacteria | Achromobacter (4) | 1NR | 0 | 3NR |
| Burkholderia (1) | 0 | 0 | 1 “None” | |
| Total # β- Proteobacteria (5) | 1 | 0 | 4 | |
| Actinobacteria | Arthrobacter (1) | 1 “None” | 0 | 0 |
| Rhodococcus (1) | 0 | 0 | 1 “None” | |
| Microbacterium (1) | 0 | 1NR | 0 | |
| Kocuria (1) | 0 | 0 | 1NR | |
| Total # Actinobacteria (4) | 1 | 1 | 2 | |
| Bacilli | Brevibacillus (8) | 3NR | 0 | 4NR, 1“None” |
| Bacillus (3) | 2NR | 0 | 1”None” | |
| Paenibacillus (1) | 1NR | 0 | 0 | |
| Lysinibacillus (2) | 0 | 0 | 1NR, 1”None” | |
| Total # Bacilli (14) | 6 | 0 | 8 | |
| Flavobacteria | Chryseobacterium (1) | 0 | 1NR | 0 |
| Total # Flavobacteria (1) | 0 | 1 | 0 | |
| Total # strains isolated | 20 | 12 | 43 | |
| Total # NR + DN | 17 | 9 | 28 | |
| #NR, #DN | 14, 3 | 9, 0 | 25, 3 |
| Taxonomic Group | Genus * (Total # of Strains, # Screened for qnorB, cnorB, nosZ) | NR a | DN b | None c |
|---|---|---|---|---|
| γ-Proteobacteria | Stenotrophomonas (27, 15) | 16 narG (2) | 1 | 10 |
| Pseudomonas (9, 7) | 6 napA (2) | 3 napA (1), nirS (2), cnorB (1), nosZ (1) | 0 | |
| Aeromonas (1, 1) | 1 | 0 | 0 | |
| Serratia (1, 1) | 1 | 0 | 0 | |
| Enterobacter (3, 1) | 3 | 0 | 0 | |
| Acinetobacter (5, 1) | 2 | 0 | 3 | |
| α-Proteobacteria | Ensifer (2, 1) | 0 | 2 napA (1), cnorB (1) | 0 |
| Bosea (2, 0) | 0 | 0 | 2 napA (2) | |
| Phenylobacterium (1, 1) | 1 | 0 | 0 | |
| β-Proteobacteria | Achromobacter (4, 2) | 4 napA (1) | 0 | 0 |
| Burkholderia (1, 0) | 0 | 0 | 1 | |
| Actinobacteria | Arthrobacter (1, 0) | 0 | 0 | 1 |
| Rhodococcus (1, 0) | 0 | 0 | 1 | |
| Microbacterium (1, 1) | 1 | 0 | 0 | |
| Kocuria (1, 1) | 1 | 0 | 0 | |
| Bacilli | Brevibacillus (8, 1) | 7 | 0 | 1 |
| Bacillus (3, 0) | 2 | 0 | 1 | |
| Paenibacillus (1, 0) | 1 | 0 | 0 | |
| Lysinibacillus (2, 0) | 1 | 0 | 1 | |
| Flavobacteria | Chryseobacterium (1, 0) | 1 | 0 | 0 |
| Total | 75 (33) | 48 | 6 | 21 |
| Strain # (Genus) | Soil Type | napA | narG | nirS | cnorB | nosZ | NR or DN * |
|---|---|---|---|---|---|---|---|
| 1 (Pseudomonas) | Brine/Oil | Yes | Yes | DN | |||
| 2 (Pseudomonas) | Brine/Oil | Yes | DN | ||||
| 4 (Ensifer) | Brine/Oil | Yes | DN | ||||
| 7 (Bosea) | Brine/Oil | Yes | None | ||||
| 8 (Bosea) | Brine/Oil | Yes | None | ||||
| 39 (Stenotrophomonas) | Prairie | Yes | NR | ||||
| 42 (Stenotrophomonas) | Prairie | Yes | NR | ||||
| 49 (Achromobacter) | Prairie | Yes | NR | ||||
| 65 (Pseudomonas) | Prairie | Yes | Yes | DN | |||
| 67 (Pseudomonas) | Prairie | Yes | NR | ||||
| 68 (Pseudomonas) | Prairie | Yes | NR | ||||
| 69 (Ensifer) | Prairie | Yes | NR |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
AbuBakr, S.M.; Najar, F.Z.; Duncan, K.E. Detection of Nitrate-Reducing/Denitrifying Bacteria from Contaminated and Uncontaminated Tallgrass Prairie Soil: Limitations of PCR Primers. Microorganisms 2024, 12, 1981. https://doi.org/10.3390/microorganisms12101981
AbuBakr SM, Najar FZ, Duncan KE. Detection of Nitrate-Reducing/Denitrifying Bacteria from Contaminated and Uncontaminated Tallgrass Prairie Soil: Limitations of PCR Primers. Microorganisms. 2024; 12(10):1981. https://doi.org/10.3390/microorganisms12101981
Chicago/Turabian StyleAbuBakr, Samer M., Fares Z. Najar, and Kathleen E. Duncan. 2024. "Detection of Nitrate-Reducing/Denitrifying Bacteria from Contaminated and Uncontaminated Tallgrass Prairie Soil: Limitations of PCR Primers" Microorganisms 12, no. 10: 1981. https://doi.org/10.3390/microorganisms12101981
APA StyleAbuBakr, S. M., Najar, F. Z., & Duncan, K. E. (2024). Detection of Nitrate-Reducing/Denitrifying Bacteria from Contaminated and Uncontaminated Tallgrass Prairie Soil: Limitations of PCR Primers. Microorganisms, 12(10), 1981. https://doi.org/10.3390/microorganisms12101981