
Effect of the 16S rRNA Gene Hypervariable Region on the Microbiome Taxonomic Profile and Diversity in the Endangered Fish Totoaba macdonaldi

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and DNA Extraction
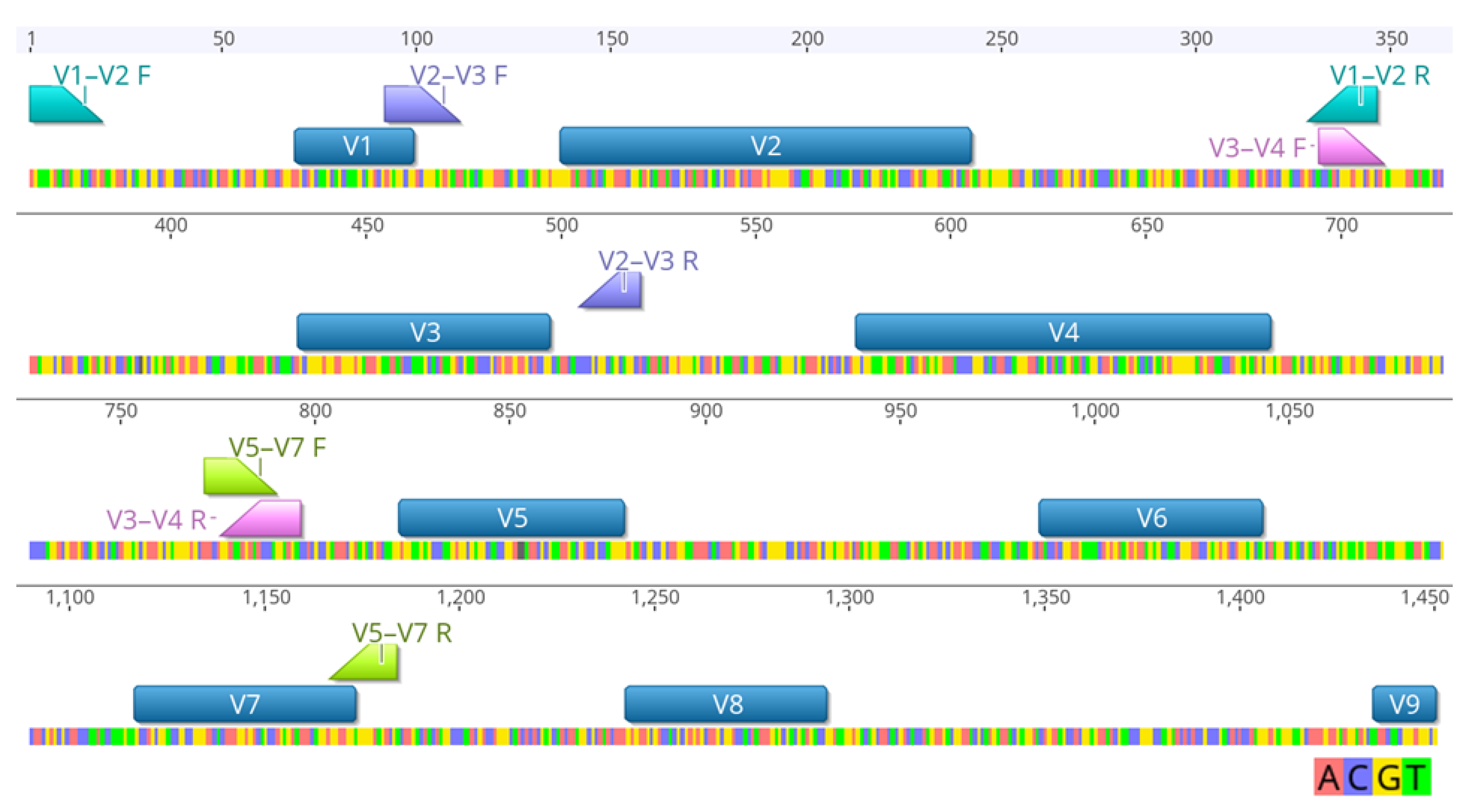
2.2. 16S rRNA Gene Amplicon Sequencing
2.3. Bioinformatic and Statistical Analysis
3. Results
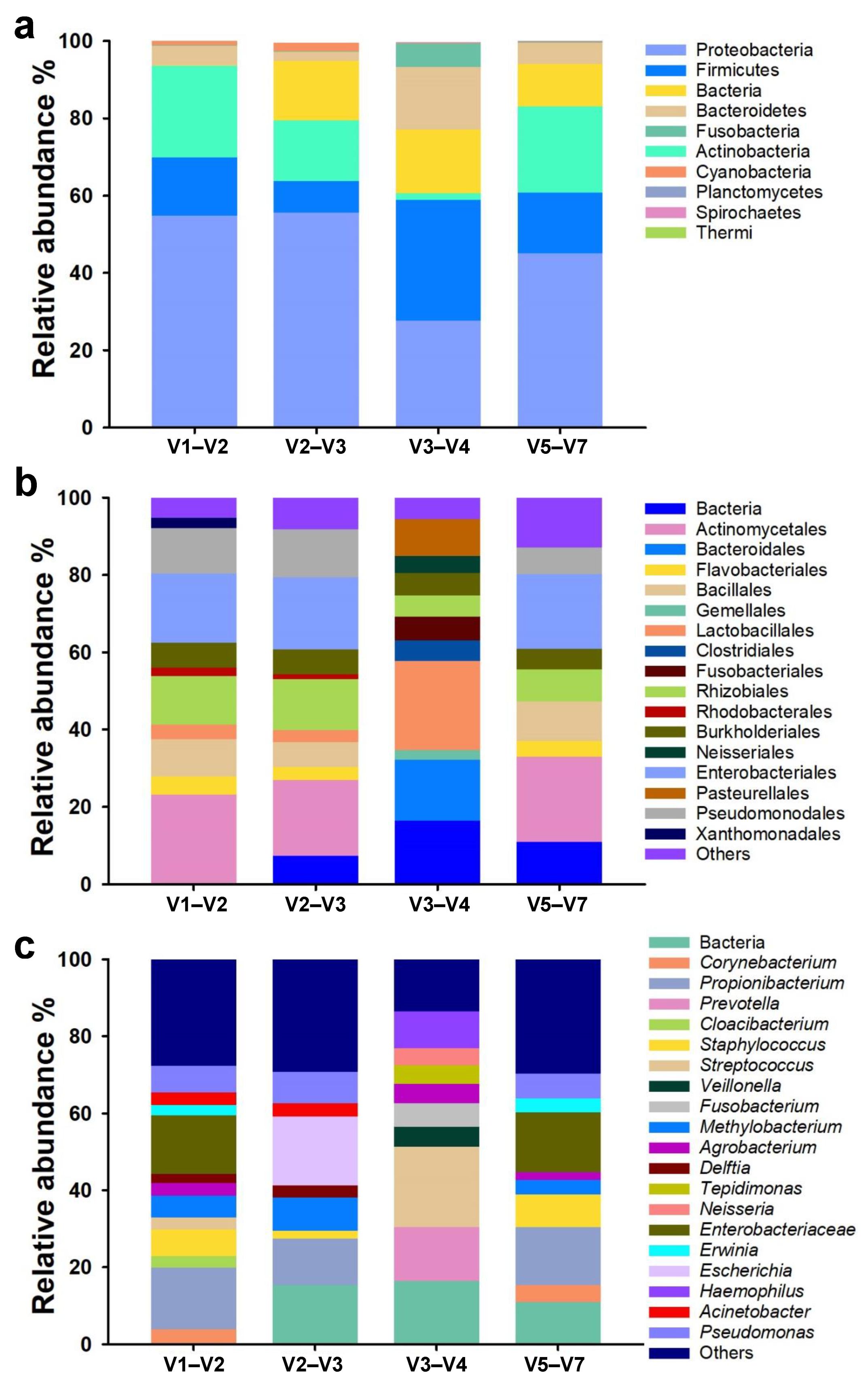
3.1. Taxonomic Composition and Relative Abundance
3.2. Number of Bacterial Taxa Detected by Each Hypervariable Region
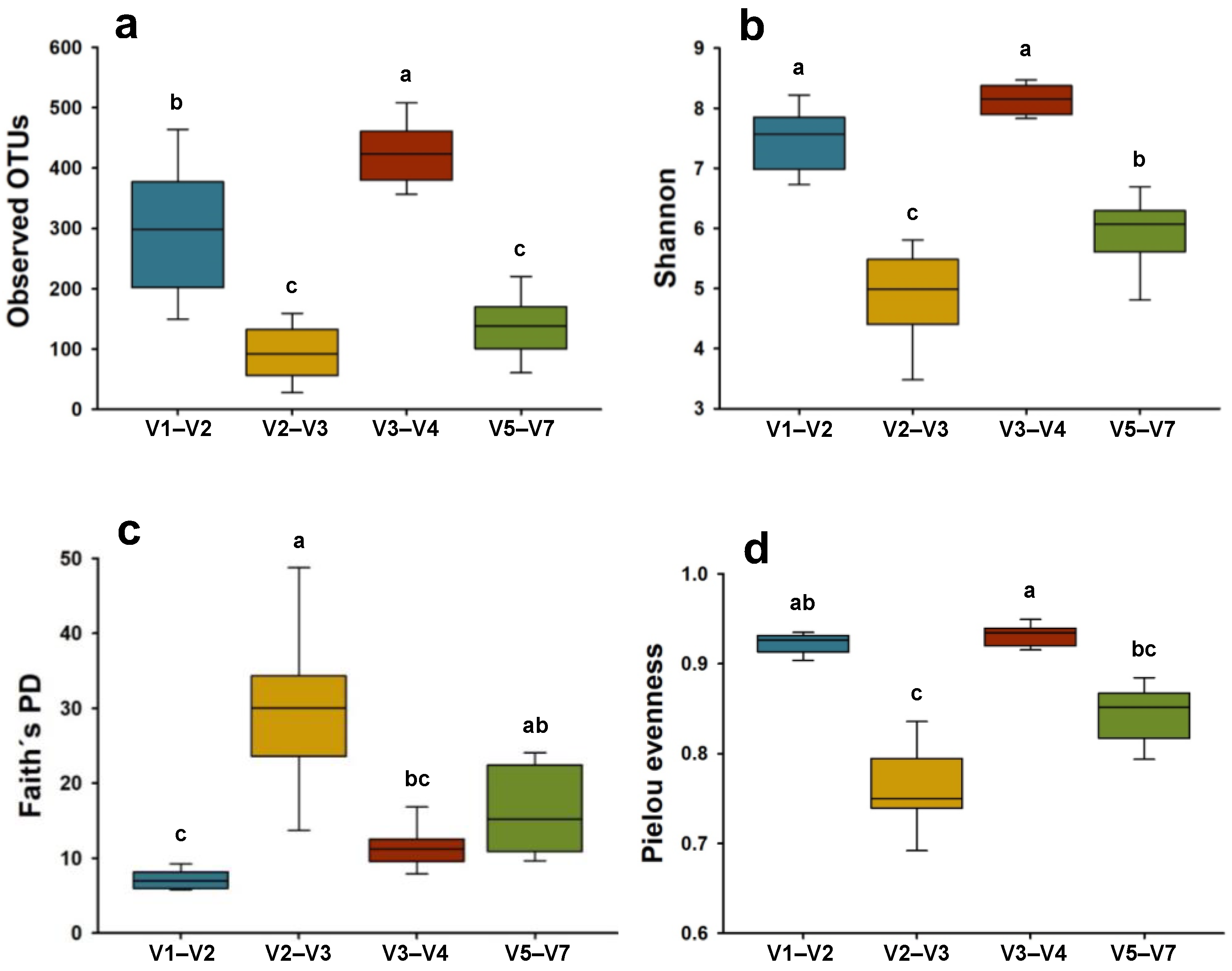
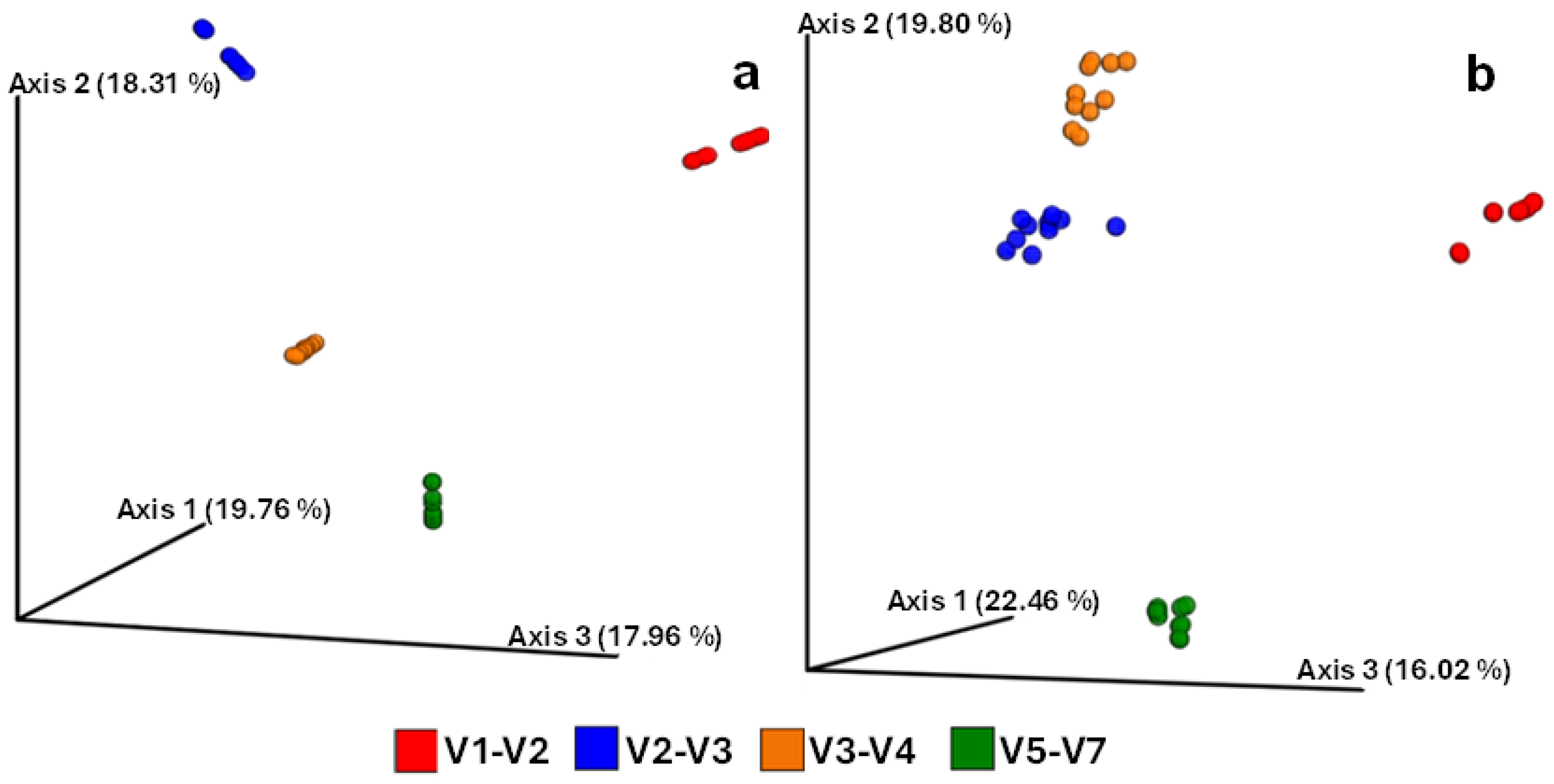
3.3. Microbial Alpha and Beta Diversity Metrics
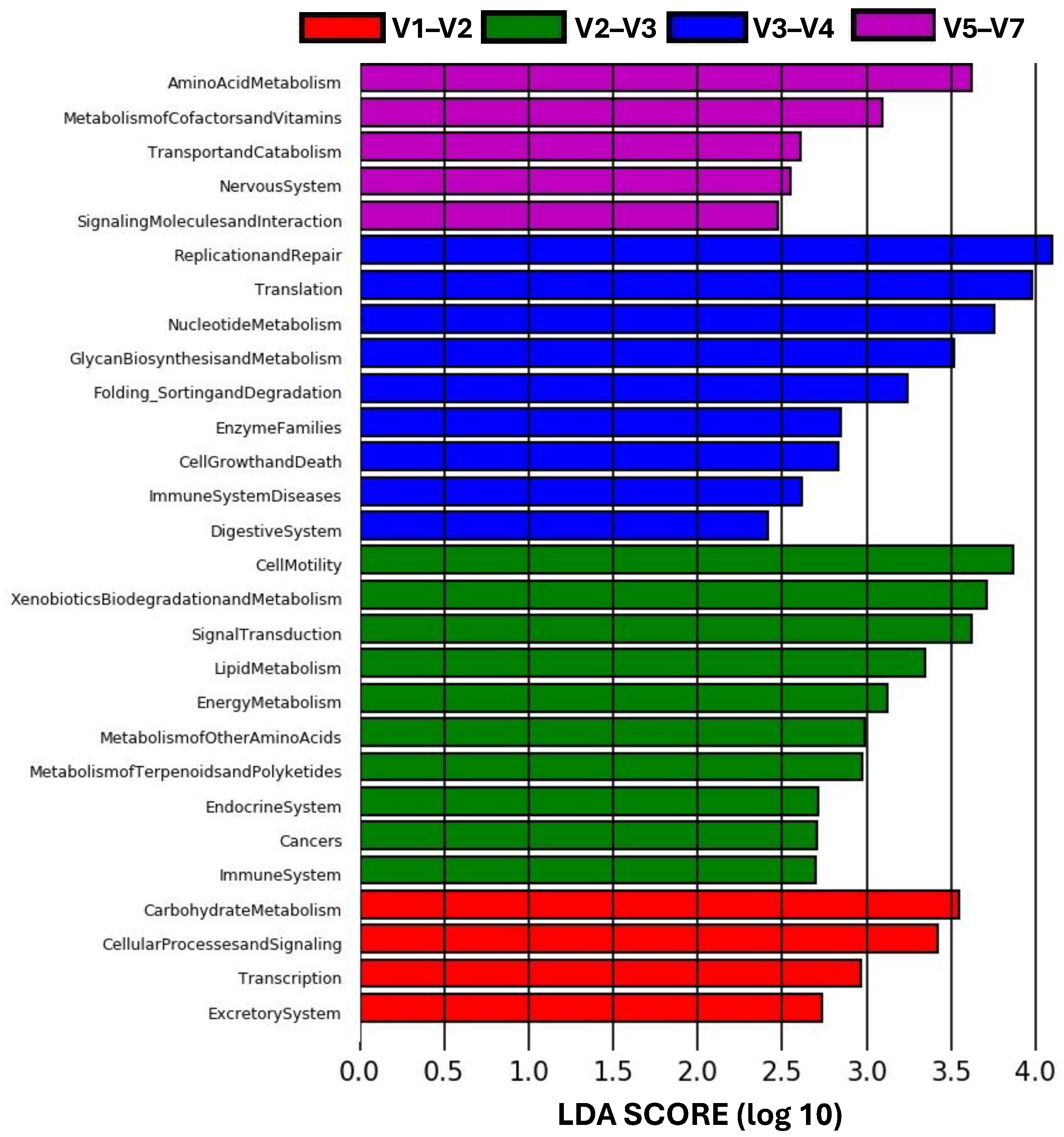
3.4. Microbiome Differential Abundance and Function Prediction
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 16S rRNA | 16S Ribosomal RNA Gene |
| V | Hypervariable Region |
| C | Conserved Region |
| NGS | Next-Generation Sequencing |
| IUCN | The International Union for Conservation of Nature |
| OTUs | Operational Taxonomic Units |
| PD | Faith’s Phylogenetic Diversity |
| PCoA | Principal Coordinate Analysis |
| LDA | Linear Discriminant Analysis |
| LEfSe | Linear Discriminant Analysis Effect Size |
| PICRUSt | Phylogenetic Investigation of Communities by Reconstruction of Unobserved States |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
References
- Srinivasan, R.; Karaoz, U.; Volegova, M.; MacKichan, J.; Kato-Maeda, M.; Miller, S.; Nadarajan, R.; Brodie, E.L.; Lynch, S.V. Use of 16S rRNA Gene for Identification of a Broad Range of Clinically Relevant Bacterial Pathogens. PLoS ONE 2015, 10, e117617. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, K.; Ogawa, M.; Taniguchi, H.; Saito, M. Molecular Approaches to Studying Microbial Communities: Targeting the 16S Ribosomal RNA Gene. J. UOEH 2016, 38, 223–232. [Google Scholar] [CrossRef]
- Chakravorty, S.; Helb, D.; Burday, M.; Connell, N.; Alland, D. A Detailed Analysis of 16S Ribosomal RNA Gene Segments for the Diagnosis of Pathogenic Bacteria. J. Microbiol. Methods 2007, 69, 330–339. [Google Scholar] [CrossRef]
- Kim, K.H.; Kim, J.M.; Baek, J.H.; Jeong, S.E.; Kim, H.; Yoon, H.S.; Jeon, C.O. Metabolic Relationships between Marine Red Algae and Algae-Associated Bacteria. Mar. Life Sci. Technol. 2024, 6, 298–314. [Google Scholar] [CrossRef] [PubMed]
- Scanes, E.; Parker, L.M.; Seymour, J.R.; Siboni, N.; Dove, M.C.; O’Connor, W.A.; Ross, P.M. Microbiomes of an Oyster Are Shaped by Metabolism and Environment. Sci. Rep. 2021, 11, 21112. [Google Scholar] [CrossRef]
- Li, M.; Ghonimy, A.; Chen, D.Q.; Li, J.T.; He, Y.Y.; López Greco, L.S.; Dyzenchauz, F.; Chang, Z.Q. Profile of the Gut Microbiota of Pacific White Shrimp under Industrial Indoor Farming System. Appl. Microbiol. Biotechnol. 2024, 108, 225. [Google Scholar] [CrossRef] [PubMed]
- Soh, M.; Tay, Y.C.; Lee, C.S.; Low, A.; Orban, L.; Jaafar, Z.; Seedorf, H. The Intestinal Digesta Microbiota of Tropical Marine Fish Is Largely Uncultured and Distinct from Surrounding Water Microbiota. NPJ Biofilms Microbiomes 2024, 10, 11. [Google Scholar] [CrossRef]
- Woese, C.R.; Kandlert, O.; Wheelis, M.L. Towards a Natural System of Organisms: Proposal for the Domains Archaea, Bacteria, and Eucarya. Proc. Natl. Acad. Sci. USA 1990, 87, 4576–4579. [Google Scholar] [CrossRef]
- Wang, Y.; Qian, P.Y. Conservative Fragments in Bacterial 16S rRNA Genes and Primer Design for 16S Ribosomal DNA Amplicons in Metagenomic Studies. PLoS ONE 2009, 4, e7401. [Google Scholar] [CrossRef]
- Gray, M.W.; Burger, G.; Lang, B.F. Mitochondrial Evolution. Science 1999, 283, 1476–1481. [Google Scholar] [CrossRef]
- Reigel, A.M.; Owens, S.M.; Hellberg, M.E. Reducing Host DNA Contamination in 16S rRNA Gene Surveys of Anthozoan Microbiomes Using PNA Clamps. Coral Reefs 2020, 39, 1817–1827. [Google Scholar] [CrossRef]
- Clarridge, J.E. Impact of 16S rRNA Gene Sequence Analysis for Identification of Bacteria on Clinical Microbiology and Infectious Diseases. Clin. Microbiol. Rev. 2004, 17, 840–862. [Google Scholar] [CrossRef]
- Janda, J.M.; Abbott, S.L. 16S rRNA Gene Sequencing for Bacterial Identification in the Diagnostic Laboratory: Pluses, Perils, and Pitfalls. J. Clin. Microbiol. 2007, 45, 2761–2764. [Google Scholar] [CrossRef]
- Baker, G.C.; Smith, J.J.; Cowan, D.A. Review and Re-Analysis of Domain-Specific 16S Primers. J. Microbiol. Methods 2003, 55, 541–555. [Google Scholar] [CrossRef]
- Patel, J. 16S rRNA Gene Sequencing for Bacterial Pathogen Identification in the Clinical Laboratory. Mol. Diagn. 2001, 6, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Sirichoat, A.; Sankuntaw, N.; Engchanil, C.; Buppasiri, P.; Faksri, K.; Namwat, W.; Chantratita, W.; Lulitanond, V. Comparison of Different Hypervariable Regions of 16S rRNA for Taxonomic Profiling of Vaginal Microbiota Using Next-Generation Sequencing. Arch. Microbiol. 2021, 203, 1159–1166. [Google Scholar] [CrossRef] [PubMed]
- Bukin, Y.S.; Galachyants, Y.P.; Morozov, I.V.; Bukin, S.V.; Zakharenko, A.S.; Zemskaya, T.I. The Effect of 16s rRNA Region Choice on Bacterial Community Metabarcoding Results. Sci. Data 2019, 6, 190007. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Wang, Y.; Qian, P.Y. Sensitivity and Correlation of Hypervariable Regions in 16S rRNA Genes in Phylogenetic Analysis. BMC Bioinform. 2016, 17, 135. [Google Scholar] [CrossRef]
- Bharti, R.; Grimm, D.G. Current challenges and best-practice protocols for microbiome analysis. Brief. Bioinform. 2021, 22, 178–193. [Google Scholar] [CrossRef]
- Kim, H.; Kim, S.; Jung, S. Instruction of Microbiome Taxonomic Profiling Based on 16S rRNA Sequencing. J. Microbiol. 2020, 58, 193–205. [Google Scholar] [CrossRef]
- Abellan-Schneyder, I.; Matchado, M.S.; Reitmeier, S.; Sommer, A.; Sewald, Z.; Baumbach, J.; List, M.; Neuhaus, K. Primer, Pipelines, Parameters: Issues in 16S rRNA Gene Sequencing. mSphere 2021, 6, e01202-20. [Google Scholar] [CrossRef] [PubMed]
- Illumina, Inc. 16S Sequencing Method Guide; Illumina, Inc.: San Diego, CA, USA, 2020. [Google Scholar]
- Sune, D.; Rydberg, H.; Augustinsson, Å.N.; Serrander, L.; Jungeström, M.B. Optimization of 16S rRNA Gene Analysis for Use in the Diagnostic Clinical Microbiology Service. J. Microbiol. Methods 2020, 170, 105854. [Google Scholar] [CrossRef] [PubMed]
- López-Aladid, R.; Fernández-Barat, L.; Alcaraz-Serrano, V.; Bueno-Freire, L.; Vázquez, N.; Pastor-Ibáñez, R.; Palomeque, A.; Oscanoa, P.; Torres, A. Determining the Most Accurate 16S rRNA Hypervariable Region for Taxonomic Identification from Respiratory Samples. Sci. Rep. 2023, 13, 3974. [Google Scholar] [CrossRef]
- Walker, S.P.; Barrett, M.; Hogan, G.; Flores Bueso, Y.; Claesson, M.J.; Tangney, M. Non-Specific Amplification of Human DNA Is a Major Challenge for 16S rRNA Gene Sequence Analysis. Sci. Rep. 2020, 10, 16356. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Flores, R.; Hernández Rodríguez, M.; Flores, J.S.O.G.; Dhar, A.K. Formalin-Fixed Paraffin-Embedded Tissues for Microbiome Analysis in Rainbow Trout (Oncorhynchus mykiss). J. Microbiol. Methods 2022, 192, 106389. [Google Scholar] [CrossRef]
- Larios-Soriano, E.; Zavala, R.C.; López, L.M.; Gómez-Gil, B.; Ramírez, D.T.; Sanchez, S.; Canales, K.; Galaviz, M.A. Soy Protein Concentrate Effects on Gut Microbiota Structure and Digestive Physiology of Totoaba macdonaldi. J. Appl. Microbiol. 2022, 132, 1384–1396. [Google Scholar] [CrossRef]
- Jang, W.J.; Kim, S.K.; Lee, S.J.; Kim, H.; Ryu, Y.W.; Shin, M.G.; Lee, J.M.; Lee, K.B.; Lee, E.W. Effect of Bacillus Sp. Supplementation Diet on Survival Rate and Microbiota Composition in Artificially Produced Eel Larvae (Anguilla japonica). Front. Microbiol. 2022, 13, 891070. [Google Scholar] [CrossRef]
- Marchesi, J.R.; Ravel, J. The Vocabulary of Microbiome Research: A Proposal. Microbiome 2015, 3, 31. [Google Scholar] [CrossRef]
- Ray, A.K.; Ghosh, K.; Ringø, E. Enzyme-Producing Bacteria Isolated from Fish Gut: A Review. Aquac. Nutr. 2012, 18, 465–492. [Google Scholar] [CrossRef]
- Merrifield, D.L.; Rodiles, A. The Fish Microbiome and Its Interactions with Mucosal Tissues. In Mucosal Health in Aquaculture; Academic Press: Cambridge, MA, USA, 2015; pp. 273–295. [Google Scholar]
- Perry, W.B.; Lindsay, E.; Payne, C.J.; Brodie, C.; Kazlauskaite, R. The Role of the Gut Microbiome in Sustainable Teleost Aquaculture. Proc. R. Soc. B Biol. Sci. 2020, 287, 20200184. [Google Scholar] [CrossRef]
- Xu, Z.; Takizawa, F.; Parra, D.; Gómez, D.; Von Gersdorff Jørgensen, L.; Lapatra, S.E.; Sunyer, J.O. Mucosal Immunoglobulins at Respiratory Surfaces Mark an Ancient Association That Predates the Emergence of Tetrapods. Nat. Commun. 2016, 7, 10728. [Google Scholar] [CrossRef]
- Arvizu, J.; Chavez, H. Sinopsis Sobre La Biología de La Totoaba, Cyoscion Macdonaldi (Gilbert, 1890); FAO: Ciudad de México, Mexico, 1972. [Google Scholar]
- Barragán-Longoria, M.F.; Hinojosa-Alvarez, S.; Hernandez-Perez, J.; Gonzalez-Cobian, L.N.; Fabian-Ortiz, E.; Garcia-Roche, A.; Chavez-Santoscoy, R.A. Supplementation of Flavonoids and Inulin in Totoaba macdonaldi: Microbiota, Liver Gene Expression and Growth Performance Responses. Aquac. Rep. 2023, 31, 101654. [Google Scholar] [CrossRef]
- SEMARNAT la Totoaba, Especie en Peligro de Extinción, Víctima del Tráfico Ilegal|Secretaría de Medio Ambiente y Recursos Naturales|Gobierno|Gob.Mx. Available online: https://www.gob.mx/semarnat/articulos/la-totoaba-especie-en-peligro-de-extincion-victima-del-trafico-ilegal?idiom=es (accessed on 17 June 2024).
- Cisneros-Mata, M.A.; True, C.; Enrique-Paredes, L.M.; Sadovy, Y.; Liu, M. Totoaba macdonaldi. The IUCN Red List of Threatened Species. 2021. Available online: https://www.iucnredlist.org/species/22003/2780880 (accessed on 17 June 2024).
- True, C.D. Desarrollo de la Biotecnia de Cultivo de Totoaba macdonaldi. Ph.D. Thesis, Universidad Autónoma de Baja California, Ensenada, Mexico, 2012. [Google Scholar]
- Cáceres-Martínez, J.; Sánchez-Serrano, S.; Vásquez-Yeomans, R. Buenas Prácticas Para el Cultivo de la Totoaba macdonaldi y Plan de Manejo Sanitario Totoaba; Universidad Autónoma de Baja California: Ensenada, Mexico, 2023. [Google Scholar]
- González-Félix, M.L.; Gatlin, D.M.; Urquidez-Bejarano, P.; de la Reé-Rodríguez, C.; Duarte-Rodríguez, L.; Sánchez, F.; Casas-Reyes, A.; Yamamoto, F.Y.; Ochoa-Leyva, A.; Perez-Velazquez, M. Effects of Commercial Dietary Prebiotic and Probiotic Supplements on Growth, Innate Immune Responses, and Intestinal Microbiota and Histology of Totoaba macdonaldi. Aquaculture 2018, 491, 239–251. [Google Scholar] [CrossRef]
- Barreto-Curiel, F.; Ramirez-Puebla, S.T.; Ringø, E.; Escobar-Zepeda, A.; Godoy-Lozano, E.; Vazquez-Duhalt, R.; Sanchez-Flores, A.; Viana, M.T. Effects of Extruded Aquafeed on Growth Performance and Gut Microbiome of Juvenile Totoaba macdonaldi. Anim. Feed. Sci. Technol. 2018, 245, 91–103. [Google Scholar] [CrossRef]
- Fadeev, E.; Cardozo-Mino, M.G.; Rapp, J.Z.; Bienhold, C.; Salter, I.; Salman-Carvalho, V.; Molari, M.; Tegetmeyer, H.E.; Buttigieg, P.L.; Boetius, A. Comparison of Two 16S rRNA Primers (V3–V4 and V4–V5) for Studies of Arctic Microbial Communities. Front. Microbiol. 2021, 12, 637526. [Google Scholar] [CrossRef]
- Kim, M.; Morrison, M.; Yu, Z. Evaluation of Different Partial 16S rRNA Gene Sequence Regions for Phylogenetic Analysis of Microbiomes. J. Microbiol. Methods 2011, 84, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Willis, C.; Desai, D.; Laroche, J. Influence of 16S rRNA Variable Region on Perceived Diversity of Marine Microbial Communities of the Northern North Atlantic. FEMS Microbiol. Lett. 2019, 366, fnz152. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An Integrated and Extendable Desktop Software Platform for the Organization and Analysis of Sequence Data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Hung, Y.M.; Lu, T.P.; Tsai, M.H.; Lai, L.C.; Chuang, E.Y. EasyMAP: A User-Friendly Online Platform for Analyzing 16S Ribosomal DNA Sequencing Data. New Biotechnol. 2021, 63, 37–44. [Google Scholar] [CrossRef]
- Heidrich, V.; Inoue, L.T.; Asprino, P.F.; Bettoni, F.; Mariotti, A.C.H.; Bastos, D.A.; Jardim, D.L.F.; Arap, M.A.; Camargo, A.A. Choice of 16S Ribosomal RNA Primers Impacts Male Urinary Microbiota Profiling. Front. Cell. Infect. Microbiol. 2022, 12, 862338. [Google Scholar] [CrossRef]
- Klemetsen, T.; Willassen, N.P.; Karlsen, C.R. Full-Length 16S rRNA Gene Classification of Atlantic Salmon Bacteria and Effects of Using Different 16S Variable Regions on Community Structure Analysis. Microbiologyopen 2019, 8, e898. [Google Scholar] [CrossRef]
- García-López, R.; Cornejo-Granados, F.; Lopez-Zavala, A.A.; Sánchez-López, F.; Cota-Huízar, A.; Sotelo-Mundo, R.R.; Guerrero, A.; Mendoza-Vargas, A.; Gómez-Gil, B.; Ochoa-Leyva, A. Doing More with Less: A Comparison of 16S Hypervariable Regions in Search of Defining the Shrimp Microbiota. Microorganisms 2020, 8, 134. [Google Scholar] [CrossRef] [PubMed]
- Egerton, S.; Culloty, S.; Whooley, J.; Stanton, C.; Ross, R.P. The Gut Microbiota of Marine Fish. Front. Microbiol. 2018, 9, 873. [Google Scholar] [CrossRef] [PubMed]
- Faith, D.P. Conservation Evaluation and Phylogenetic Diversity. Biol. Conserv. 1992, 61, 1–10. [Google Scholar] [CrossRef]
- Faith, D.P. The Role of the Phylogenetic Diversity Measure, PD, in Bio-Informatics: Getting the Defi Nition Right. Evol. Bioinform. 2006, 2, 277–283. [Google Scholar] [CrossRef]
- Kers, J.G.; Saccenti, E. The Power of Microbiome Studies: Some Considerations on Which Alpha and Beta Metrics to Use and How to Report Results. Front. Microbiol. 2022, 12, 796025. [Google Scholar] [CrossRef]
- Shannon, C.E.; Weaver, W. The Mathematical Theory of Communication. Phys. Today 1949, 3, 31–32. [Google Scholar] [CrossRef]
- Pielou, E.C. The Measurement of Diversity in Different Types of Biological Collections. J. Theor. Biol. 1966, 13, 131–144. [Google Scholar] [CrossRef]
- Ram, J.L.; Karim, A.S.; Sendler, E.D.; Kato, I. Strategy for Microbiome Analysis Using 16S rRNA Gene Sequence Analysis on the Illumina Sequencing Platform. Syst. Biol. Reprod. Med. 2011, 57, 162–170. [Google Scholar] [CrossRef]
- Van De Peer, Y.; Chapelle, S.; De Wachter, R. A Quantitative Map of Nucleotide Substitution Rates in Bacterial rRNA. Nucleic Acids Res. 1996, 24, 3381–3391. [Google Scholar] [CrossRef]
- Linhart, C.; Shamir, R. The Degenerate Primer Design Problem: Theory and Applications. J. Comput. Electron. 2005, 12, 431–456. [Google Scholar] [CrossRef] [PubMed]
- Sambo, F.; Finotello, F.; Lavezzo, E.; Baruzzo, G.; Masi, G.; Peta, E.; Falda, M.; Toppo, S.; Barzon, L.; Di Camillo, B. Optimizing PCR Primers Targeting the Bacterial 16S Ribosomal RNA Gene. BMC Bioinform. 2018, 19, 343. [Google Scholar] [CrossRef]
- Wenger, A.M.; Peluso, P.; Rowell, W.J.; Chang, P.C.; Hall, R.J.; Concepcion, G.T.; Ebler, J.; Fungtammasan, A.; Kolesnikov, A.; Olson, N.D.; et al. Accurate Circular Consensus Long-Read Sequencing Improves Variant Detection and Assembly of a Human Genome. Nat. Biotechnol. 2019, 37, 1155–1162. [Google Scholar] [CrossRef]
- Rhoads, A.; Au, K.F. PacBio Sequencing and Its Applications. Genom. Proteom. Bioinform. 2015, 13, 278–289. [Google Scholar] [CrossRef]
- Whon, T.W.; Chung, W.H.; Lim, M.Y.; Song, E.J.; Kim, P.S.; Hyun, D.W.; Shin, N.R.; Bae, J.W.; Nam, Y. Do Data Descriptor: The Effects of Sequencing Platforms on Phylogenetic Resolution in 16 S rRNA Gene Profiling of Human Feces. Sci. Data 2018, 5, 180068. [Google Scholar] [CrossRef] [PubMed]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic Biomarker Discovery and Explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive Functional Profiling of Microbial Communities Using 16S rRNA Marker Gene Sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Vargas-Albores, F.; Martínez-Córdova, L.R.; Hernández-Mendoza, A.; Cicala, F.; Lago-Lestón, A.; Martínez-Porchas, M. Therapeutic Modulation of Fish Gut Microbiota, a Feasible Strategy for Aquaculture? Aquaculture 2021, 544, 737050. [Google Scholar] [CrossRef]
- López-Carvallo, J.A.; Cruz-Flores, R.; Dhar, A.K. The Emerging Pathogen Enterocytozoon Hepatopenaei Drives a Degenerative Cyclic Pattern in the Hepatopancreas Microbiome of the Shrimp (Penaeus vannamei). Sci. Rep. 2022, 12, 14766. [Google Scholar] [CrossRef]
- Tan, Z.; Dong, W.; Ding, Y.; Ding, X.; Zhang, Q.; Jiang, L. Changes in Cecal Microbiota Community of Suckling Piglets Infected with Porcine Epidemic Diarrhea Virus. PLoS ONE 2019, 14, e219868. [Google Scholar] [CrossRef] [PubMed]
- Cornuault, J.K.; Byatt, G.; Paquet, M.E.; De Koninck, P.; Moineau, S. Zebrafish: A Big Fish in the Study of the Gut Microbiota. Curr. Opin. Biotechnol. 2022, 73, 308–313. [Google Scholar] [CrossRef] [PubMed]
- Torge, D.; Bernardi, S.; Ciciarelli, G.; Macchiarelli, G.; Bianchi, S. Dedicated Protocol for Ultrastructural Analysis of Farmed Rainbow Trout (Oncorhynchus mykiss) Tissues with Red Mark Syndrome: The Skin—Part One. Methods Protoc. 2024, 7, 37. [Google Scholar] [CrossRef] [PubMed]
- Watteau, F.; Villemin, G. Soil Microstructures Examined through Transmission Electron Microscopy Reveal Soil-Microorganisms Interactions. Front. Environ. Sci. 2018, 6, 106. [Google Scholar] [CrossRef]
- McCallum, G.; Tropini, C. The Gut Microbiota and Its Biogeography. Nat. Rev. Microbiol. 2024, 22, 105–118. [Google Scholar] [CrossRef]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of General 16S Ribosomal RNA Gene PCR Primers for Classical and Next-Generation Sequencing-Based Diversity Studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef]
- Beckers, B.; Op De Beeck, M.; Thijs, S.; Truyens, S.; Weyens, N.; Boerjan, W.; Vangronsveld, J. Performance of 16s RDNA Primer Pairs in the Study of Rhizosphere and Endosphere Bacterial Microbiomes in Metabarcoding Studies. Front. Microbiol. 2016, 7, 650. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pérez-Bustamante, I.S.; Cruz-Flores, R.; López-Carvallo, J.A.; Sánchez-Serrano, S. Effect of the 16S rRNA Gene Hypervariable Region on the Microbiome Taxonomic Profile and Diversity in the Endangered Fish Totoaba macdonaldi. Microorganisms 2024, 12, 2119. https://doi.org/10.3390/microorganisms12112119
Pérez-Bustamante IS, Cruz-Flores R, López-Carvallo JA, Sánchez-Serrano S. Effect of the 16S rRNA Gene Hypervariable Region on the Microbiome Taxonomic Profile and Diversity in the Endangered Fish Totoaba macdonaldi. Microorganisms. 2024; 12(11):2119. https://doi.org/10.3390/microorganisms12112119
Chicago/Turabian StylePérez-Bustamante, Itzel Soledad, Roberto Cruz-Flores, Jesús Antonio López-Carvallo, and Samuel Sánchez-Serrano. 2024. "Effect of the 16S rRNA Gene Hypervariable Region on the Microbiome Taxonomic Profile and Diversity in the Endangered Fish Totoaba macdonaldi" Microorganisms 12, no. 11: 2119. https://doi.org/10.3390/microorganisms12112119
APA StylePérez-Bustamante, I. S., Cruz-Flores, R., López-Carvallo, J. A., & Sánchez-Serrano, S. (2024). Effect of the 16S rRNA Gene Hypervariable Region on the Microbiome Taxonomic Profile and Diversity in the Endangered Fish Totoaba macdonaldi. Microorganisms, 12(11), 2119. https://doi.org/10.3390/microorganisms12112119