_Di_Marco.png)
Ionizing Radiation Dose Differentially Affects the Host–Microbe Relationship over Time

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
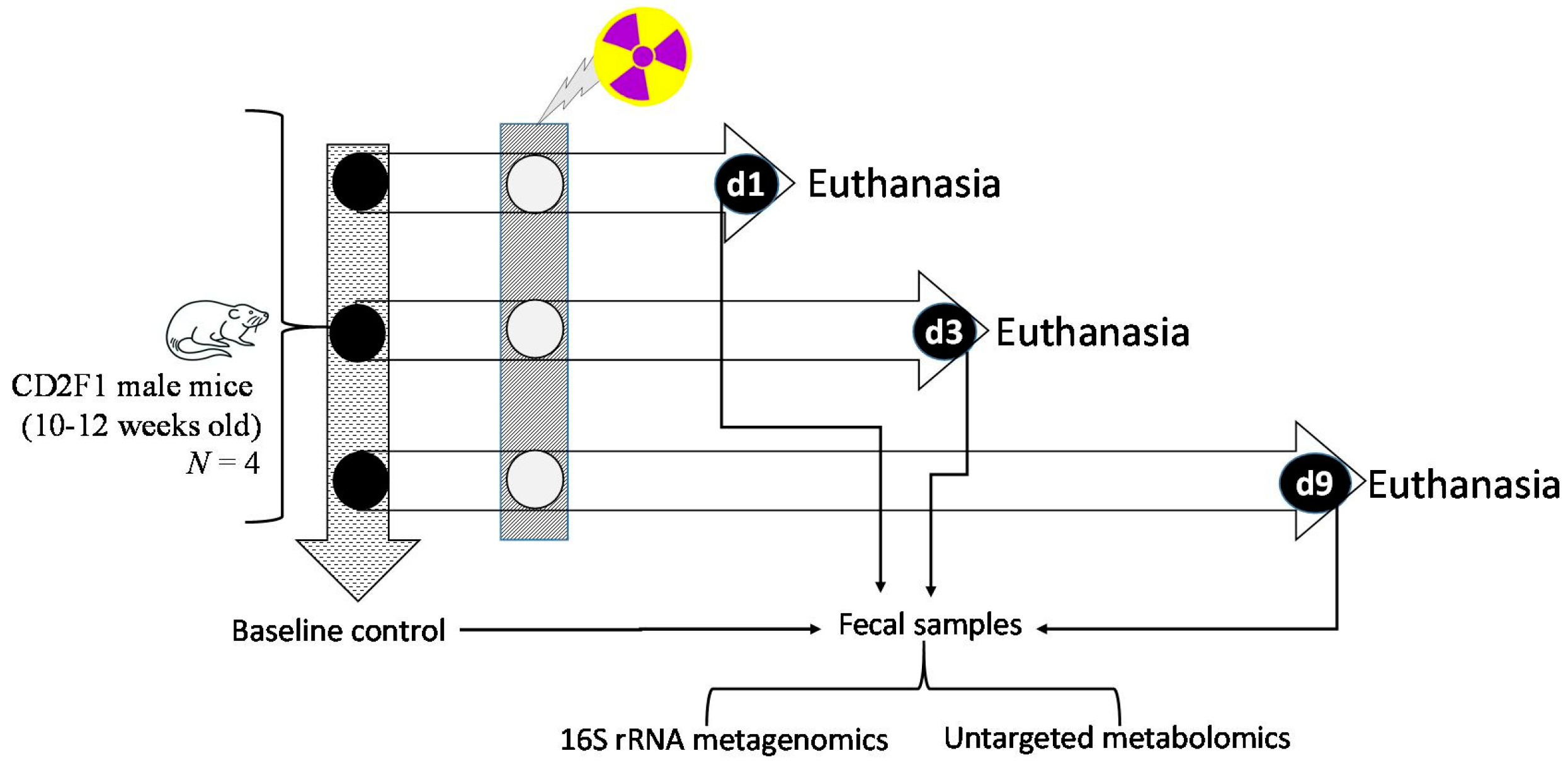
2.1. Animals, and Veterinary Care
2.2. Total Body Gamma Irradiation Exposure and Monitoring Clinical Signs of Radiation
2.3. Sample Collection Post-Euthanasia
2.4. Descending Colon Contents 16S rRNA Gene Sequencing Assay
2.5. Untargeted Metabolomics of Descending Colon Contents
2.6. Quantitative Detection of Lipopolysaccharide Binding Protein (LBP) in Liver
2.7. Statistical Analysis and Data Integration
3. Results
3.1. Animal Health Status Following Lethal Doses of Ionizing Radiation
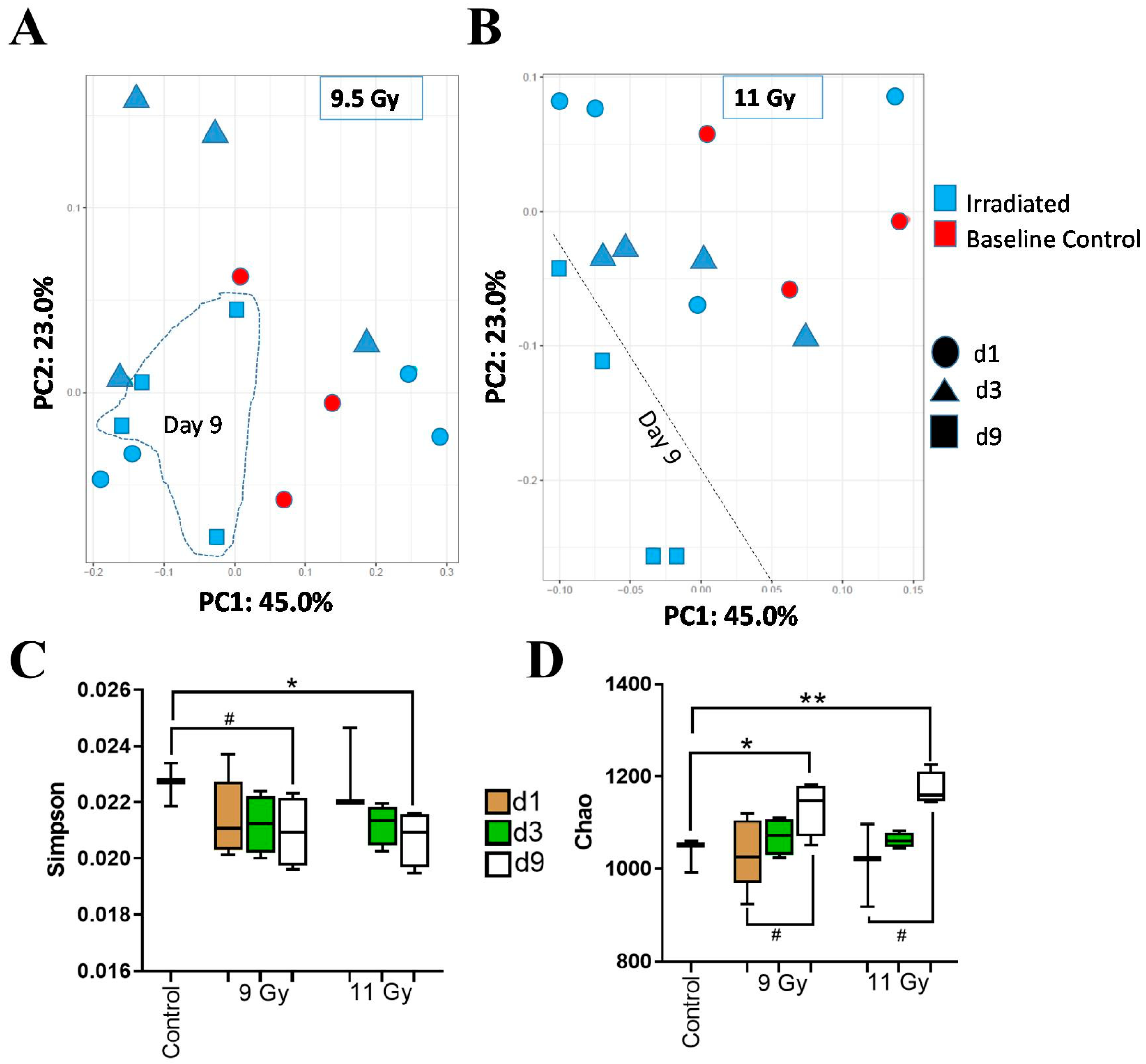
3.2. Descending Colon Contents’ Bacterial Diversity Showed Dose-Dependent Longitudinal Alterations
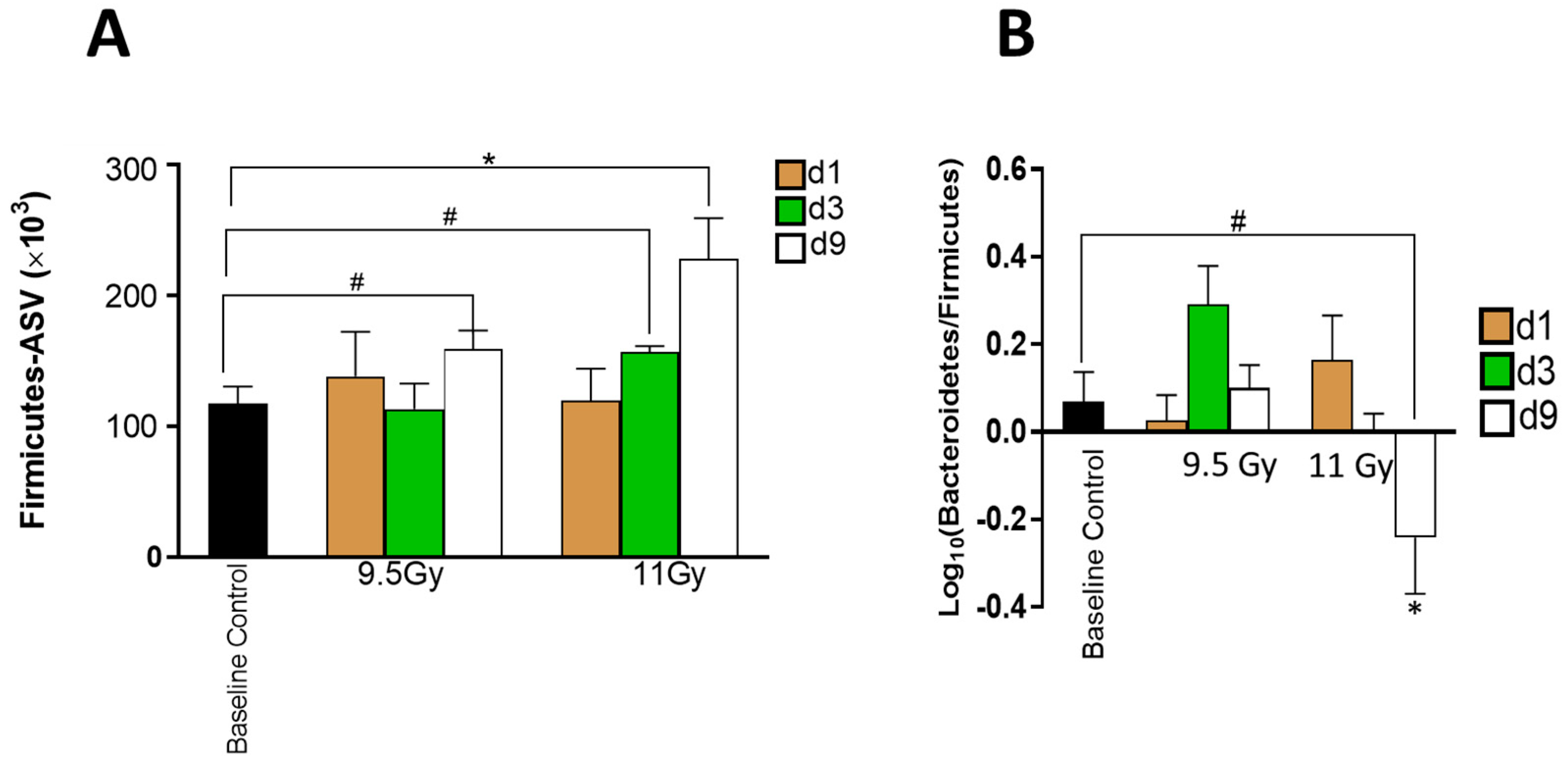
3.3. Shifts in Bacterial Abundances Depended on Radiation Dose and Time since Irradiation
3.4. Bionetworks Potentially Linked to Bacterial Shifted with Time since Irradiation
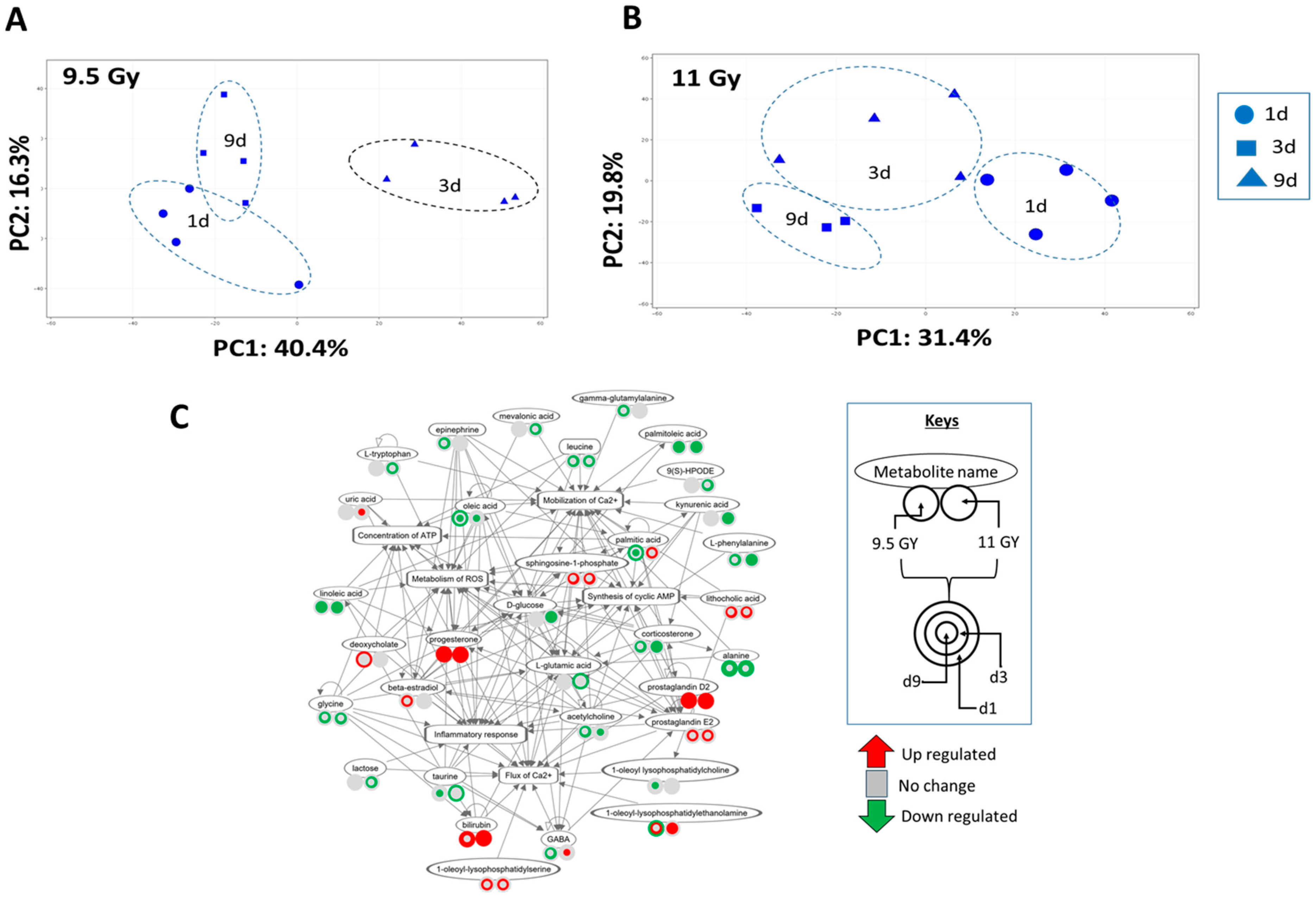
3.5. Descending Colon Contents’ Metabolite Expression Profile Shifted with Time since Irradiation
3.6. Differential Expression Analysis Identified Putative Early and Time-Independent Metabolite Markers of Irradiation
3.7. Regulation Profile of Metabolite-Enriched Networks Shifted Due to Radiation Dose and Time since Irradiation
3.8. LBP Loads in the Liver Samples Were Exclusively Increased by 11 Gy Irradiation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Heslet, L.; Bay, C.; Nepper-Christensen, S. Acute radiation syndrome (ARS)—Treatment of the reduced host defense. Int. J. Gen. Med. 2012, 5, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Booth, C.; Tudor, G.; Tudor, J.; Katz, B.P.; MacVittie, T.J. Acute Gastrointestinal Syndrome in High-Dose Irradiated Mice. Health Physics 2012, 103, 383–399. [Google Scholar] [CrossRef]
- Cheema, A.K.; Li, Y.; Singh, J.; Johnson, R.; Girgis, M.; Wise, S.Y.; Fatanmi, O.O.; Kaytor, M.D.; Singh, V.K. Microbiome study in irradiated mice treated with BIO 300, a promising radiation countermeasure. Anim. Microbiome 2021, 3, 71. [Google Scholar] [CrossRef] [PubMed]
- Xiong, W. Intestinal microbiota in various animals. Integr. Zool. 2022, 17, 331–332. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D. Human gut microbiome: Hopes, threats and promises. Gut 2018, 67, 1716–1725. [Google Scholar] [CrossRef]
- Sender, R.; Fuchs, S.; Milo, R. Revised estimates for the number of human and bacteria cells in the body. PLoS Biol. 2016, 14, e1002533. [Google Scholar] [CrossRef]
- Kim, J.Y.; Whon, T.W.; Lim, M.Y.; Kim, Y.B.; Kim, N.; Kwon, M.-S.; Kim, J.; Lee, S.H.; Choi, H.-J.; Nam, I.-H. The human gut archaeome: Identification of diverse haloarchaea in Korean subjects. Microbiome 2020, 8, 114. [Google Scholar] [CrossRef]
- Shkoporov, A.N.; Hill, C. Bacteriophages of the human gut: The “known unknown” of the microbiome. Cell Host Microbe 2019, 25, 195–209. [Google Scholar] [CrossRef]
- Hollingsworth, B.A.; Cassatt, D.R.; DiCarlo, A.L.; Rios, C.I.; Satyamitra, M.M.; Winters, T.A.; Taliaferro, L.P. Acute Radiation Syndrome and the Microbiome: Impact and Review. Front. Pharmacol. 2021, 12, 643283. [Google Scholar] [CrossRef]
- Backhed, F.; Ley, R.E.; Sonnenburg, J.L.; Peterson, D.A.; Gordon, J.I. Host-bacterial mutualism in the human intestine. Science 2005, 307, 1915–1920. [Google Scholar] [CrossRef]
- Hooper, L.V.; Macpherson, A.J. Immune adaptations that maintain homeostasis with the intestinal microbiota. Nat. Rev. Immunol. 2010, 10, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Stecher, B.; Hardt, W.D. Mechanisms controlling pathogen colonization of the gut. Curr. Opin. Microbiol. 2011, 14, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.R. Survival Studies of Whole-Body X-Irradiated Germfree (Axenic) Mice. Radiat. Res. 1963, 20, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Crawford, P.A.; Gordon, J.I. Microbial regulation of intestinal radiosensitivity. Proc. Natl. Acad. Sci. USA 2005, 102, 13254–13259. [Google Scholar] [CrossRef]
- Gerassy-Vainberg, S.; Blatt, A.; Danin-Poleg, Y.; Gershovich, K.; Sabo, E.; Nevelsky, A.; Daniel, S.; Dahan, A.; Ziv, O.; Dheer, R.; et al. Radiation induces proinflammatory dysbiosis: Transmission of inflammatory susceptibility by host cytokine induction. Gut 2018, 67, 97–107. [Google Scholar] [CrossRef]
- Guo, H.; Chou, W.C.; Lai, Y.; Liang, K.; Tam, J.W.; Brickey, W.J.; Chen, L.; Montgomery, N.D.; Li, X.; Bohannon, L.M.; et al. Multi-omics analyses of radiation survivors identify radioprotective microbes and metabolites. Science 2020, 370, eaay9097. [Google Scholar] [CrossRef]
- van Horssen, J.; van Schaik, P.; Witte, M. Inflammation and mitochondrial dysfunction: A vicious circle in neurodegenerative disorders? Neurosci. Lett. 2019, 710, 132931. [Google Scholar] [CrossRef] [PubMed]
- Daisley, B.A.; Koenig, D.; Engelbrecht, K.; Doney, L.; Hards, K.; Al, K.F.; Reid, G.; Burton, J.P. Emerging connections between gut microbiome bioenergetics and chronic metabolic diseases. Cell Rep. 2021, 37, 110087. [Google Scholar] [CrossRef]
- Liu, T.F.; Brown, C.M.; El Gazzar, M.; McPhail, L.; Millet, P.; Rao, A.; Vachharajani, V.T.; Yoza, B.K.; McCall, C.E. Fueling the flame: Bioenergy couples metabolism and inflammation. J. Leukoc. Biol. 2012, 92, 499–507. [Google Scholar] [CrossRef]
- Kieser, S.; Zdobnov, E.M.; Trajkovski, M. Comprehensive mouse microbiota genome catalog reveals major difference to its human counterpart. PLoS Comput. Biol. 2022, 18, e1009947. [Google Scholar] [CrossRef]
- Krautkramer, K.A.; Fan, J.; Backhed, F. Gut microbial metabolites as multi-kingdom intermediates. Nat. Rev. Microbiol. 2021, 19, 77–94. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, N. Metabolites: A converging node of host and microbe to explain meta-organism. Front. Microbiol. 2024, 15, 1337368. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Winters, T.A.; Marzella, L.; Molinar-Inglis, O.; Price, P.W.; Han, N.C.; Cohen, J.E.; Wang, S.J.; Fotenos, A.F.; Sullivan, J.M.; Esker, J.I.; et al. Gastrointestinal Acute Radiation Syndrome: Mechanisms, Models, Markers, and Medical Countermeasures. Radiat. Res. 2024, 201, 628–646. [Google Scholar] [CrossRef] [PubMed]
- Cook, J.A.; Sowers, A.L.; Choudhuri, R.; Gadisetti, C.; Edmondson, E.F.; Gohain, S.; Krishna, M.C.; Mitchell, J.B. The effect of modulation of gut microbiome profile on radiation-induced carcinogenesis and survival. J. Radiat. Res. 2023, 64, 24–32. [Google Scholar] [CrossRef]
- Kumar, V.P.; Holmes-Hampton, G.P.; Biswas, S.; Stone, S.; Sharma, N.K.; Hritzo, B.; Guilfoyle, M.; Eichenbaum, G.; Guha, C.; Ghosh, S.P. Mitigation of total body irradiation-induced mortality and hematopoietic injury of mice by a thrombopoietin mimetic (JNJ-26366821). Sci. Rep. 2022, 12, 3485. [Google Scholar] [CrossRef]
- Bene, B.J.; Blakely, W.F.; Burmeister, D.M.; Cary, L.; Chhetri, S.J.; Davis, C.M.; Ghosh, S.P.; Holmes-Hampton, G.P.; Iordanskiy, S.; Kalinich, J.F.; et al. Celebrating 60 Years of Accomplishments of the Armed Forces Radiobiology Research Institute1. Radiat. Res. 2021, 196, 129–146. [Google Scholar] [CrossRef]
- Kumar, V.P.; Biswas, S.; Sharma, N.K.; Stone, S.; Fam, C.M.; Cox, G.N.; Ghosh, S.P. PEGylated IL-11 (BBT-059): A Novel Radiation Countermeasure for Hematopoietic Acute Radiation Syndrome. Health Phys. 2018, 115, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Koch, A.; Gulani, J.; King, G.; Hieber, K.; Chappell, M.; Ossetrova, N. Establishment of Early Endpoints in Mouse Total-Body Irradiation Model. PLoS ONE 2016, 11, e0161079. [Google Scholar] [CrossRef]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef]
- Chakraborty, N.; Zamarioli, A.; Gautam, A.; Campbell, R.; Mendenhall, S.K.; Childress, P.J.; Dimitrov, G.; Sowe, B.; Tucker, A.; Zhao, L.; et al. Gene-metabolite networks associated with impediment of bone fracture repair in spaceflight. Comput. Struct. Biotechnol. J. 2021, 19, 3507–3520. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, N.; Gautam, A.; Holmes-Hampton, G.P.; Kumar, V.P.; Biswas, S.; Kumar, R.; Hamad, D.; Dimitrov, G.; Olabisi, A.O.; Hammamieh, R.; et al. microRNA and Metabolite Signatures Linked to Early Consequences of Lethal Radiation. Sci. Rep. 2020, 10, 5424. [Google Scholar] [CrossRef] [PubMed]
- Libiseller, G.; Dvorzak, M.; Kleb, U.; Gander, E.; Eisenberg, T.; Madeo, F.; Neumann, S.; Trausinger, G.; Sinner, F.; Pieber, T.; et al. IPO: A tool for automated optimization of XCMS parameters. BMC Bioinform. 2015, 16, 118. [Google Scholar] [CrossRef]
- Hoke, A.; Chakraborty, N.; Gautam, A.; Hammamieh, R.; Jett, M. Acute and Delayed Effects of Stress Eliciting Post-Traumatic Stress-Like Disorder Differentially Alters Fecal Microbiota Composition in a Male Mouse Model. Front. Cell Infect. Microbiol. 2022, 12, 810815. [Google Scholar] [CrossRef] [PubMed]
- Karl, J.P.; Margolis, L.M.; Madslien, E.H.; Murphy, N.E.; Castellani, J.W.; Gundersen, Y.; Hoke, A.V.; Levangie, M.W.; Kumar, R.; Chakraborty, N.; et al. Changes in intestinal microbiota composition and metabolism coincide with increased intestinal permeability in young adults under prolonged physiological stress. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 312, G559–G571. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004, 5, R80. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Lozupone, C.A.; Hamady, M.; Kelley, S.T.; Knight, R. Quantitative and qualitative beta diversity measures lead to different insights into factors that structure microbial communities. Appl. Environ. Microbiol. 2007, 73, 1576–1585. [Google Scholar] [CrossRef]
- Anderson, M.J. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 2001, 26, 32–46. [Google Scholar]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McFlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.; Solymos, P.; et al. Vegan: Community Ecology Package. 2018. Available online: https://cran.r-project.org/web/packages/vegan/index.html (accessed on 28 August 2024).
- Simpson, E.H. Measurement of diversity. Nature 1949, 163, 688. [Google Scholar] [CrossRef]
- Chao, A. Non-Parametric Estimation of the Number of Classes in a Population. Scand. J. Stat. 1984, 11, 265–270. [Google Scholar]
- Shannon, C.E.; Weaver, W. The Mathematical Theory of Communication; University of Illonois Press: Champaign, IL, USA, 1949. [Google Scholar]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Gregory Caporaso, J. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine learning in python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- McDonald, D.; Price, M.N.; Goodrich, J.; Nawrocki, E.P.; DeSantis, T.Z.; Probst, A.; Andersen, G.L.; Knight, R.; Hugenholtz, P. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012, 6, 610–618. [Google Scholar] [CrossRef]
- Caspi, R.; Altman, T.; Billington, R.; Dreher, K.; Foerster, H.; Fulcher, C.A.; Holland, T.A.; Keseler, I.M.; Kothari, A.; Kubo, A.; et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome Databases. Nucleic Acids Res. 2014, 42, D459–D471. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Barbera, P.; Kozlov, A.M.; Czech, L.; Morel, B.; Darriba, D.; Flouri, T.; Stamatakis, A. EPA-ng: Massively Parallel Evolutionary Placement of Genetic Sequences. Syst. Biol. 2019, 68, 365–369. [Google Scholar] [CrossRef]
- Czech, L.; Barbera, P.; Stamatakis, A. Genesis and Gappa: Processing, analyzing, and visualizing phylogenetic (placement) data. Bioinformatics 2020, 36, 3263–3265. [Google Scholar] [CrossRef]
- Louca, S.; Doebeli, M. Efficient comparative phylogenetics on large trees. Bioinformatics 2018, 34, 1053–1055. [Google Scholar] [CrossRef]
- Ye, Y.; Doak, T.G. A parsimony approach to biological pathway reconstruction/inference for genomes and metagenomes. PLoS Comput. Biol. 2009, 5, e1000465. [Google Scholar] [CrossRef] [PubMed]
- Holmes-Hampton, G.P.; Kumar, V.P.; Biswas, S.; Stone, S.; Sharma, N.K.; Legesse, B.; Vercellino, J.; Guha, C.; Eichenbaum, G.; Ghosh, S.P. PEGylated thrombopoietin mimetic, JNJ-26366821 a novel prophylactic radiation countermeasure for acute radiation injury. Sci. Rep. 2023, 13, 15211. [Google Scholar] [CrossRef] [PubMed]
- Parida, S.; Sharma, D. Microbial Alterations and Risk Factors of Breast Cancer: Connections and Mechanistic Insights. Cells 2020, 9, 1091. [Google Scholar] [CrossRef] [PubMed]
- Harmon, G.S.; Lam, M.T.; Glass, C.K. PPARs and lipid ligands in inflammation and metabolism. Chem. Rev. 2011, 111, 6321–6340. [Google Scholar] [CrossRef] [PubMed]
- Oppolzer, D.; Barroso, M.; Gallardo, E. Bioanalytical procedures and developments in the determination of alcohol biomarkers in biological specimens. Bioanalysis 2016, 8, 229–251. [Google Scholar] [CrossRef]
- Mure, K.; Tomono, S.; Mure, M.; Horinaka, M.; Mutoh, M.; Sakai, T.; Ishikawa, H.; Wakabayashi, K. The Combination of Cigarette Smoking and Alcohol Consumption Synergistically Increases Reactive Carbonyl Species in Human Male Plasma. Int. J. Mol. Sci. 2021, 22, 16. [Google Scholar] [CrossRef]
- Creeden, J.F.; Gordon, D.M.; Stec, D.E.; Hinds, T.D., Jr. Bilirubin as a metabolic hormone: The physiological relevance of low levels. Am. J. Physiol. Endocrinol. Metab. 2021, 320, E191–E207. [Google Scholar] [CrossRef]
- Guo, J.; Yang, Y.; Buettner, R.; Rosen, S.T. Targeting the methionine-methionine adenosyl transferase 2A- S -adenosyl methionine axis for cancer therapy. Curr. Opin. Oncol. 2022, 34, 546–551. [Google Scholar] [CrossRef]
- Ullah, H.; Khan, A.; Rengasamy, K.R.; Di Minno, A.; Sacchi, R.; Daglia, M. The Efficacy of S-Adenosyl Methionine and Probiotic Supplementation on Depression: A Synergistic Approach. Nutrients 2022, 14, 2751. [Google Scholar] [CrossRef]
- Jung, H.R.; Oh, Y.; Jang, D.; Shin, S.; Lee, S.J.; Kim, J.; Lee, S.E.; Oh, J.; Jang, G.; Kwon, O.; et al. Gut bacteria-derived 3-phenylpropionylglycine mitigates adipocyte differentiation of 3T3-L1 cells by inhibiting adiponectin-PPAR pathway. Genes Genomics. 2023, 45, 71–81. [Google Scholar] [CrossRef]
- Theis, K.R.; Dheilly, N.M.; Klassen, J.L.; Brucker, R.M.; Baines, J.F.; Bosch, T.C.; Cryan, J.F.; Gilbert, S.F.; Goodnight, C.J.; Lloyd, E.A.; et al. Getting the Hologenome Concept Right: An Eco-Evolutionary Framework for Hosts and Their Microbiomes. mSystems 2016, 1, e00028-16. [Google Scholar] [CrossRef] [PubMed]
- Muhamad Rizal, N.S.; Neoh, H.M.; Ramli, R.; Periyasamy, P.R.A.L.K.; Hanafiah, A.; Abdul Samat, M.N.; Tan, T.L.; Wong, K.K.; Nathan, S.; Chieng, S.; et al. Advantages and Limitations of 16S rRNA Next-Generation Sequencing for Pathogen Identification in the Diagnostic Microbiology Laboratory: Perspectives from a Middle-Income Country. Diagnostics 2020, 10, 816. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.S.; Muthyala, S.D.V.; Klemashevich, C.; Ufondu, A.U.; Menon, R.; Chen, Z.; Devaraj, S.; Jayaraman, A.; Sun, Y. Age-dependent remodeling of gut microbiome and host serum metabolome in mice. Aging 2021, 13, 6330–6345. [Google Scholar] [CrossRef] [PubMed]
- Schlomann, B.H.; Parthasarathy, R. Timescales of gut microbiome dynamics. Curr. Opin. Microbiol. 2019, 50, 56–63. [Google Scholar] [CrossRef]
- Baczkowski, A.J.; Joanes, D.N.; Shamia, G.M. Range of validity of alpha and beta for a generalized diversity index H (alpha, beta) due to Good. Math. Biosci. 1998, 148, 115–128. [Google Scholar] [CrossRef]
- Calle, M.L. Statistical Analysis of Metagenomics Data. Genom. Inform. 2019, 17, e6. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, S.; Xu, L. The pivotal roles of gut microbiota in insect plant interactions for sustainable pest management. NPJ Biofilms Microbiomes 2023, 9, 66. [Google Scholar] [CrossRef]
- Faith, D.P.; Peter, R. Minchin, and Lee Belbin. Compositional dissimilarity as a robust measure of ecological distance. Vegetatio 1987, 69, 57–68. [Google Scholar] [CrossRef]
- Kers, J.G.; Saccenti, E. The power of microbiome studies: Some considerations on which alpha and beta metrics to use and how to report results. Front. Microbiol. 2022, 12, 796025. [Google Scholar]
- Mariat, D.; Firmesse, O.; Levenez, F.; Guimaraes, V.; Sokol, H.; Dore, J.; Corthier, G.; Furet, J.P. The Firmicutes/Bacteroidetes ratio of the human microbiota changes with age. BMC Microbiol. 2009, 9, 123. [Google Scholar] [CrossRef]
- de Oliveira, G.L.V.; Cardoso, C.R.B.; Taneja, V.; Fasano, A. Editorial: Intestinal Dysbiosis in Inflammatory Diseases. Front. Immunol. 2021, 12, 727485. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, A.; Harty, S.; Lehto, S.M.; Moeller, A.H.; Dinan, T.G.; Dunbar, R.I.M.; Cryan, J.F.; Burnet, P.W.J. The Microbiome in Psychology and Cognitive Neuroscience. Trends Cogn. Sci. 2018, 22, 611–636. [Google Scholar] [CrossRef] [PubMed]
- Parker, A.; Fonseca, S.; Carding, S.R. Gut microbes and metabolites as modulators of blood-brain barrier integrity and brain health. Gut Microbes 2020, 11, 135–157. [Google Scholar] [CrossRef] [PubMed]
- De Bandt, J.P.; Waligora-Dupriet, A.J.; Butel, M.J. Intestinal microbiota in inflammation and insulin resistance: Relevance to humans. Curr. Opin. Clin. Nutr. Metab. Care 2011, 14, 334–340. [Google Scholar] [CrossRef]
- Zou, Y.; Ju, X.; Chen, W.; Yuan, J.; Wang, Z.; Aluko, R.E.; He, R. Rice bran attenuated obesity via alleviating dyslipidemia, browning of white adipocytes and modulating gut microbiota in high-fat diet-induced obese mice. Food Funct. 2020, 11, 2406–2417. [Google Scholar] [CrossRef]
- Palma, D.; Pickles, T.; Tyldesley, S.; Prostate Cohort Outcomes Initiative. Obesity as a predictor of biochemical recurrence and survival after radiation therapy for prostate cancer. BJU Int. 2007, 100, 315–319. [Google Scholar] [CrossRef]
- Alqahtani, S.J.M.; Welbourn, R.; Meakin, J.R.; Palfrey, R.M.; Rimes, S.J.; Thomson, K.; Knapp, K.M. Increased radiation dose and projected radiation-related lifetime cancer risk in patients with obesity due to projection radiography. J. Radiol. Prot. 2019, 39, 38–53. [Google Scholar] [CrossRef]
- Fernandes, A.; Oliveira, A.; Soares, R.; Barata, P. The Effects of Ionizing Radiation on Gut Microbiota: What Can Animal Models Tell Us?-A Systematic Review. Curr. Issues Mol. Biol. 2023, 45, 3877–3910. [Google Scholar] [CrossRef]
- Magne, F.; Gotteland, M.; Gauthier, L.; Zazueta, A.; Pesoa, S.; Navarrete, P.; Balamurugan, R. The Firmicutes/Bacteroidetes Ratio: A Relevant Marker of Gut Dysbiosis in Obese Patients? Nutrients 2020, 12, 1474. [Google Scholar] [CrossRef]
- Gao, X.; Lin, S.H.; Ren, F.; Li, J.T.; Chen, J.J.; Yao, C.B.; Yang, H.B.; Jiang, S.X.; Yan, G.Q.; Wang, D.; et al. Acetate functions as an epigenetic metabolite to promote lipid synthesis under hypoxia. Nat. Commun. 2016, 7, 11960. [Google Scholar] [CrossRef]
- Blandford, L.E.; Johnston, E.L.; Sanderson, J.D.; Wade, W.G.; Lax, A.J. Promoter orientation of the immunomodulatory Bacteroides fragilis capsular polysaccharide A (PSA) is off in individuals with inflammatory bowel disease (IBD). Gut Microbes 2019, 10, 569–577. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.A.; Worobey, M. Geographical variation of human gut microbial composition. Biol. Lett. 2014, 10, 20131037. [Google Scholar] [CrossRef] [PubMed]
- Louis, P.; Flint, H.J. Formation of propionate and butyrate by the human colonic microbiota. Environ. Microbiol. 2017, 19, 29–41. [Google Scholar] [CrossRef]
- Chevalier, C.; Stojanovic, O.; Colin, D.J.; Suarez-Zamorano, N.; Tarallo, V.; Veyrat-Durebex, C.; Rigo, D.; Fabbiano, S.; Stevanovic, A.; Hagemann, S.; et al. Gut Microbiota Orchestrates Energy Homeostasis during Cold. Cell 2015, 163, 1360–1374. [Google Scholar] [CrossRef]
- Stojanov, S.; Berlec, A.; Strukelj, B. The Influence of Probiotics on the Firmicutes/Bacteroidetes Ratio in the Treatment of Obesity and Inflammatory Bowel disease. Microorganisms 2020, 8, 1715. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.Y.; Xu, H.; Cao, G.Z.; Tian, L.L.; Wang, L.H.; Zhu, N.Q.; Zhang, J.J.; Yang, H.J. Multi-omics reveals Dengzhan Shengmai formulation ameliorates cognitive impairments in D-galactose-induced aging mouse model by regulating CXCL12/CXCR4 and gut microbiota. Front. Pharmacol. 2023, 14, 1175970. [Google Scholar] [CrossRef]
- Jian, Y.; Zhang, D.; Liu, M.; Wang, Y.; Xu, Z.X. The Impact of Gut Microbiota on Radiation-Induced Enteritis. Front. Cell. Infect. Microbiol. 2021, 11, 586392. [Google Scholar] [CrossRef]
- Seethaler, B.; Basrai, M.; Neyrinck, A.M.; Nazare, J.A.; Walter, J.; Delzenne, N.M.; Bischoff, S.C. Biomarkers for assessment of intestinal permeability in clinical practice. Am. J. Physiol. Gastrointest. Liver Physiol. 2021, 321, G11–G17. [Google Scholar] [CrossRef]
- Sukumaran, P.; Nascimento Da Conceicao, V.; Sun, Y.; Ahamad, N.; Saraiva, L.R.; Selvaraj, S.; Singh, B.B. Calcium Signaling Regulates Autophagy and Apoptosis. Cells 2021, 10, 2125. [Google Scholar] [CrossRef]
- Hardie, D.G. Sensing of energy and nutrients by AMP-activated protein kinase. Am. J. Clin. Nutr. 2011, 93, 891S–896. [Google Scholar] [CrossRef] [PubMed]
- Carling, D. AMPK signalling in health and disease. Curr. Opin. Cell Biol. 2017, 45, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.J.; Chandrasekaran, K.; Morgan, W.F. Mitochondrial dysfunction, persistently elevated levels of reactive oxygen species and radiation-induced genomic instability: A review. Mutagenesis 2006, 21, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S. Metabolomics for Investigating Physiological and Pathophysiological Processes. Physiol. Rev. 2019, 99, 1819–1875. [Google Scholar] [CrossRef]
- Shakyawar, S.K.; Mishra, N.K.; Vellichirammal, N.N.; Cary, L.; Helikar, T.; Powers, R.; Oberley-Deegan, R.E.; Berkowitz, D.B.; Bayles, K.W.; Singh, V.K.; et al. A Review of Radiation-Induced Alterations of Multi-Omic Profiles, Radiation Injury Biomarkers, and Countermeasures. Radiat. Res. 2023, 199, 89–111. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 9.5 Gy | 11 Gy | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Day 1 | Day 3 | Day 9 | Day 1 | Day 3 | Day 9 | |||||||
| Biofunctions’ Superfamily | Metagenome | Metabolome | Metagenome | Metabolome | Metagenome | Metabolome | Metagenome | Metabolome | Metagenome | Metabolome | Metagenome | Metabolome |
| Amino acid metabolism | 4/1 | 0/0 | 0/0 | 3/2 | 1/2 | 1/0 | 3/2 | 0/0 | 3/1 | 4/0 | 3/2 | 0/0 |
| Biosynthesis | 0/2 | 1/1 | 0/1 | 0/1 | 4/3 | 0/0 | 0/1 | 0/0 | 1/8 | 1/0 | 7/1 | 1/2 |
| Bioenergy | 0/0 | 1/0 | 0/0 | 1/1 | 2/0 | 1/0 | 2/0 | 1/0 | 1/0 | 3/1 | 4/0 | 2/4 |
| Immune functions | 0/0 | 0/0 | 0/0 | 7/1 | 0/0 | 0/3 | 0/0 | 0/1 | 0/0 | 2/0 | 1/0 | 0/3 |
| Lipid metabolism | 1/0 | 0/4 | 0/0 | 6/1 | 0/0 | 2/4 | 1/1 | 0/0 | 2/2 | 0/1 | 2/0 | 0/9 |
| Metabolite ID | Metabolite Name | Functional Relevance |
|---|---|---|
| All data points are consistently upregulated | ||
| HMDB0240259 | Stercobilin/L-urobilin | A pro-inflammatory microbial metabolite, which is generated via the reduction in bilirubin by intestinal microbiota |
| HMDB0001830 | Progesterone | Cancer-related metabolite and gut microbes metabolize and regulate the bioavailability of progesterone [55] |
| HMDB0001898 | Mesobilirubinogen | Closely related to stercobilin, as they have the same parent compound, namely urobilinogen |
| HMDB0006059 | 20-Carboxy-leukotriene B4 | An oxidized metabolite of leukotriene B4 (LTB4), which is released from polymorphonuclear granulocytes of severely burned patients |
| HMDB0062552 | Goralatide | Protector of hematopoietic progenitors |
| HMDB0060095 | Prostaglandin-c2 | Associated with inflammation [56] |
| All but one data points are consistently upregulated | ||
| HMDB0013200 | 5-Hydroxytryptophol glucuronide (GTOL) | A biomarker of alcohol load in body fluid [57] |
| HMDB0031039 | Heptadecanal | Linked to smoking [58] |
| HMDB0011563 | 1-pentadecanoylglycerol | -- |
| HMDB0000054 | Bilirubin | Its elevation is linked to several diseases and disorders [59] |
| All but one data points are consistently downregulated | ||
| HMDB0011538 | 2-Linoleoylglycerol | -- |
| HMDB0060987 | 2-Hydroxymethylolanzapine | Derivative of olanzapine, an atypical antipsychotic agent |
| HMDB0000988 | S-Adenosylmethioninamine or dadomet | Linked to cancer [60] and psychotic deficiencies [61] |
| HMDB0000860 | Phenylpropionylglycine | Associated with bacteria-driven metabolism [62] |
| Early markers: Consistently upregulated at d1 and d3 post-TBI | ||
| HMDB0001403 | Prostaglandin D2 (PGD2) | Associated with inflammation [56] |
| HMDB0004161 | Urobilin | Generated through the degradation of heme |
| HMDB0062389 | A sterol lipid molecule | A member to the class of cholesterols and derivatives |
| HMDB0006888 | 5b-Cyprinol sulfate | An intermediate of bile acid biosynthesis |
| Early markers: Consistently downregulated at d1 and d3 post-TBI | ||
| HMDB0062251 | Alanine | A member to the class of an alanine or an alanine derivative |
| HMDB0010727 | 3-Oxododecanoic acid | An intermediate in fatty acid biosynthesis |
| HMDB0000860 | Phenylpropionylglycine | A fatty acid metabolite that could be a marker of mitochondrial dysfunction |
| 9.5 Gy | 11 Gy | |||||
|---|---|---|---|---|---|---|
| Subnetworks | d1 | d3 | d9 | d1 | d3 | d9 |
| Flux of Ca+2 | Up | Up | Up | Up | Up | Up |
| Synthesis of cAMP | Up | Up | Up | Up | Up | Up |
| Metabolism of ROS | Down | Up | Down | Up | Up | Down |
| Inflammatory response | Down | Down | Down | Down | Down | Down |
| Conc. Of ATP | Down | Down | Down | Down | Down | Down |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chakraborty, N.; Hoke, A.; Campbell, R.; Holmes-Hampton, G.; Kumar, V.P.; Moyler, C.; Gautam, A.; Hammamieh, R.; Ghosh, S.P. Ionizing Radiation Dose Differentially Affects the Host–Microbe Relationship over Time. Microorganisms 2024, 12, 1995. https://doi.org/10.3390/microorganisms12101995
Chakraborty N, Hoke A, Campbell R, Holmes-Hampton G, Kumar VP, Moyler C, Gautam A, Hammamieh R, Ghosh SP. Ionizing Radiation Dose Differentially Affects the Host–Microbe Relationship over Time. Microorganisms. 2024; 12(10):1995. https://doi.org/10.3390/microorganisms12101995
Chicago/Turabian StyleChakraborty, Nabarun, Allison Hoke, Ross Campbell, Gregory Holmes-Hampton, Vidya P. Kumar, Candace Moyler, Aarti Gautam, Rasha Hammamieh, and Sanchita P. Ghosh. 2024. "Ionizing Radiation Dose Differentially Affects the Host–Microbe Relationship over Time" Microorganisms 12, no. 10: 1995. https://doi.org/10.3390/microorganisms12101995