
One-Step Soft Agar Enrichment and Isolation of Human Lung Bacteria Inhibiting the Germination of Aspergillus fumigatus Conidia

 ,
,  , , , , , and
, , , , , and
Abstract
:1. Introduction
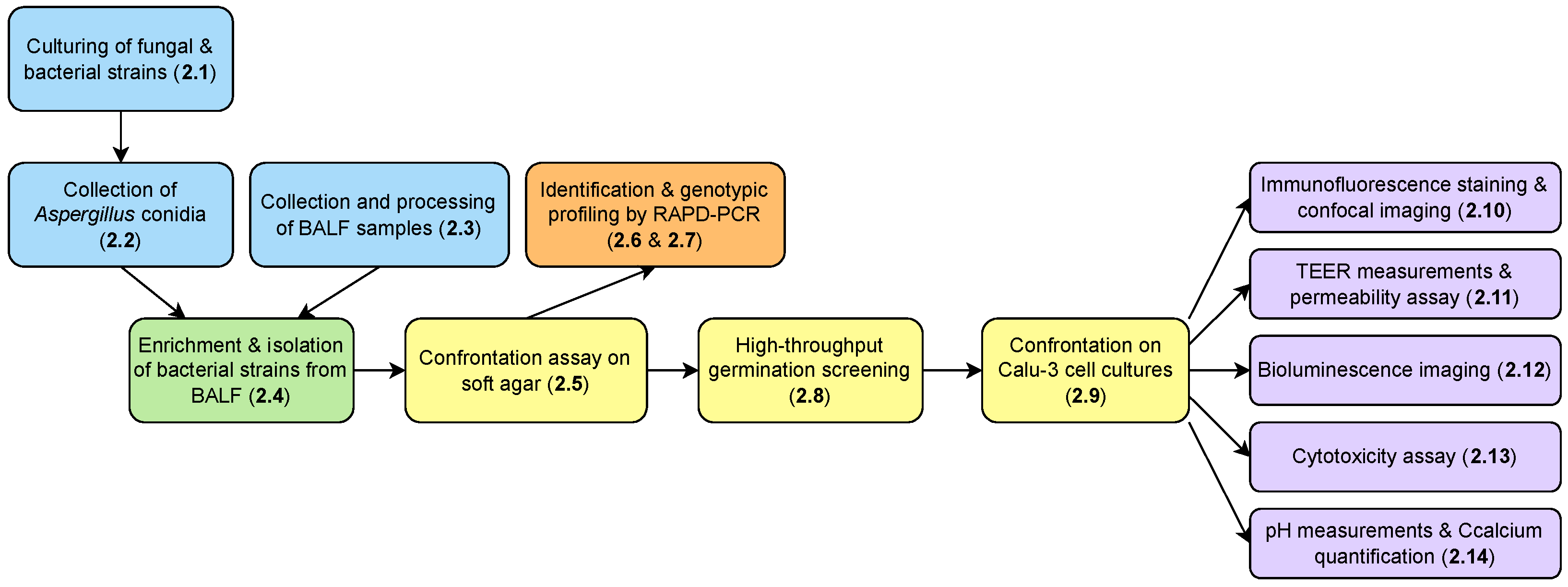
2. Materials and Methods
2.1. Culturing of Fungal and Bacterial Strains
2.2. Collection of Aspergillus spp. Conidia
2.3. Collection and Processing of Bronchoalveolar Lavage Fluid (BALF) Samples
2.4. Enrichment and Isolation of Antagonistic Bacterial Strains from BALF Samples
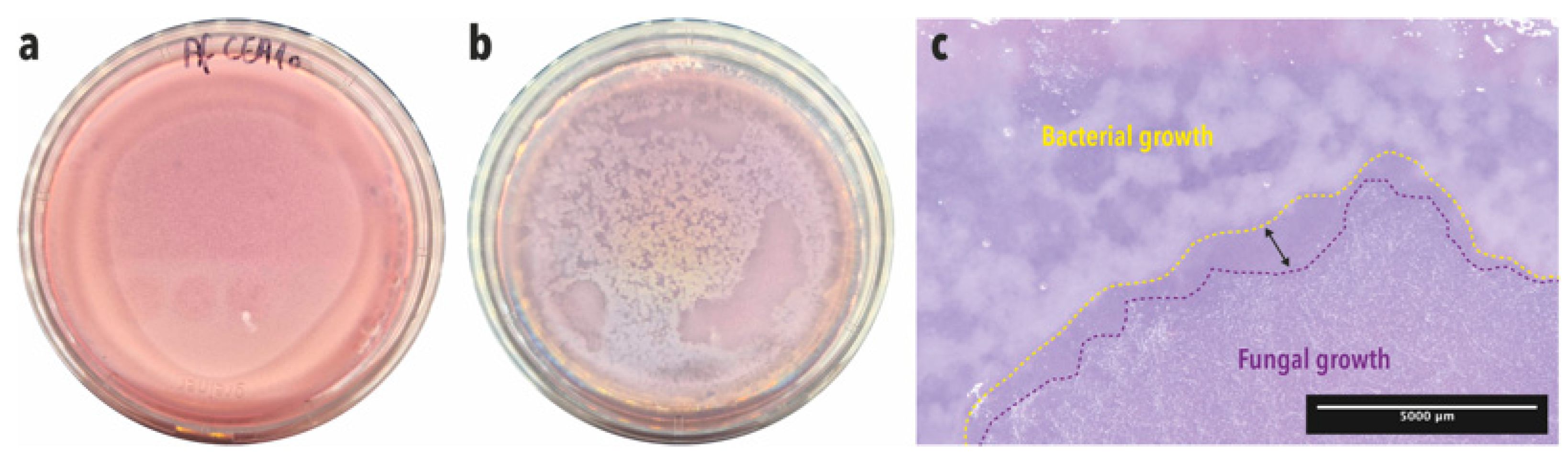
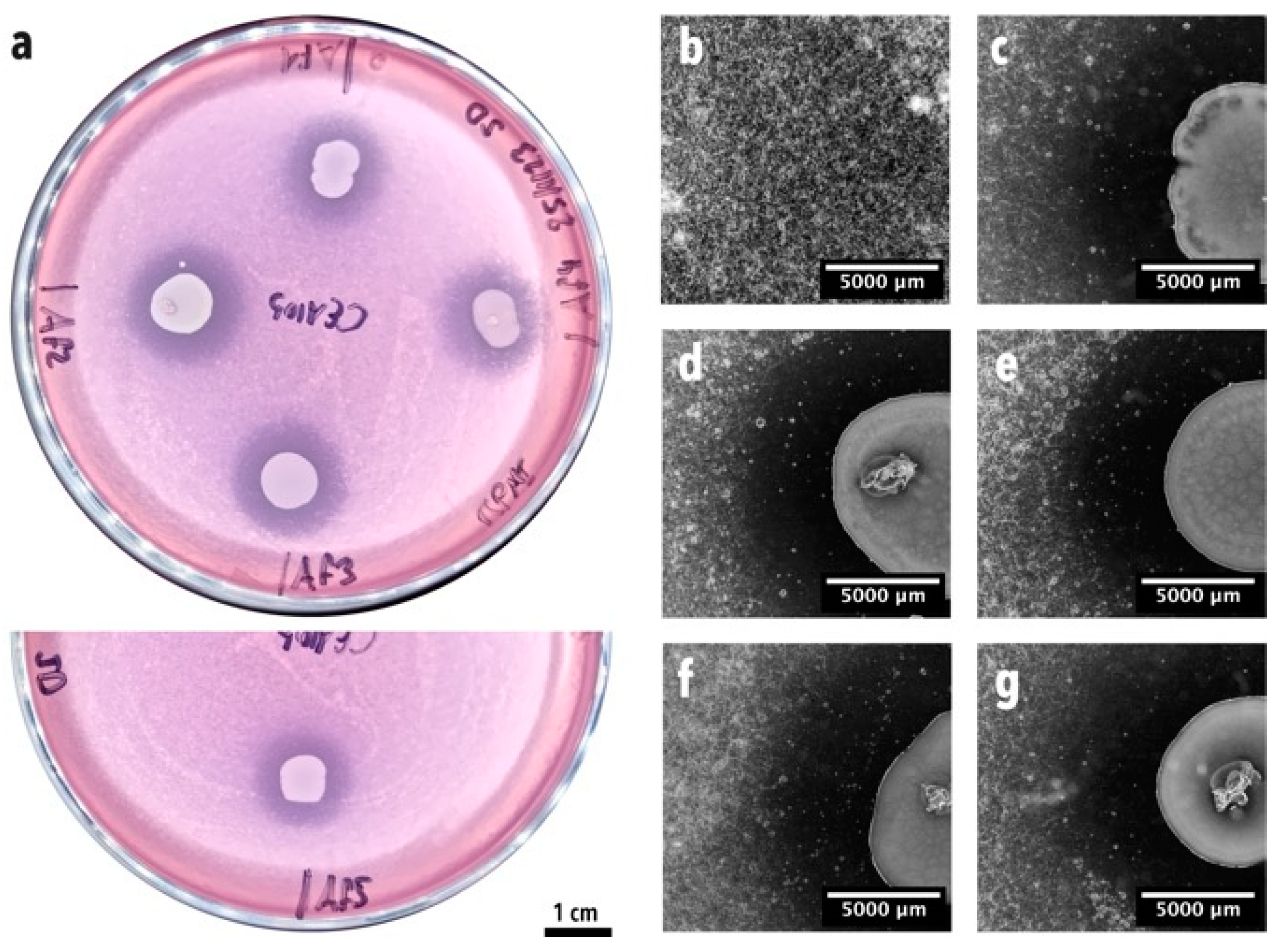
2.5. Confrontation Assay on a Soft Agar Overlay Assay Plate
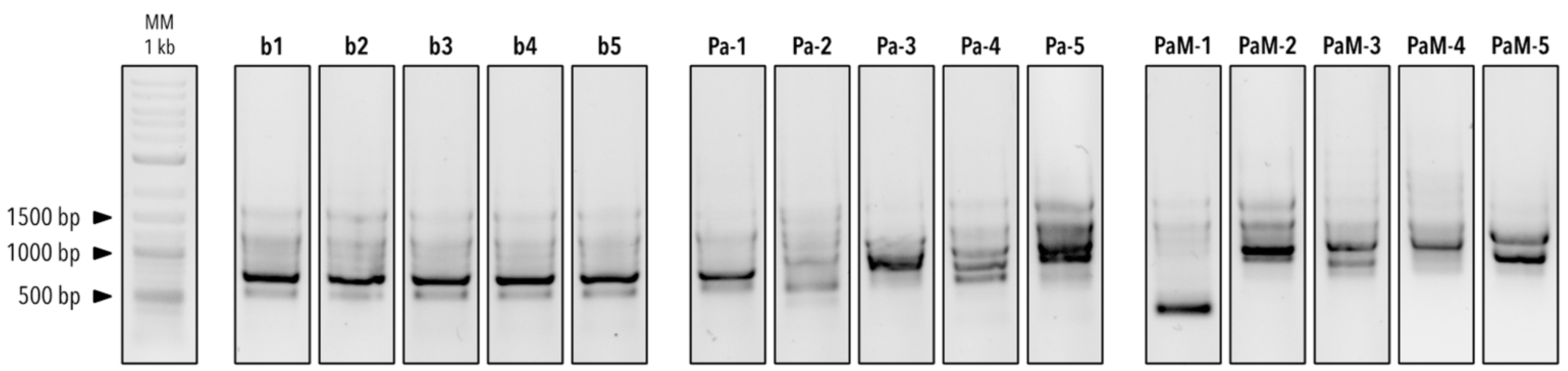
2.6. Identification of the Bacterial Isolates
2.7. Genotypic Profiling of the Isolated P. aeruginosa Strains by Random Amplified Polymorphic DNA PCR (RAPD–PCR)
2.8. High-Throughput Germination Screening Assay
2.9. Confrontation on Calu-3 Cell Cultures on Permeable Transwell® Inserts
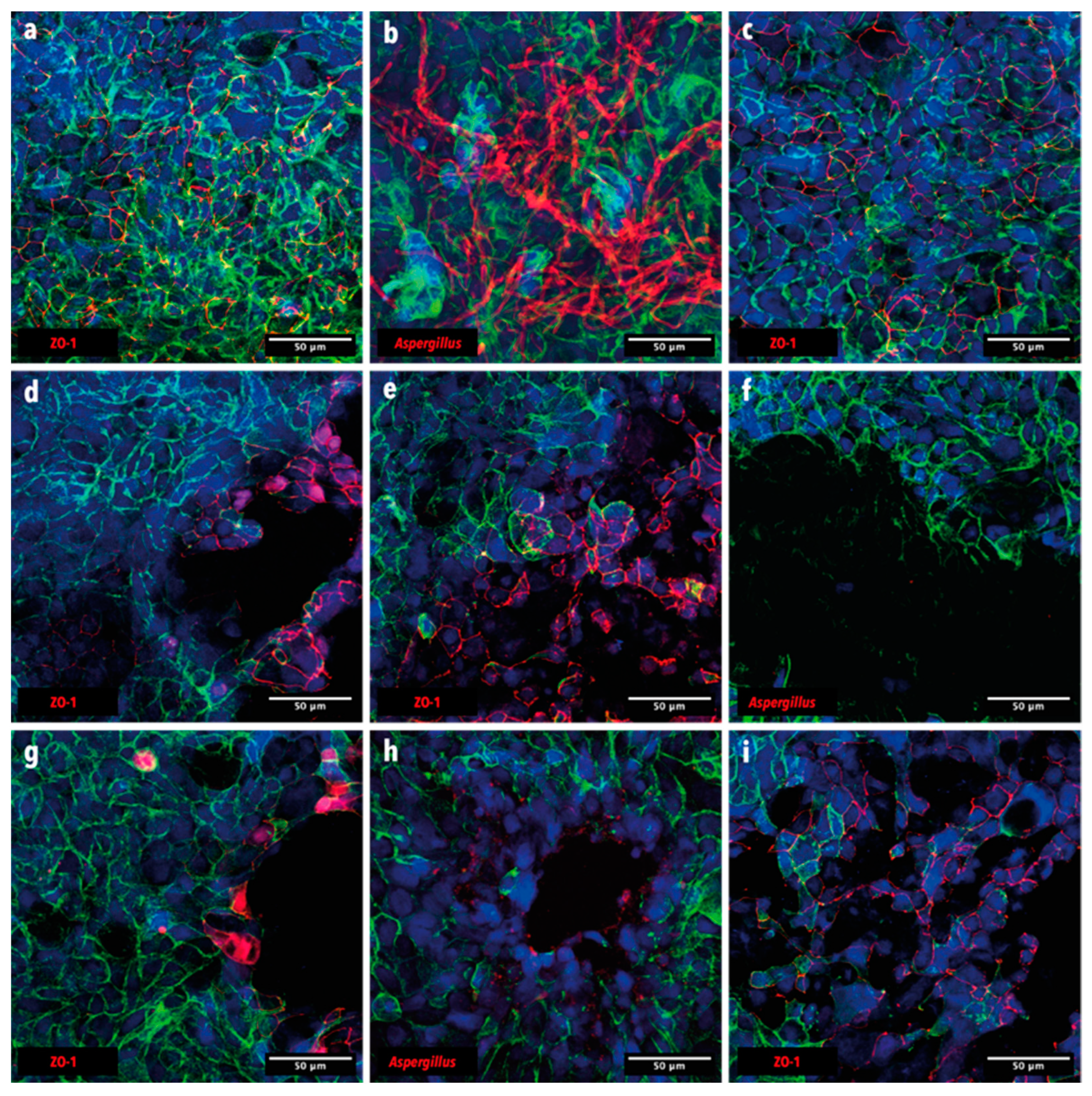
2.10. Immunofluorescence Staining and Confocal Imaging
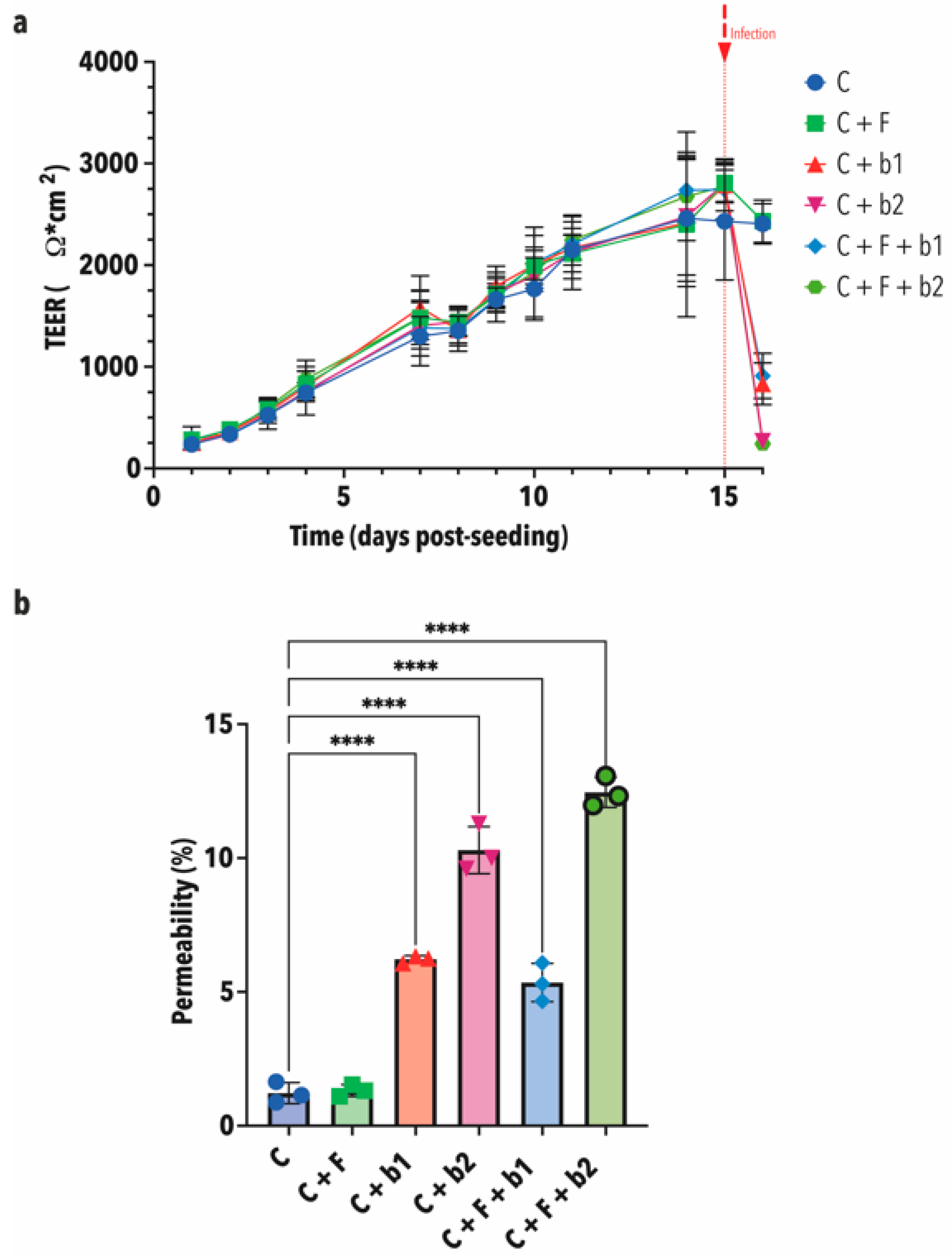
2.11. Assessment of Epithelial Barrier Function: TEER Measurements and Lucifer Yellow Permeability Assay
2.12. Bioluminescence Imaging
2.13. Cytotoxicity Assay
2.14. pH Measurements and Quantification of Calcium
2.15. Statistical Analyses
3. Results
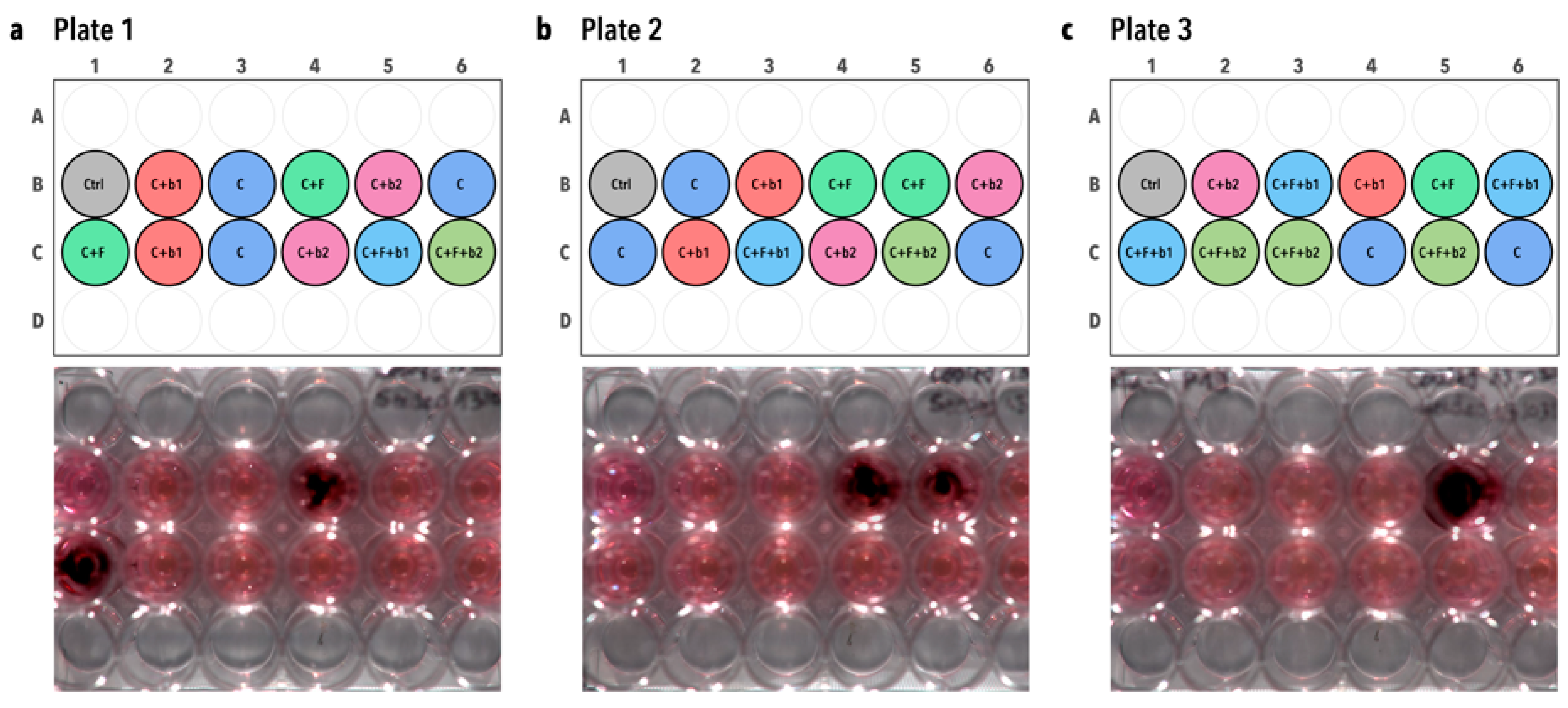
3.1. Enrichment and Isolation of Lung Bacteria Inhibiting A. fumigatus Conidia Germination and Growth in One-Step Soft agar Plate Assay
3.2. Genotyping of the Five BALF P. aeruginosa Isolates
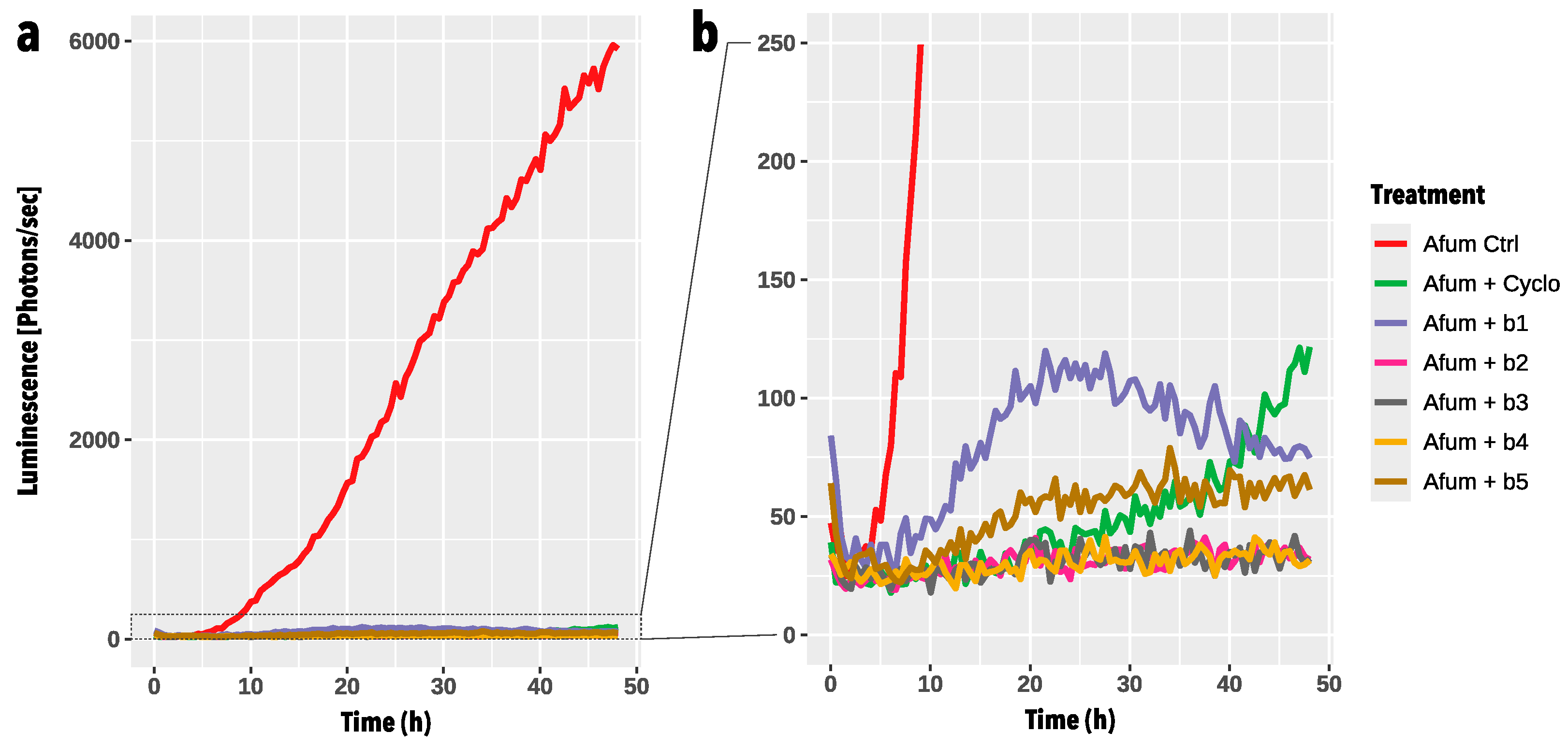
3.3. High-Throughput Germination Screening Assay
3.4. In-Vitro Confrontation of A. fumigatus with BALF P. aeruginosa Isolates on Calu-3 Cell Monolayers
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Denning, D.W. Global incidence and mortality of severe fungal disease. Lancet Infect. Dis. 2024, 24, e428–e438. [Google Scholar] [CrossRef] [PubMed]
- Paulussen, C.; Hallsworth, J.E.; Álvarez-Pérez, S.; Nierman, W.C.; Hamill, P.G.; Blain, D.; Rediers, H.; Lievens, B. Ecology of aspergillosis: Insights into the pathogenic potency of Aspergillus fumigatus and some other Aspergillus species. Microb. Biotechnol. 2017, 10, 296–322. [Google Scholar] [CrossRef] [PubMed]
- Latgé, J.P.; Chamilos, G. Aspergillus fumigatus and Aspergillosis in 2019. Clin. Microbiol. Rev. 2019, 33. [Google Scholar] [CrossRef] [PubMed]
- Gago, S.; Denning, D.W.; Bowyer, P. Pathophysiological aspects of Aspergillus colonization in disease. Med. Mycol. 2019, 57, S219–S227. [Google Scholar] [CrossRef]
- WHO. WHO Fungal Priority Pathogens List to Guide Research, Development and Public Health Action; World Health Organization: Geneva, Switzerland, 2022; Available online: https://www.who.int/publications/i/item/9789240060241 (accessed on 24 September 2024).
- Arastehfar, A.; Carvalho, A.; Houbraken, J.; Lombardi, L.; Garcia-Rubio, R.; Jenks, J.D.; Rivero-Menendez, O.; Aljohani, R.; Jacobsen, I.D.; Berman, J.; et al. Aspergillus fumigatus and aspergillosis: From basics to clinics. Stud. Mycol. 2021, 100, 100115. [Google Scholar] [CrossRef]
- Palmieri, F.; Koutsokera, A.; Bernasconi, E.; Junier, P.; von Garnier, C.; Ubags, N. Recent Advances in Fungal Infections: From Lung Ecology to Therapeutic Strategies With a Focus on Aspergillus spp. Front. Med. 2022, 9, 832510. [Google Scholar] [CrossRef]
- Ortiz, S.C.; Pennington, K.; Thomson, D.D.; Bertuzzi, M. Novel Insights into Aspergillus fumigatus Pathogenesis and Host Response from State-of-the-Art Imaging of Host-Pathogen Interactions during Infection. J. Fungi 2022, 8, 264. [Google Scholar] [CrossRef]
- Barda, O.; Maor, U.; Sadhasivam, S.; Bi, Y.; Zakin, V.; Prusky, D.; Sionov, E. The pH-Responsive Transcription Factor PacC Governs Pathogenicity and Ochratoxin A Biosynthesis in Aspergillus carbonarius. Front. Microbiol. 2020, 11, 210. [Google Scholar] [CrossRef]
- Bertuzzi, M.; Schrettl, M.; Alcazar-Fuoli, L.; Cairns, T.C.; Munoz, A.; Walker, L.A.; Herbst, S.; Safari, M.; Cheverton, A.M.; Chen, D.; et al. The pH-responsive PacC transcription factor of Aspergillus fumigatus governs epithelial entry and tissue invasion during pulmonary aspergillosis. PLoS Pathog. 2014, 10, e1004413. [Google Scholar] [CrossRef]
- Bignell, E.; Negrete-Urtasun, S.; Calcagno, A.M.; Haynes, K.; Arst, H.N., Jr.; Rogers, T. The Aspergillus pH-responsive transcription factor PacC regulates virulence. Mol. Microbiol. 2005, 55, 1072–1084. [Google Scholar] [CrossRef]
- Blatzer, M.; Schrettl, M.; Sarg, B.; Lindner, H.H.; Pfaller, K.; Haas, H. SidL, an Aspergillus fumigatus transacetylase involved in biosynthesis of the siderophores ferricrocin and hydroxyferricrocin. Appl. Environ. Microbiol. 2011, 77, 4959–4966. [Google Scholar] [CrossRef]
- Haas, H.; Eisendle, M.; Turgeon, B.G. Siderophores in fungal physiology and virulence. Annu. Rev. Phytopathol. 2008, 46, 149–187. [Google Scholar] [CrossRef] [PubMed]
- Gresnigt, M.S.; Rekiki, A.; Rasid, O.; Savers, A.; Jouvion, G.; Dannaoui, E.; Parlato, M.; Fitting, C.; Brock, M.; Cavaillon, J.-M.; et al. Reducing hypoxia and inflammation during invasive pulmonary aspergillosis by targeting the Interleukin-1 receptor. Sci. Rep. 2016, 6, 26490. [Google Scholar] [CrossRef]
- Hérivaux, A.; Willis, J.R.; Mercier, T.; Lagrou, K.; Gonçalves, S.M.; Gonçales, R.A.; Maertens, J.; Carvalho, A.; Gabaldón, T.; Cunha, C. Lung microbiota predict invasive pulmonary aspergillosis and its outcome in immunocompromised patients. Thorax 2021, 77, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Ao, Z.; Xu, H.; Li, M.; Liu, H.; Deng, M.; Liu, Y. Clinical characteristics, diagnosis, outcomes and lung microbiome analysis of invasive pulmonary aspergillosis in the community-acquired pneumonia patients. BMJ Open Respir. Res. 2023, 10, e001358. [Google Scholar] [CrossRef] [PubMed]
- Kolwijck, E.; van de Veerdonk, F.L. The potential impact of the pulmonary microbiome on immunopathogenesis of Aspergillus-related lung disease. Eur. J. Immunol. 2014, 44, 3156–3165. [Google Scholar] [CrossRef]
- Li, R.; Li, J.; Zhou, X. Lung microbiome: New insights into the pathogenesis of respiratory diseases. Signal Transduct. Target. Ther. 2024, 9, 19. [Google Scholar] [CrossRef]
- Zhao, L.; Luo, J.L.; Ali, M.K.; Spiekerkoetter, E.; Nicolls, M.R. The Human Respiratory Microbiome: Current Understandings and Future Directions. Am. J. Respir. Cell Mol. Biol. 2023, 68, 245–255. [Google Scholar] [CrossRef]
- Dickson, R.P.; Erb-Downward, J.R.; Martinez, F.J.; Huffnagle, G.B. The Microbiome and the Respiratory Tract. Annu. Rev. Physiol. 2016, 78, 481–504. [Google Scholar] [CrossRef]
- Lynch, S.V. The Lung Microbiome and Airway Disease. Ann. Am. Thorac. Soc. 2016, 13, S462–S465. [Google Scholar] [CrossRef]
- Debourgogne, A.; Monpierre, L.; Sy, K.A.; Valsecchi, I.; Decousser, J.-W.; Botterel, F. Interactions between Bacteria and Aspergillus fumigatus in Airways: From the Mycobiome to Molecular Interactions. J. Fungi 2023, 9, 900. [Google Scholar] [CrossRef] [PubMed]
- Margalit, A.; Carolan, J.C.; Kavanagh, K. Bacterial Interactions with Aspergillus fumigatus in the Immunocompromised Lung. Microorganisms 2021, 9, 435. [Google Scholar] [CrossRef] [PubMed]
- Bertuzzi, M.; van Rhijn, N.; Krappmann, S.; Bowyer, P.; Bromley, M.J.; Bignell, E.M. On the lineage of Aspergillus fumigatus isolates in common laboratory use. Med. Mycol. 2020, 59, 7–13. [Google Scholar] [CrossRef]
- Bowyer, P.; Currin, A.; Delneri, D.; Fraczek, M.G. Telomere-to-telomere genome sequence of the model mould pathogen Aspergillus fumigatus. Nat. Commun. 2022, 13, 5394. [Google Scholar] [CrossRef]
- Resendiz-Sharpe, A.; da Silva, R.P.; Geib, E.; Vanderbeke, L.; Seldeslachts, L.; Hupko, C.; Brock, M.; Lagrou, K.; Vande Velde, G. Longitudinal multimodal imaging-compatible mouse model of triazole-sensitive and -resistant invasive pulmonary aspergillosis. Dis. Model. Mech. 2022, 15, dmm049165. [Google Scholar] [CrossRef]
- Das, S.; Bernasconi, E.; Koutsokera, A.; Wurlod, D.-A.; Tripathi, V.; Bonilla-Rosso, G.; Aubert, J.-D.; Derkenne, M.-F.; Mercier, L.; Pattaroni, C.; et al. A prevalent and culturable microbiota links ecological balance to clinical stability of the human lung after transplantation. Nat. Commun. 2021, 12, 2126. [Google Scholar] [CrossRef] [PubMed]
- Muyzer, G.; Teske, A.; Wirsen, C.O.; Jannasch, H.W. Phylogenetic relationships of Thiomicrospira species and their identification in deep-sea hydrothermal vent samples by denaturing gradient gel-electrophoresis of 16S rDNA fragments. Arch. Microbiol. 1995, 164, 165–172. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Hematzadeh, A.; Haghkhah, M. Biotyping of isolates of Pseudomonas aeruginosa isolated from human infections by RAPD and ERIC-PCR. Heliyon 2021, 7, e07967. [Google Scholar] [CrossRef]
- Nanvazadeh, F.; Khosravi, A.D.; Zolfaghari, M.R.; Parhizgari, N. Genotyping of Pseudomonas aeruginosa strains isolated from burn patients by RAPD-PCR. Burns 2013, 39, 1409–1413. [Google Scholar] [CrossRef]
- Brock, M.; Jouvion, G.; Droin-Bergère, S.; Dussurget, O.; Nicola, M.A.; Ibrahim-Granet, O. Bioluminescent Aspergillus fumigatus, a new tool for drug efficiency testing and in vivo monitoring of invasive aspergillosis. Appl. Environ. Microbiol. 2008, 74, 7023–7035. [Google Scholar] [CrossRef] [PubMed]
- Arefin, A.; McCulloch, Q.; Martinez, R.; Martin, S.A.; Singh, R.; Ishak, O.M.; Higgins, E.M.; Haffey, K.E.; Huang, J.H.; Iyer, S.; et al. Micromachining of Polyurethane Membranes for Tissue Engineering Applications. ACS Biomater. Sci. Eng. 2018, 4, 3522–3533. [Google Scholar] [CrossRef]
- Quast, J. ggplate: Create Layout Plots of Biological Culture Plates and Microplates. 2023. Available online: https://github.com/jpquast/ggplate (accessed on 24 September 2024).
- Sass, G.; Nazik, H.; Penner, J.; Shah, H.; Ansari, S.R.; Clemons, K.V.; Groleau, M.C.; Dietl, A.M.; Visca, P.; Haas, H.; et al. Aspergillus-Pseudomonas interaction, relevant to competition in airways. Med. Mycol. 2019, 57 (Suppl. S2), S228–S232. [Google Scholar] [CrossRef]
- Anith, K.N.; Nysanth, N.S.; Natarajan, C. Novel and rapid agar plate methods for in vitro assessment of bacterial biocontrol isolates’ antagonism against multiple fungal phytopathogens. Lett. Appl. Microbiol. 2021, 73, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, C.Y.; Zhou, L.; Yi, Y.; Kuipers, O.P. Biocontrol properties from phyllospheric bacteria isolated from Solanum lycopersicum and Lactuca sativa and genome mining of antimicrobial gene clusters. BMC Genom. 2022, 23, 152. [Google Scholar] [CrossRef]
- Estoppey, A.; Weisskopf, L.; Di Francesco, E.; Vallat-Michel, A.; Bindschedler, S.; Chain, P.S.; Junier, P. Improved methods to assess the effect of bacteria on germination of fungal spores. FEMS Microbiol. Lett. 2022, 369, fnac034. [Google Scholar] [CrossRef]
- Verburg, K.; van Neer, J.; Duca, M.; de Cock, H. Novel Treatment Approach for Aspergilloses by Targeting Germination. J. Fungi 2022, 8, 758. [Google Scholar] [CrossRef]
- Stjärne Aspelund, A.; Hammarström, H.; Inghammar, M.; Larsson, H.; Hansson, L.; Riise, G.C.; Friman, V.; Christensson, B.; Påhlman, L.I. Microbiological findings in bronchoalveolar lavage fluid from lung transplant patients in Sweden. Transpl. Infect. Dis. 2018, 20, e12973. [Google Scholar] [CrossRef] [PubMed]
- Zeglen, S.; Wojarski, J.; Wozniak-Grygiel, E.; Siola, M.; Jastrzebski, D.; Kucewicz-Czech, E.; Zembala, M. Frequency of Pseudomonas aeruginosa colonizations/infections in lung transplant recipients. Transplant. Proc. 2009, 41, 3222–3224. [Google Scholar] [CrossRef]
- Sanchez-Rosario, Y.; Johnson, M.D.L. Media Matters, Examining Historical and Modern Streptococcus pneumoniae Growth Media and the Experiments They Affect. Front. Cell Infect. Microbiol. 2021, 11, 613623. [Google Scholar] [CrossRef]
- Leonidou, N.; Ostyn, L.; Coenye, T.; Crabbe, A.; Drager, A. Genome-scale model of Rothia mucilaginosa predicts gene essentialities and reveals metabolic capabilities. Microbiol. Spectr. 2024, 12, e0400623. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Santos, C.A.Q. Management and prophylaxis of bacterial and mycobacterial infections among lung transplant recipients. Ann. Transl. Med. 2020, 8, 413. [Google Scholar] [CrossRef] [PubMed]
- Patel, T.S.; Eschenauer, G.A.; Stuckey, L.J.; Carver, P.L. Antifungal Prophylaxis in Lung Transplant Recipients. Transplantation 2016, 100, 1815–1826. [Google Scholar] [CrossRef] [PubMed]
- Keown, K.; Reid, A.; Moore, J.E.; Taggart, C.C.; Downey, D.G. Coinfection with Pseudomonas aeruginosa and Aspergillus fumigatus in cystic fibrosis. Eur. Respir. Rev. 2020, 29, 200011. [Google Scholar] [CrossRef] [PubMed]
- Fourie, R.; Pohl, C.H. Beyond Antagonism: The Interaction Between Candida Species and Pseudomonas aeruginosa. J. Fungi 2019, 5, 34. [Google Scholar] [CrossRef]
- Mayer, F.L.; Kronstad, J.W. The Spectrum of Interactions between Cryptococcus neoformans and Bacteria. J. Fungi 2019, 5, 31. [Google Scholar] [CrossRef]
- Wood, S.J.; Goldufsky, J.W.; Seu, M.Y.; Dorafshar, A.H.; Shafikhani, S.H. Pseudomonas aeruginosa Cytotoxins: Mechanisms of Cytotoxicity and Impact on Inflammatory Responses. Cells 2023, 12, 195. [Google Scholar] [CrossRef]
- Braakhuis, H.M.; He, R.; Vandebriel, R.J.; Gremmer, E.R.; Zwart, E.; Vermeulen, J.P.; Fokkens, P.; Boere, J.; Gosens, I.; Cassee, F.R.; et al. An Air-liquid Interface Bronchial Epithelial Model for Realistic, Repeated Inhalation Exposure to Airborne Particles for Toxicity Testing. JoVE 2020, 159, e61210. [Google Scholar] [CrossRef]
- Grainger, C.I.; Greenwell, L.L.; Lockley, D.J.; Martin, G.P.; Forbes, B. Culture of Calu-3 Cells at the Air Interface Provides a Representative Model of the Airway Epithelial Barrier. Pharm. Res. 2006, 23, 1482–1490. [Google Scholar] [CrossRef]
- Li, J.; Ramezanpour, M.; Fong, S.A.; Cooksley, C.; Murphy, J.; Suzuki, M.; Psaltis, A.J.; Wormald, P.J.; Vreugde, S. Pseudomonas aeruginosa Exoprotein-Induced Barrier Disruption Correlates With Elastase Activity and Marks Chronic Rhinosinusitis Severity. Front. Cell. Infect. Microbiol. 2019, 9, 38. [Google Scholar] [CrossRef]
- Srinivasan, B.; Kolli, A.R.; Esch, M.B.; Abaci, H.E.; Shuler, M.L.; Hickman, J.J. TEER Measurement Techniques for In Vitro Barrier Model Systems. J. Lab. Autom. 2015, 20, 107–126. [Google Scholar] [CrossRef] [PubMed]
- Navalkele, B.; Chopra, T. Clinical Application of Live Biotherapeutic Products in Infectious Diseases. Front. Microbiomes 2024, 3, 1415083. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Strain Name | Reference |
|---|---|---|
| CEA10 | Aspergillus fumigatus CEA10, WT | [25] |
| CEA10b | Aspergillus fumigatus CEA10, red-shifted bioluminescent strain | [26] |
| Strain Code | Species | Sample Origin |
|---|---|---|
| Pa-1 | Pseudomonas aeruginosa | Expectoration |
| Pa-2 | Pseudomonas aeruginosa | Expectoration |
| Pa-3 | Pseudomonas aeruginosa | Expectoration |
| Pa-4 | Pseudomonas aeruginosa | Expectoration |
| Pa-5 | Pseudomonas aeruginosa | Expectoration |
| PaM-1 | Pseudomonas aeruginosa, cystic fibrosis | Expectoration |
| PaM-2 | Pseudomonas aeruginosa, cystic fibrosis | Expectoration |
| PaM-3 | Pseudomonas aeruginosa, cystic fibrosis | Expectoration |
| PaM-4 | Pseudomonas aeruginosa, cystic fibrosis | Expectoration |
| PaM-5 | Pseudomonas aeruginosa, cystic fibrosis | Expectoration |
| ID | Antibacterial Exposure | Antibacterial/Antifungal Drugs Administered |
|---|---|---|
| BAL1 | Yes | Colifin (colistimethate) |
| BAL2 | Yes | Bactrim (sulfamethoxazole + trimethoprim) |
| BAL3 | Yes | Colifin (colistimethate) |
| BAL4 | No | Ambisome (amphotericin B) |
| BAL5 | No | Ambisome (amphotericin B) |
| Strain Code | Species | Query Cover | Per. Ident |
|---|---|---|---|
| b1 | Pseudomonas aeruginosa | 100% | 99.21% |
| b2 | Pseudomonas aeruginosa | 100% | 97.40% |
| b3 | Pseudomonas aeruginosa | 100% | 98.64% |
| b4 | Pseudomonas aeruginosa | 100% | 98.72% |
| b5 | Pseudomonas aeruginosa | 100% | 99.48% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palmieri, F.; Diserens, J.; Gresse, M.; Magnin, M.; Helle, J.; Salamin, B.; Bisanti, L.; Bernasconi, E.; Pernot, J.; Shanmuganathan, A.; et al. One-Step Soft Agar Enrichment and Isolation of Human Lung Bacteria Inhibiting the Germination of Aspergillus fumigatus Conidia. Microorganisms 2024, 12, 2025. https://doi.org/10.3390/microorganisms12102025
Palmieri F, Diserens J, Gresse M, Magnin M, Helle J, Salamin B, Bisanti L, Bernasconi E, Pernot J, Shanmuganathan A, et al. One-Step Soft Agar Enrichment and Isolation of Human Lung Bacteria Inhibiting the Germination of Aspergillus fumigatus Conidia. Microorganisms. 2024; 12(10):2025. https://doi.org/10.3390/microorganisms12102025
Chicago/Turabian StylePalmieri, Fabio, Jérémy Diserens, Manon Gresse, Margo Magnin, Julina Helle, Benoît Salamin, Lorenzo Bisanti, Eric Bernasconi, Julie Pernot, Apiha Shanmuganathan, and et al. 2024. "One-Step Soft Agar Enrichment and Isolation of Human Lung Bacteria Inhibiting the Germination of Aspergillus fumigatus Conidia" Microorganisms 12, no. 10: 2025. https://doi.org/10.3390/microorganisms12102025