
Microbial Basis for Suppression of Soil-Borne Disease in Crop Rotation


Abstract
:1. Introduction
2. Materials and Methods
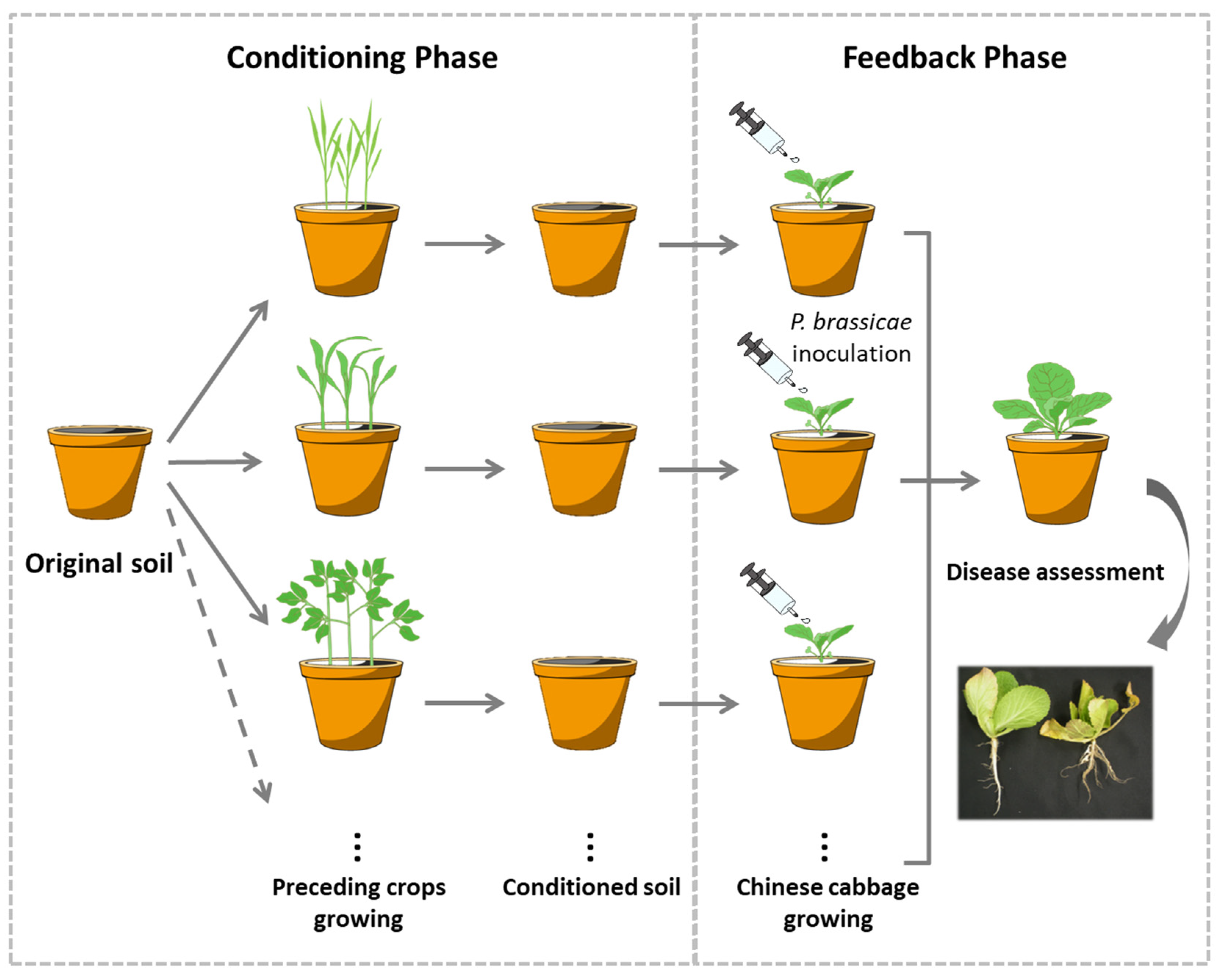
2.1. Experimental Design
2.2. Soil and Plant Materials
2.3. Chinese Cabbage Cultivation and Clubroot Disease Inoculation
2.4. Sampling and Disease Assessment
2.5. Sample Preparation and DNA Extraction
2.6. Quantification of P. brassicae Pathogen
2.7. PCR Amplification and Next-Generation Sequencing
2.8. Sequence Processing
2.9. Statistical Analysis and Visualization
3. Results
3.1. Effects of Preceding Crops on Clubroot Damage
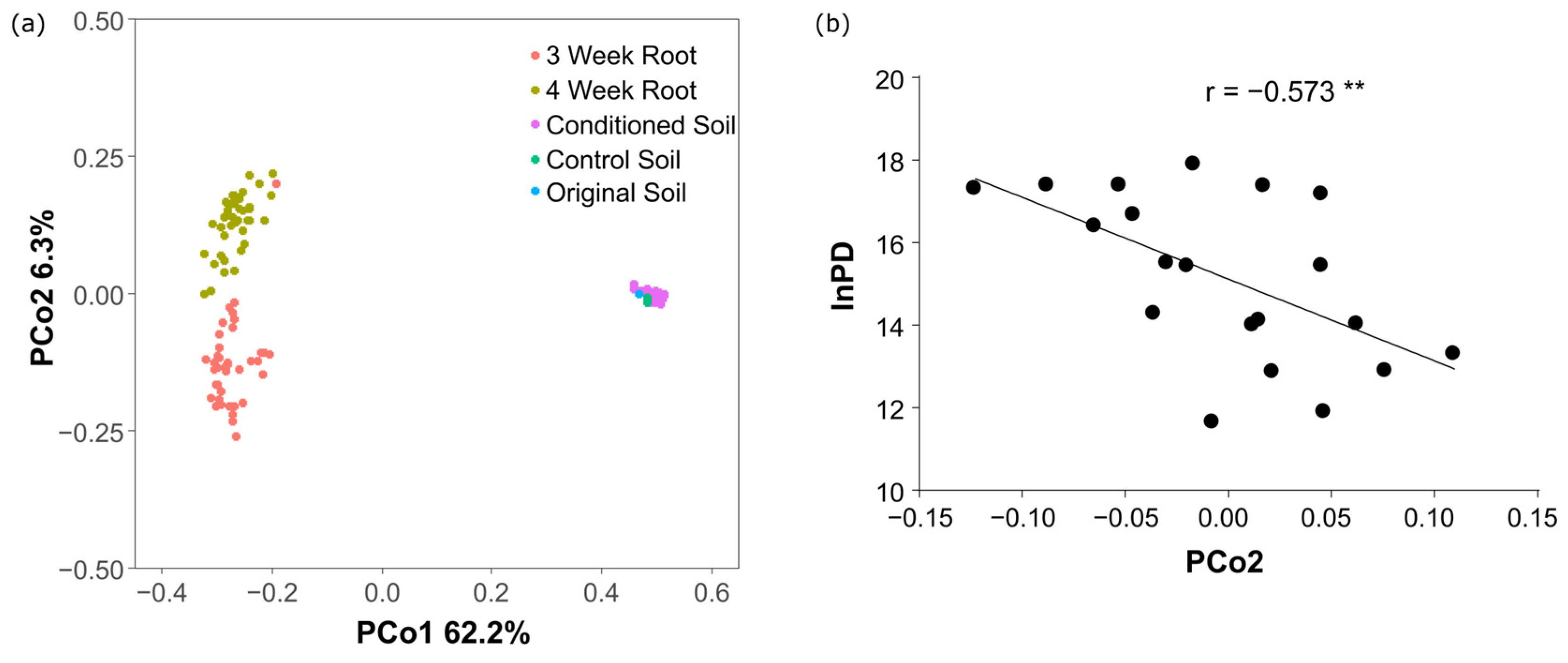
3.2. Changes in Bacterial Communities Due to Soil Conditioning
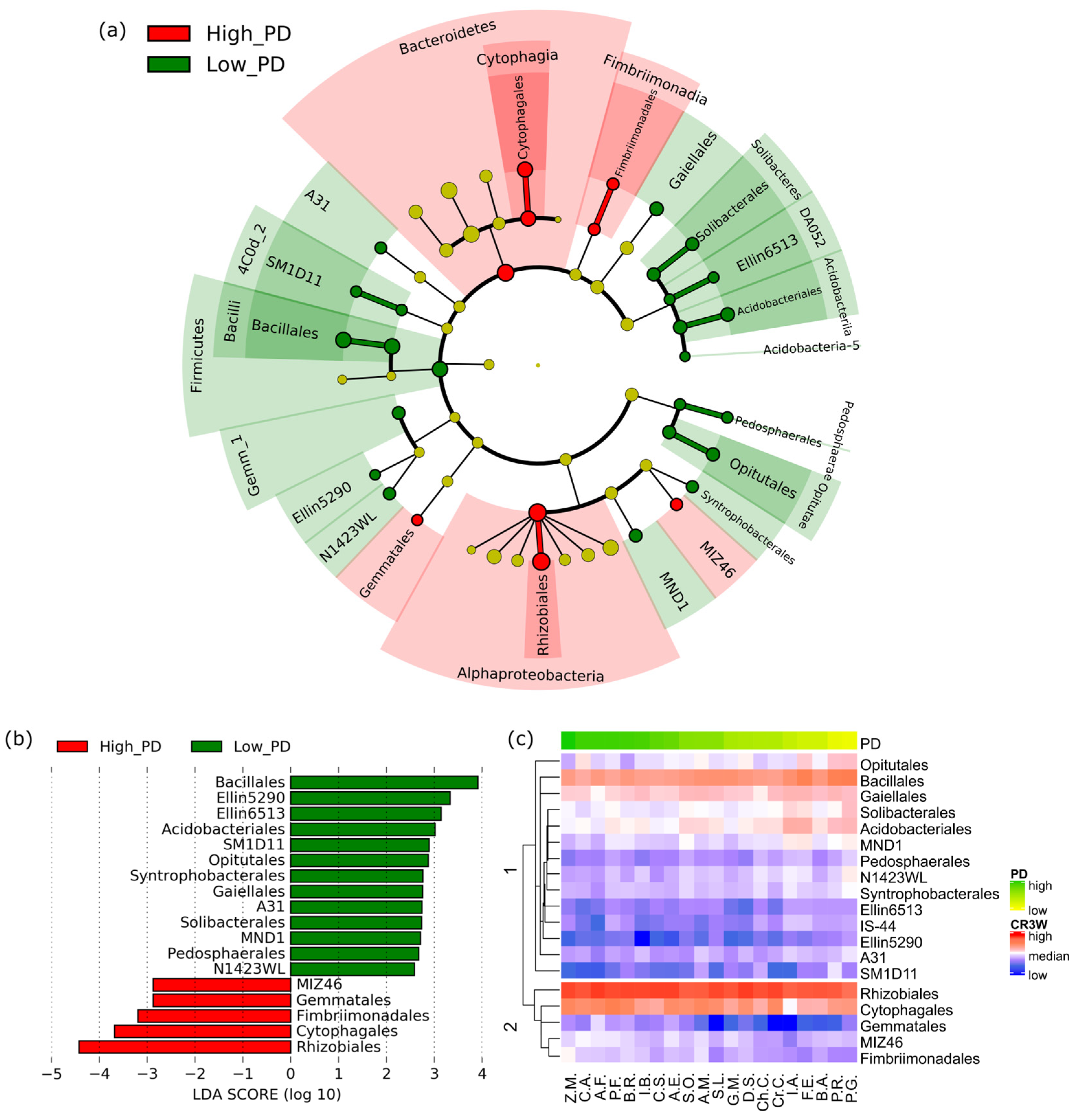
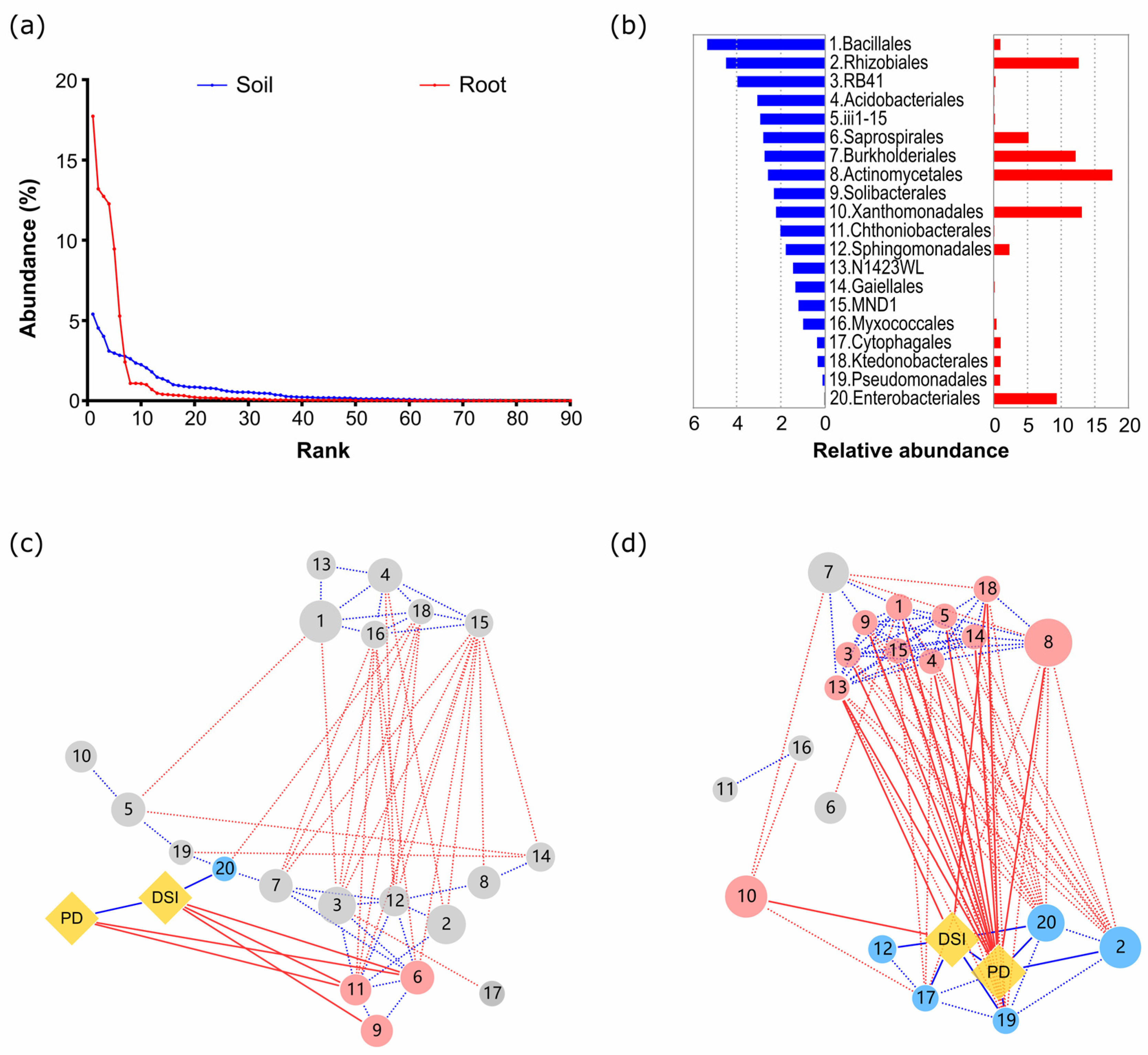
3.3. Microbial Basis for Suppression of Clubroot Damage
3.4. Legacy Effects of Preceding Crops
4. Discussion
4.1. Microbial Basis for Disease Suppression
4.2. Legacy Effects of Conditioned Soil on Disease Suppression
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tilman, D.; Reich, P.B.; Knops, M.H. Biodiversity and ecosystem stability in a decade-long grassland experiment. Nature 2006, 441, 629–632. [Google Scholar] [CrossRef] [PubMed]
- Schnitzer, S.A.; Klironomos, J.N.; HilleRisLambers, J.; Kinkel, L.L.; Reich, P.B.; Xiao, K.; Rillig, M.C.; Sikes, B.A.; Callaway, R.M.; Mangan, S.A.; et al. Soil microbes drive the classic plant diversity–productivity pattern. Ecology 2011, 92, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.S.; Hill, J.D.; Chase, C.A.; Johanns, A.M.; Liebman, M. Increasing cropping system diversity balances productivity, profitability and environmental health. PLoS ONE 2012, 7, e47149. [Google Scholar] [CrossRef] [PubMed]
- Cook, R.J. Toward cropping systems that enhance productivity and sustainability. Proc. Natl. Acad. Sci. USA 2006, 103, 18389–18394. [Google Scholar] [CrossRef]
- Yang, X.-X.; Huang, X.-Q.; Wu, W.-X.; Xiang, Y.-J.; Du, L.; Zhang, L.; Liu, Y. Effects of different rotation patterns on the occurrence of clubroot disease and diversity of rhizosphere microbes. J. Integr. Agric. 2020, 19, 2265–2273. [Google Scholar] [CrossRef]
- Peralta, A.L.; Sun, Y.M.; McDaniel, M.D.; Lennon, J.T. Crop rotational diversity increases disease suppressive capacity of soil microbiomes. Ecosphere 2018, 9, e02235. [Google Scholar] [CrossRef]
- Fitzpatrick, C.R.; Copeland, J.; Wang, P.W.; Guttman, D.S.; Kotanen, P.M.; Johnson, M.T.J. Assembly and ecological function of the root microbiome across angiosperm plant species. Proc. Natl. Acad. Sci. USA 2018, 115, E1157–E1165. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, D.S.; Lebeis, S.L.; Paredes, S.H.; Yourstone, S.; Gehring, J.; Malfatti, S.; Tremblay, J.; Engelbrektson, A.; Kunin, V.; Rio, T.G.d.; et al. Defining the core arabidopsis thaliana root microbiome. Nature 2012, 488, 86–90. [Google Scholar] [CrossRef]
- Bulgarelli, D.; Rott, M.; Schlaeppi, K.; Ver Loren van Themaat, E.; Ahmadinejad, N.; Assenza, F.; Rauf, P.; Huettel, B.; Reinhardt, R.; Schmelzer, E.; et al. Revealing structure and assembly cues for arabidopsis root-inhabiting bacterial microbiota. Nature 2012, 488, 91–95. [Google Scholar] [CrossRef]
- Wang, B.; Sugiyama, S. Phylogenetic signal of host plants in the bacterial and fungal root microbiomes of cultivated angiosperms. Plant J. 2020, 104, 522–531. [Google Scholar] [CrossRef]
- Niu, Y.N.; Bainard, L.D.; May, W.E.; Hossain, Z.; Hamel, C.; Gan, Y.T. Intensified pulse rotations buildup pea rhizosphere pathogens in cereal and pulse based cropping systems. Front. Microbiol. 2018, 9, 1909. [Google Scholar] [CrossRef] [PubMed]
- Bever, J.D. Soil community feedback and the coexistence of competitors: Conceptual frameworks and empirical tests. New Phytol. 2003, 157, 465–473. [Google Scholar] [CrossRef] [PubMed]
- van der Putten, W.H.; Bardgett, R.D.; Bever, J.D.; Bezemer, T.M.; Casper, B.B.; Fukami, T.; Kardol, P.; Klironomos, J.N.; Kulmatiski, A.; Schweitzer, J.A.; et al. Plant–soil feedbacks: The past, the present and future challenges. J. Ecol. 2013, 101, 265–276. [Google Scholar] [CrossRef]
- Bennett, J.A.; Klironomos, J. Mechanisms of plant–soil feedback: Interactions among biotic and abiotic drivers. New Phytol. 2019, 222, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Exposito, R.G.; de Bruijn, I.; Postma, J.; Raaijmakers, J.M. Current insights into the role of rhizosphere bacteria in disease suppressive soils. Front. Microbiol. 2017, 8, 2529. [Google Scholar]
- Doornbos, R.F.; van Loon, L.C.; Bakker, P.A.H.M. Impact of root exudates and plant defense signaling on bacterial communities in the rhizosphere. A review. Agron. Sustain. Dev. 2012, 32, 227–243. [Google Scholar] [CrossRef]
- Liégard, B.; Baillet, V.; Etcheverry, M.; Joseph, E.; Lariagon, C.; Lemoine, J.; Evrard, A.; Colot, V.; Gravot, A.; Manzanares-Dauleux, M.J.; et al. Quantitative resistance to clubroot infection mediated by transgenerational epigenetic variation in arabidopsis. New Phytol. 2019, 222, 468–479. [Google Scholar] [CrossRef]
- Mendes, L.W.; Raaijmakers, J.M.; de Hollander, M.; Mendes, R.; Tsai, S.M. Influence of resistance breeding in common bean on rhizosphere microbiome composition and function. ISME J. 2017, 12, 212. [Google Scholar] [CrossRef]
- Wei, Z.; Yang, T.; Friman, V.-P.; Xu, Y.; Shen, Q.; Jousset, A. Trophic network architecture of root-associated bacterial communities determines pathogen invasion and plant health. Nat. Commun. 2015, 6, 8413. [Google Scholar] [CrossRef]
- Mendes, R.; Kruijt, M.; Bruijn, I.; Dekkers, E.; Voort, M.; Schneider, J.H.M. Deciphering the rhizosphere microbiome for disease-suppressive bacteria. Science 2011, 332, 1097–1100. [Google Scholar] [CrossRef]
- Wang, J.; Huang, Y.; Lin, S.; Liu, F.; Song, Q.; Peng, Y.; Zhao, L. A strain of Streptomyces griseoruber isolated from rhizospheric soil of chinese cabbage as antagonist to Plasmodiophora brassicae. Ann. Microbiol. 2011, 62, 247–253. [Google Scholar] [CrossRef]
- Sundelin, T.; Christensen, C.B.; Larsen, J.; Moller, K.; Lubeck, M.; Bodker, L.; Jensen, B. In planta quantification of Plasmodiophora brassicae using signature fatty acids and real-time pcr. Plant Dis. 2010, 94, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, D.S.; Yourstone, S.; Mieczkowski, P.; Jones, C.D.; Dangl, J.L. Practical innovations for high-throughput amplicon sequencing. Nat. Methods 2013, 10, 999–1002. [Google Scholar] [CrossRef]
- Magoc, T.; Salzberg, S.L. Flash: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. Uchime improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than blast. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16s rrna gene database and workbench compatible with arb. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, J. Multivariate Analysis of Ecological Communities in R: Vegan Tutorial, R Package Version 1; ResearchGate GmbH: Berlin, Germany, 2007. [Google Scholar]
- Paradis, E.; Schliep, K. Ape 5.0: An environment for modern phylogenetics and evolutionary analyses in r. Bioinformatics 2019, 35, 526–528. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef]
- Peng, G.; Pageau, D.; Strelkov, S.E.; Gossen, B.D.; Hwang, S.F.; Lahlali, R. A > 2-year crop rotation reduces resting spores of Plasmodiophora brassicae in soil and the impact of clubroot on canola. Eur. J. Agron. 2015, 70, 78–84. [Google Scholar] [CrossRef]
- Ernst, T.W.; Kher, S.; Stanton, D.; Rennie, D.C.; Hwang, S.F.; Strelkov, S.E. Plasmodiophora brassicae resting spore dynamics in clubroot resistant canola (Brassica napus) cropping systems. Plant Pathol. 2019, 68, 399–408. [Google Scholar] [CrossRef]
- Anders, J.; Katarzyna, M.S.; Gunnar, B.; Ann-Charlotte, W. Quantitative pcr shows propagation of Plasmodiophora brassicae in swedish long term field trials. Eur. J. Plant Pathol. 2016, 145, 573–581. [Google Scholar] [CrossRef]
- Gossen, B.D.; Kasinathan, H.; Deora, A.; Peng, G.; McDonald, M.R. Effect of soil type, organic matter content, bulk density and saturation on clubroot severity and biofungicide efficacy. Plant Pathol. 2016, 65, 1238–1245. [Google Scholar] [CrossRef]
- Massalha, H.; Korenblum, E.; Malitsky, S.; Shapiro, O.H.; Aharoni, A. Live imaging of root–bacteria interactions in a microfluidics setup. Proc. Natl. Acad. Sci. USA 2017, 114, 4549–4554. [Google Scholar] [CrossRef]
- Jourand, P.; Giraud, E.; Bena, G.; Sy, A.; Willems, A.; Gillis, M.; Dreyfus, B.; de Lajudie, P. Methylobacterium nodulans sp. Nov., for a group of aerobic, facultatively methylotrophic, legume root-nodule-forming and nitrogen-fixing bacteria. Int. J. Syst. Evol. Microbiol. 2004, 54, 2269–2273. [Google Scholar] [CrossRef]
- Hirsch, P.R.; Mauchline, T.M. Mutualism: Plant-microorganism interactions. In Microbial Ecological Theory–Current Perspectives; Ogilvie, L.A., Hirsch, P.R., Eds.; Caister Academic Press: Poole, UK, 2012; pp. 43–55. [Google Scholar]
- Ivanova, E.G.; Doronina, N.V.; Shepelyakovskaya, A.O.; Laman, A.G.; Brovko, F.A.; Trotsenko, Y.A. Facultative and obligate aerobic methylobacteria synthesize cytokinins. Microbiology 2000, 69, 646–651. [Google Scholar] [CrossRef]
- Delmotte, N.; Knief, C.; Chaffron, S.; Innerebner, G.; Roschitzki, B.; Schlapbach, R.; von Mering, C.; Vorholt, J.A. Community proteogenomics reveals insights into the physiology of phyllosphere bacteria. Proc. Natl. Acad. Sci. USA 2009, 106, 16428–16433. [Google Scholar] [CrossRef]
- Wang, B.; Adachi, Y.; Sugiyama, S. Soil productivity and structure of bacterial and fungal communities in unfertilized arable soil. PLoS ONE 2018, 13, e0204085. [Google Scholar] [CrossRef]
- Hegewald, H.; Wensch-Dorendorf, M.; Sieling, K.; Christen, O. Impacts of break crops and crop rotations on oilseed rape productivity: A review. Eur. J. Agron. 2018, 101, 63–77. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Abbreviation | Preceding Crops | Scientific Name | DSI (SE) | lnPD (SE) |
|---|---|---|---|---|---|
| 1 | Z.M. | Maize | Zea mays subsp. mays | 77.8 (0.00) | 17.9 (0.16) |
| 2 | A.F. | Welsh onion | Allium fistulosum | 73.3 (8.31) | 17.4 (0.25) |
| 3 | S.O. | Spinach | Spinacia oleracea | 73.3 (4.44) | 15.5 (0.18) |
| 4 | P.F. | Perilla | Perilla frutescens | 64.4 (8.89) | 17.4 (0.34) |
| 5 | S.L. | Tomato | Solanum lycopersicum | 60.0 (4.44) | 15.4 (0.42) |
| 6 | A.M. | Snapdragon | Antirrhinum majus | 55.6 (0.00) | 15.4 (0.08) |
| 7 | A.E. | Okra | Abelmoschus esculentus | 55.6 (7.03) | 16.4 (0.25) |
| 8 | B.R. | Chinese cabbage | Brassica rapa subsp. pekinensis | 55.6 (9.94) | 17.3 (1.91) |
| 9 | C.A. | Plumed cockscomb | Celosia argentea | 51.1 (4.44) | 17.4 (0.17) |
| 10 | C.S. | Cucumber | Cucumis sativus | 48.9 (12.96) | 16.7 (0.11) |
| 11 | I.B. | Garden balsam | Impatiens balsamina | 42.2 (5.44) | 17.2 (0.09) |
| 12 | B.A. | Creeping spinach | Basella alba | 37.8 (8.31) | 12.9 (2.14) |
| 13 | I.A. | Water spinach | Ipomoea aquatica | 37.8 (4.44) | 13.3 (0.25) |
| 14 | F.E. | Buckwheat | Fagopyrum esculentum | 37.8 (4.44) | 12.9 (0.58) |
| 15 | D.S. | Fringed pink | Dianthus superbus | 37.8 (4.44) | 14.1 (0.84) |
| 16 | P.R. | Corn poppy | Papaver rhoeas | 33.3 (7.03) | 11.9 (2.41) |
| 17 | Ch.C. | Crown daisy | Chrysanthemum coronarium | 28.9 (4.44) | 14.0 (0.20) |
| 18 | G.M. | Soybean | Glycine max | 26.7 (9.69) | 14.3 (0.08) |
| 19 | P.G. | Platycodon | Platycodon grandiflorus | 24.4 (5.44) | 11.6 (0.07) |
| 20 | Cr.C. | Canadian honewort | Cryptotaenia canadensis | 11.1 (0.00) | 14.0 (0.13) |
| Significance | p < 0.001 | p < 0.001 | |||
| Order | Family | Genus | DSI (p) | PD (p) | ||
|---|---|---|---|---|---|---|
| Bacillales | Paenibacillaceae | Cohnella | 0.01 | −0.19 | ||
| Paenibacillaceae | Ammoniphilus | 0.04 | −0.29 | |||
| Paenibacillaceae | Brevibacillus | −0.16 | −0.32 | |||
| Staphylococcaceae | Staphylococcus | −0.17 | −0.22 | |||
| Paenibacillaceae | Paenibacillus | 0.19 | −0.14 | |||
| Planococcaceae | Solibacillus | −0.22 | −0.52 | * | ||
| Alicyclobacillaceae | Alicyclobacillus | −0.28 | −0.49 | * | ||
| Bacillaceae | Bacillus | −0.45 | * | −0.77 | *** | |
| Rhizobiales | Bradyrhizobiaceae | Balneimonas | 0.16 | 0.38 | ||
| Bradyrhizobiaceae | Bosea | −0.31 | −0.55 | * | ||
| Brucellaceae | Ochrobactrum | −0.33 | −0.15 | |||
| Hyphomicrobiaceae | Devosia | 0.50 | * | 0.64 | ** | |
| Hyphomicrobiaceae | Hyphomicrobium | 0.25 | 0.09 | |||
| Hyphomicrobiaceae | Pedomicrobium | −0.07 | −0.18 | |||
| Hyphomicrobiaceae | Rhodoplanes | 0.04 | −0.19 | |||
| Methylobacteriaceae | Methylobacterium | −0.39 | −0.22 | |||
| Phyllobacteriaceae | Mesorhizobium | 0.60 | ** | 0.69 | *** | |
| Rhizobiaceae | Agrobacterium | 0.48 | * | 0.72 | *** | |
| Rhizobiaceae | Kaistia | 0.42 | 0.71 | *** | ||
| Rhizobiaceae | Rhizobium | −0.02 | 0.25 | |||
| Rhodobiaceae | Afifella | 0.47 | * | 0.46 | * | |
| Xanthobacteraceae | Labrys | 0.54 | * | 0.58 | ** | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, B.; Sugiyama, S. Microbial Basis for Suppression of Soil-Borne Disease in Crop Rotation. Microorganisms 2024, 12, 2290. https://doi.org/10.3390/microorganisms12112290
Wang B, Sugiyama S. Microbial Basis for Suppression of Soil-Borne Disease in Crop Rotation. Microorganisms. 2024; 12(11):2290. https://doi.org/10.3390/microorganisms12112290
Chicago/Turabian StyleWang, Boxi, and Shuichi Sugiyama. 2024. "Microbial Basis for Suppression of Soil-Borne Disease in Crop Rotation" Microorganisms 12, no. 11: 2290. https://doi.org/10.3390/microorganisms12112290
APA StyleWang, B., & Sugiyama, S. (2024). Microbial Basis for Suppression of Soil-Borne Disease in Crop Rotation. Microorganisms, 12(11), 2290. https://doi.org/10.3390/microorganisms12112290