
Metagenomic Analyses of Water Samples of Two Urban Freshwaters in Berlin, Germany, Reveal New Highly Diverse Invertebrate Viruses


Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling and Virus Enrichment
2.2. RNA Preparation and Illumina Sequencing
2.3. Sequence Data Processing and Sequence Analyses
3. Results
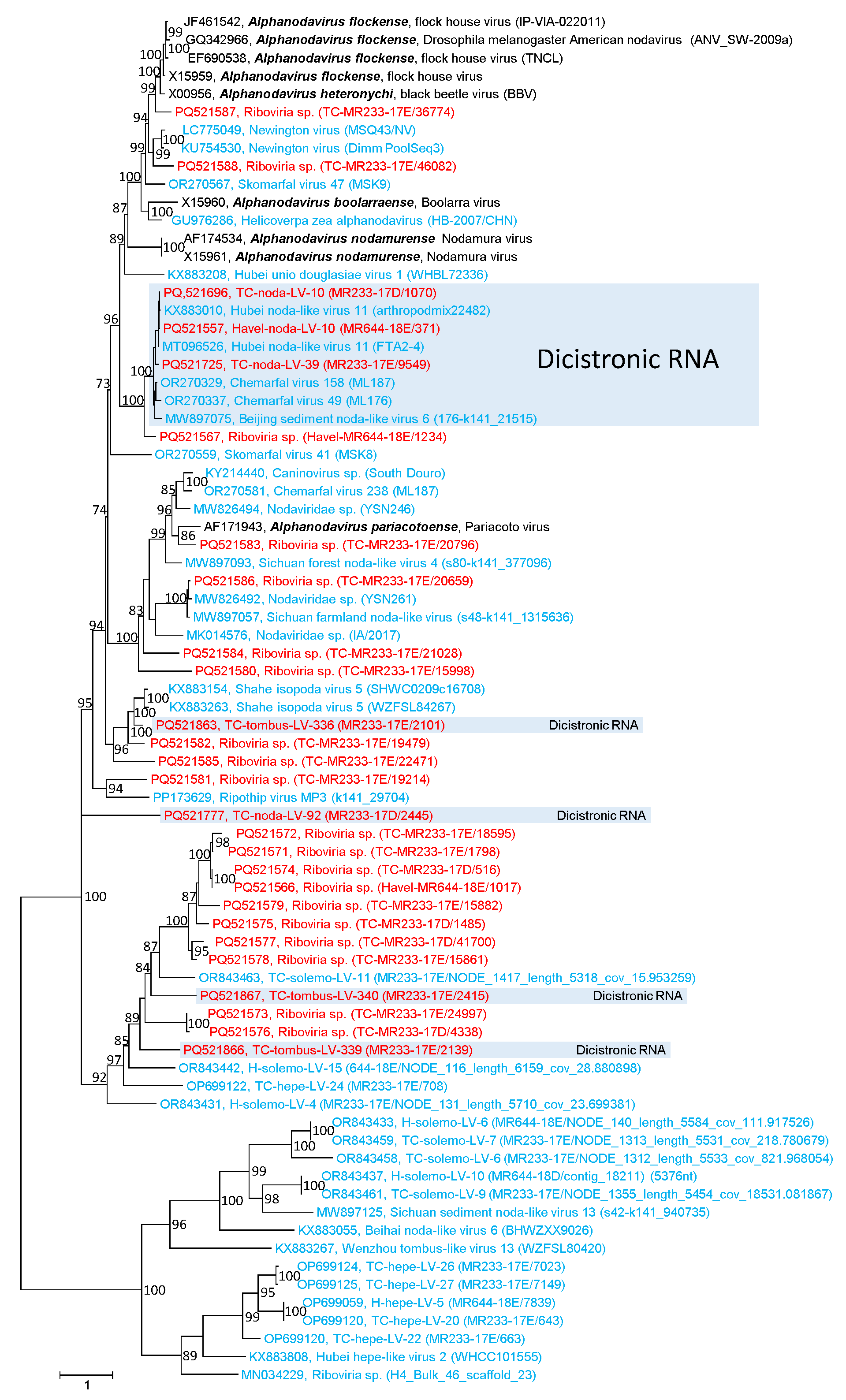
3.1. Noda-like Viruses
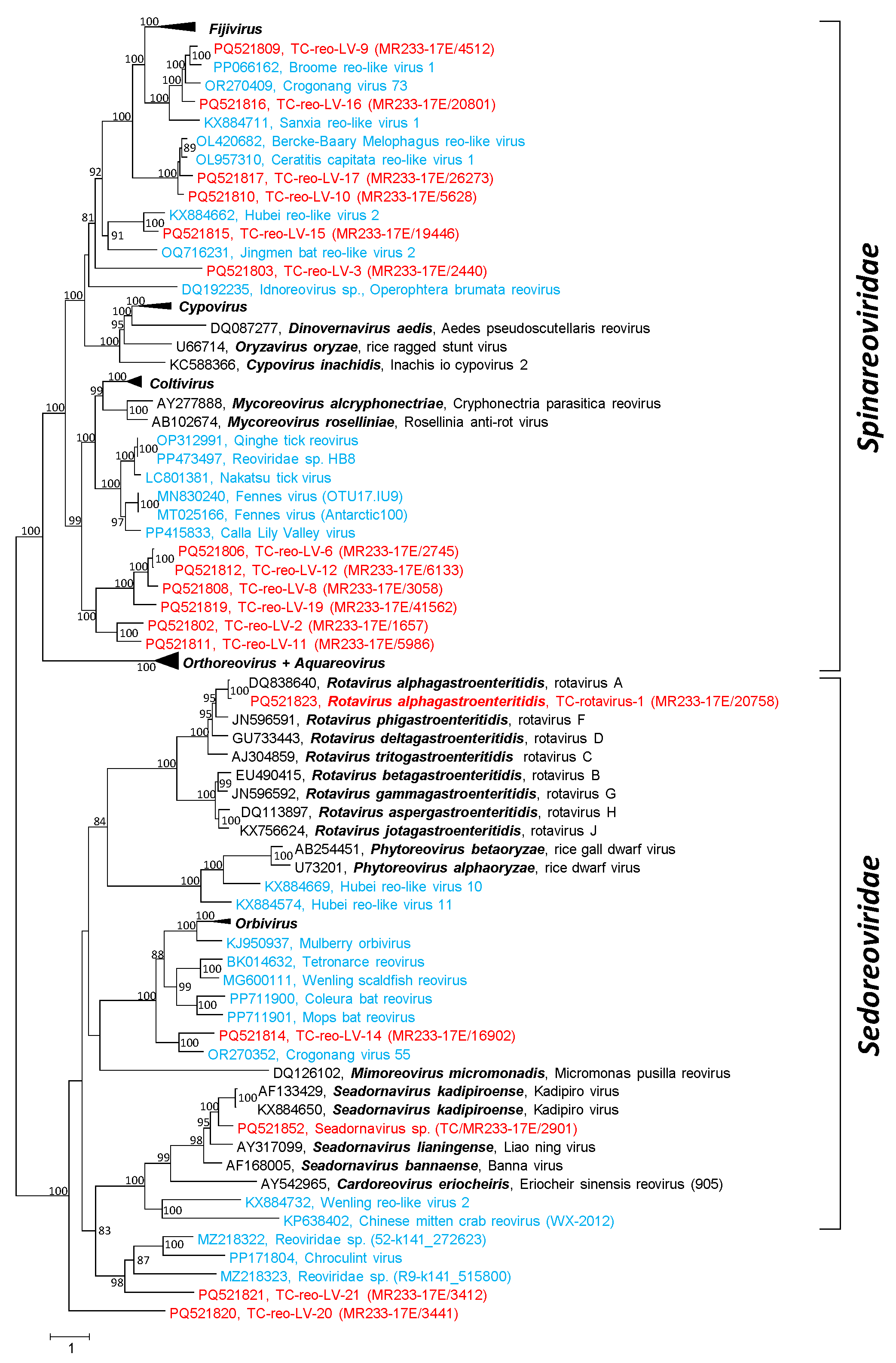
3.2. Reo-like Viruses
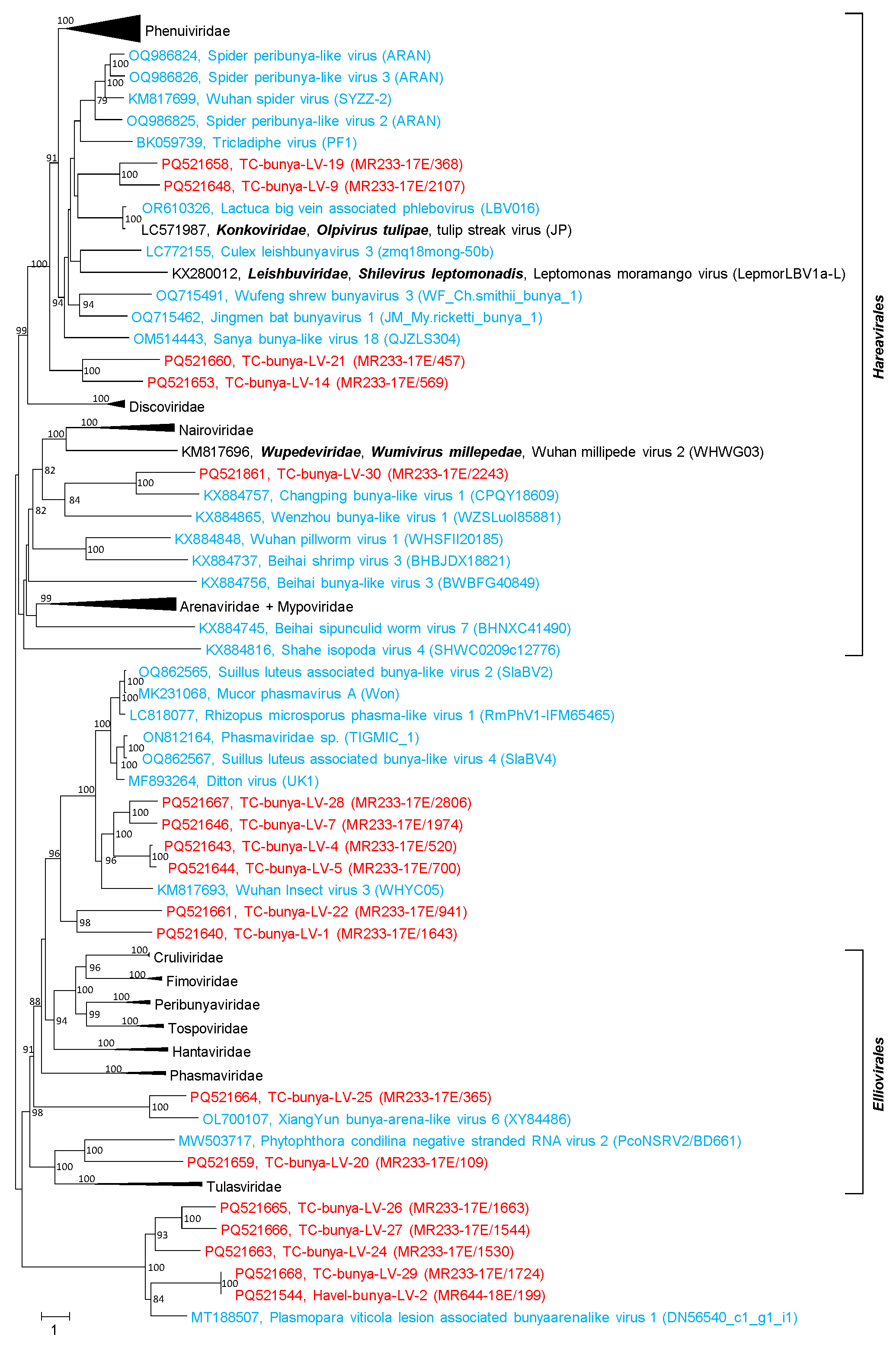
3.3. Bunya-like Viruses
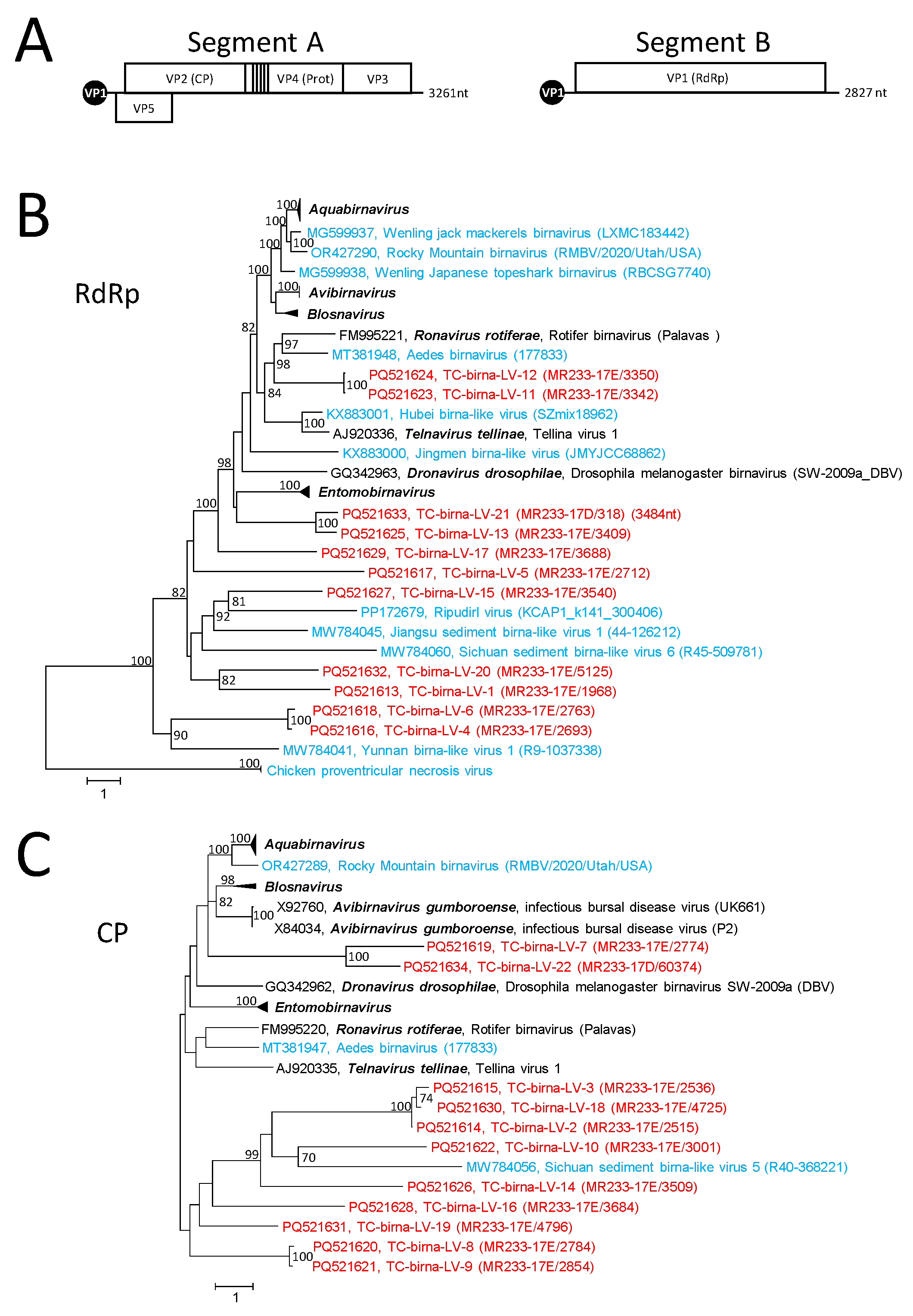
3.4. Birna-like Viruses
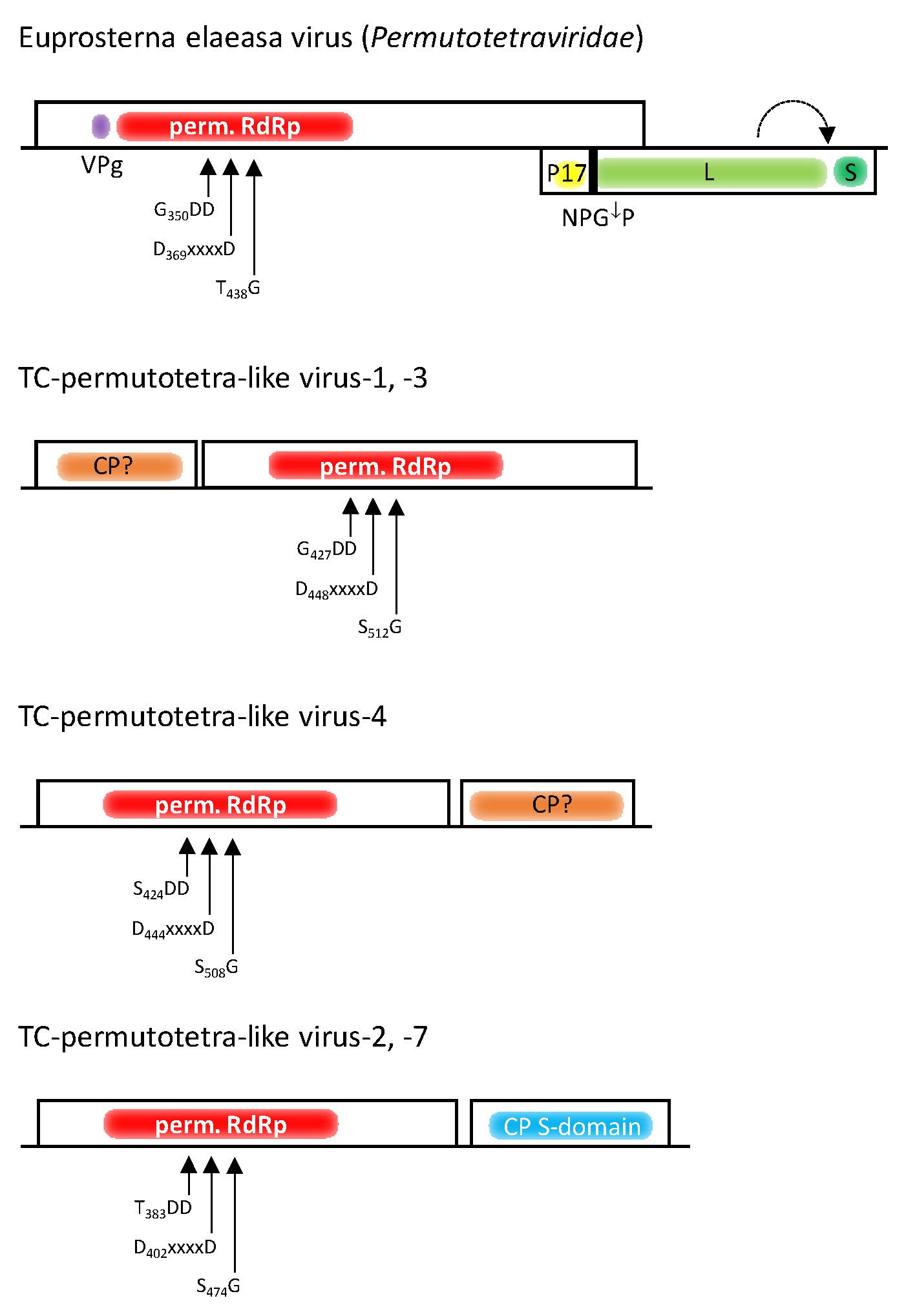
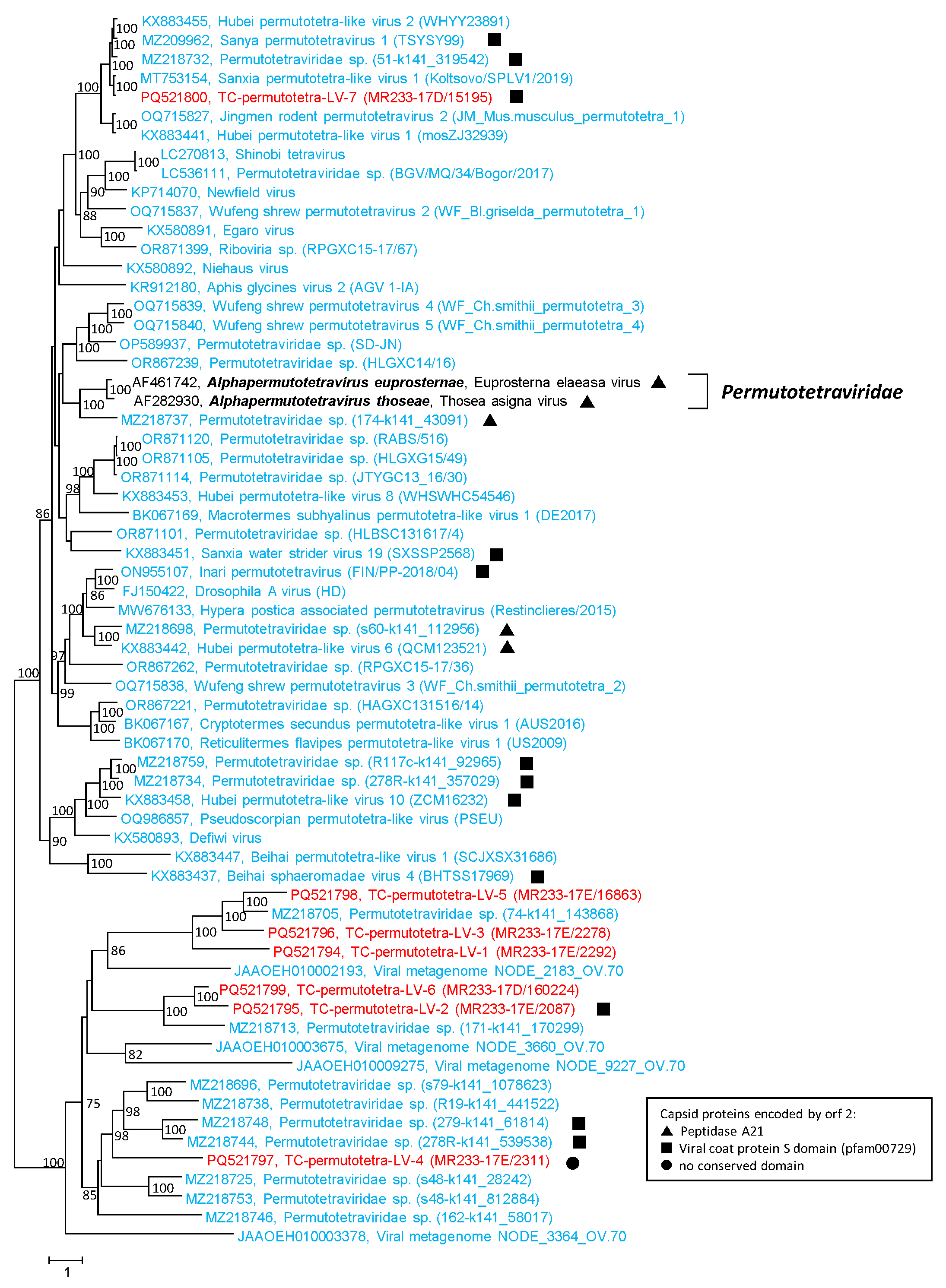
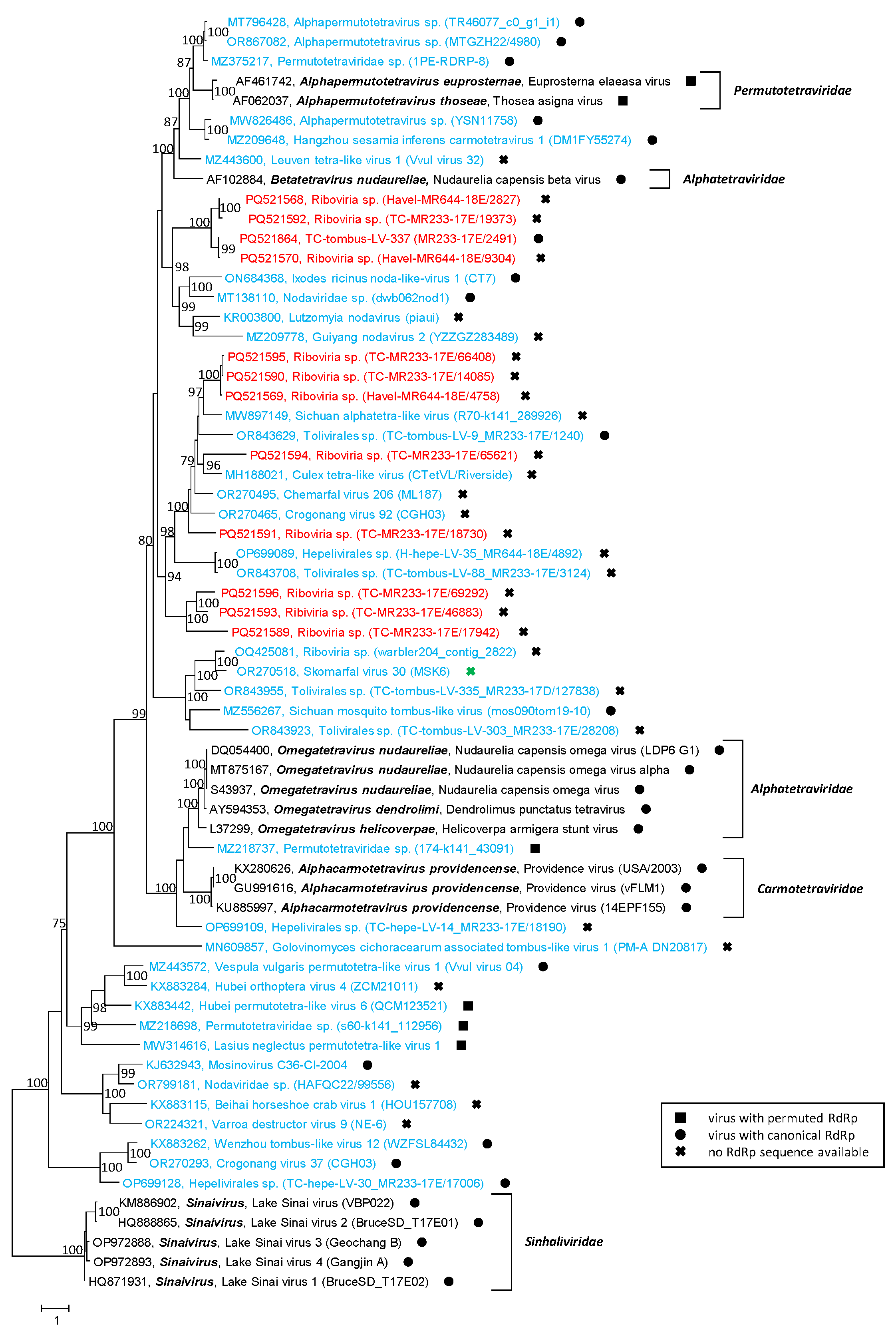
3.5. Permutotetra-like Viruses
3.6. Nido-like Viruses
3.7. Flavivirus Supergroup
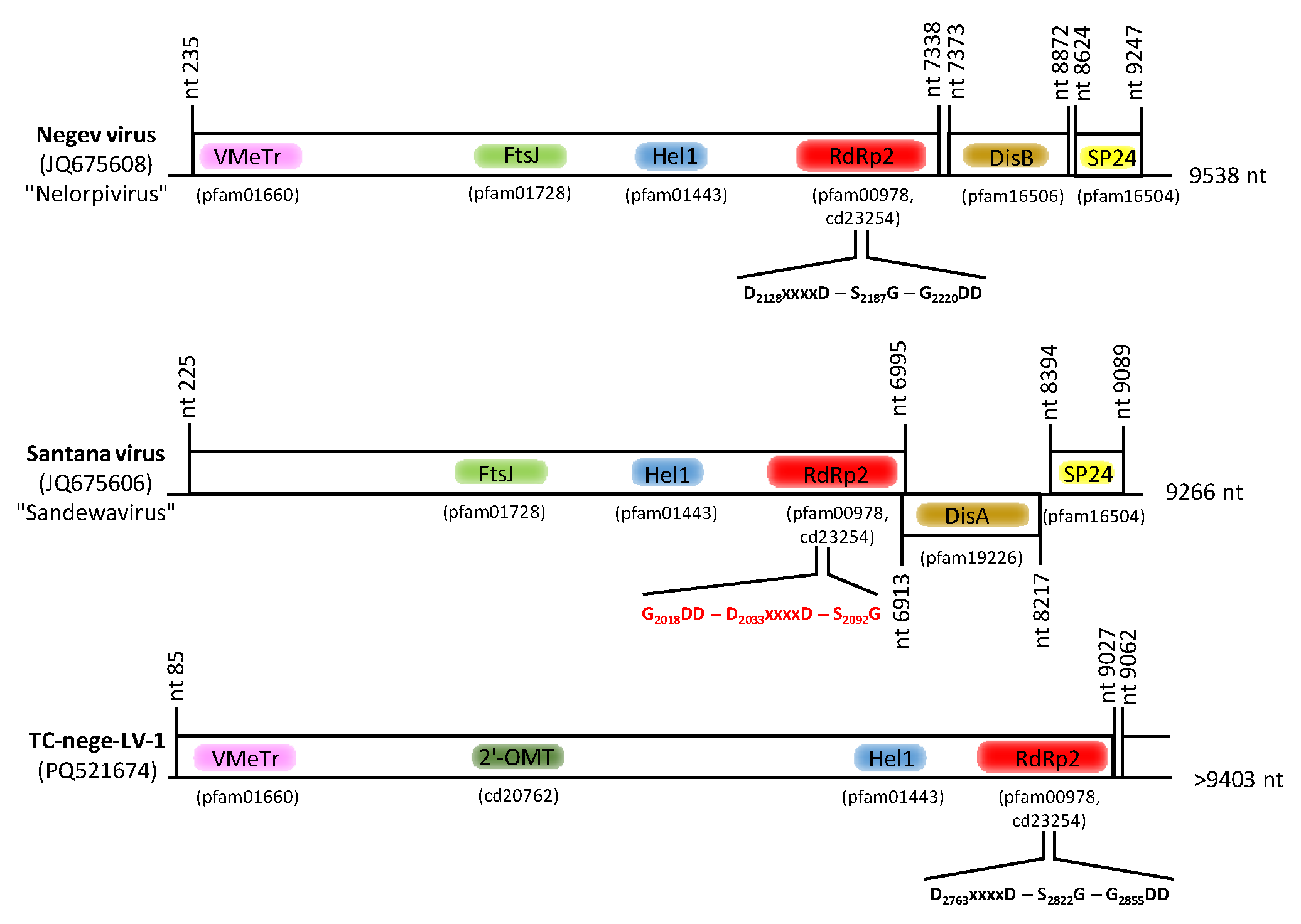
3.8. Nege-like Virus
3.9. Rhabdoviridae
3.10. Chuviridae
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wallace, J.B.; Webster, J.R. The role of macroinvertebrates in stream ecosystem function. Annu. Rev. Entomol. 1996, 41, 115–139. [Google Scholar] [CrossRef] [PubMed]
- Dudgeon, D. The ecology of tropical Asian rivers and streams in relation to biodiversity conservation. Annu. Rev. Ecol. Syst. 2000, 31, 239–263. [Google Scholar] [CrossRef]
- Malmqvist, B. Aquatic invertebrates in riverine landscapes. Freshw. Biol. 2002, 47, 679–694. [Google Scholar] [CrossRef]
- Strayer, D.L. Challenges for freshwater invertebrate conservation. J. N. Am. Benthol. Soc. 2006, 25, 271–287. [Google Scholar] [CrossRef]
- Wommack, K.E.; Colwell, R.R. Virioplankton: Viruses in aquatic ecosystems. Microbiol. Mol. Biol. Rev. 2000, 64, 69–114. [Google Scholar] [CrossRef]
- Parrat, S.R.; Laine, A.L. The role of hyperparasitism in microbial pathogen ecology and evolution. ISME J. 2016, 10, 1815–1822. [Google Scholar] [CrossRef]
- Richard, J.C.; Leis, E.M.; Dunn, C.D.; Harris, C.; Agbalog, R.E.; Campbell, L.J.; Knowles, S.; Waller, D.L.; Putnam, J.G.; Goldberg, T.L. Freshwater mussels show elevated viral richness and intensity during a mortality event. Viruses 2022, 14, 2603. [Google Scholar] [CrossRef]
- Culley, A.I.; Lang, A.S.; Suttle, C.A. High diversity of unknown picorna-like viruses in the sea. Nature 2003, 424, 1054–1057. [Google Scholar] [CrossRef]
- Fuhrmann, J.A. Marine viruses and their biogeochemical and ecological effects. Nature 1999, 399, 541–548. [Google Scholar] [CrossRef]
- Wilhelm, S.W.; Suttle, C.A. Viruses and nutrient cycles in the sea: Viruses play critical roles in the structure and function of aquatic food webs. BioScience 1999, 49, 781–788. [Google Scholar] [CrossRef]
- Maranger, R.; Bird, D.F. Viral abundance in aquatic systems: A comparison between marine and fresh waters. Mar. Ecol. Prog. Ser. 1995, 121, 217–226. [Google Scholar] [CrossRef]
- Lemke, M.J.; Wickstrom, C.E.; Leff, L.G. A preliminary study on the distribution of viruses and bacteria in lotic habitats. Arch. Hydrobiol. 1997, 141, 67–74. [Google Scholar] [CrossRef]
- Pollard, P.C.; Ducklow, H. Ultrahigh bacterial production in a eutrophic subtropical Australian river: Does viral lysis short-circuit the microbial loop? Limnol. Oceanogr. 2011, 56, 1115–1129. [Google Scholar] [CrossRef]
- Shi, M.; Lin, X.D.; Tian, J.H.; Chen, L.J.; Chen, X.; Li, C.X.; Qin, X.C.; Li, J.; Cao, J.P.; Eden, J.S.; et al. Redefining the invertebrate RNA virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Yinda, C.K.; Zell, R.; Deboutte, W.; Zeller, M.; Conceicao-Neto, N.; Heylen, E.; Maes, P.; Knowles, N.J.; Ghogomu, S.M.; Van Ranst, M.; et al. Highly diverse population of Picornaviridae and other members of the Picornavirales, in Cameroonian fruit bats. BMC Genom. 2017, 18, 249. [Google Scholar] [CrossRef]
- Roberts, J.M.K.; Anderson, D.L.; Durr, P.A. Metagenomic analysis of Varroa-fress Australian honey bees (Apis mellifera) shows a diverse Picornavirales genome. J. Gen. Virol. 2018, 99, 818–826. [Google Scholar] [CrossRef]
- Chen, Y.M.; Sadiq, S.; Tian, J.H.; Chen, X.; Lin, X.D.; Shen, J.J.; Chen, H.; Hao, Z.Y.; Wille, M.; Zhou, Z.C.; et al. RNA viromes from terrestrial sites across China expand environmental viral diversity. Nat. Microbiol. 2022, 7, 1312–1323. [Google Scholar] [CrossRef]
- Zhao, M.; Yue, C.; Yang, Z.; Li, Y.; Zhang, D.; Zhang, J.; Yang, S.; Shen, Q.; Su, X.; Qi, D.; et al. Viral metagenomics unveiled extensive communications of viruses within giant pandas and their associated organisms in the same ecosystem. Sci. Total Environm. 2022, 820, 153317. [Google Scholar] [CrossRef]
- Richard, J.C.; Blevins, E.; Dunn, C.D.; Leis, E.M.; Goldberg, T.L. Viruses of freshwater mussels during mass mortality events in Oregon and Washington, USA. Viruses 2023, 15, 1719. [Google Scholar] [CrossRef]
- Lu, X.; Ji, L.; Wang, H.; Zhang, Q.; Wang, X.; Liu, Y.; Shen, Q.; Yang, S.; Ma, X.; Zhang, W.; et al. Highly diverse RNA viruses and phage sequences concealed within birds. Microbiol. Sprectrum 2024, 12, e00802-24. [Google Scholar] [CrossRef]
- Zell, R.; Groth, M.; Selinka, L.; Selinka, H.C. Picorna-like viruses of the Havel River, Germany. Front. Microbiol. 2022, 13, 865287. [Google Scholar] [CrossRef] [PubMed]
- Zell, R.; Groth, M.; Selinka, L.; Selinka, H.C. Diversity of picorna-like viruses in the Teltow Canal, Berlin, Germany. Viruses 2024, 16, 1020. [Google Scholar] [CrossRef] [PubMed]
- Ram, A.S.P.; Palesse, S.; Colombet, J.; Thouvenot, A.; Sime-Ngando, T. The relative importance of viral lysis and nanoflagellate grazing for prokaryote mortality in temperate lakes. Freshw. Biol. 2014, 59, 300–311. [Google Scholar] [CrossRef]
- Bistolas, K.S.I.; Rudstam, L.G.; Hewson, I. Gene expression of benthic amphipods (genus: Diporeia) in relation to a circular ssDNA virus across to Laurentian Great Lakes. PeerJ 2017, 5, e3810. [Google Scholar] [CrossRef] [PubMed]
- Lumsden, J.S.; Morrison, B.; Yason, C.; Russell, S.; Young, K.; Yazdanpanah, A.; Huber, P.; Al-Hussinee, L.; Stone, D.; Way, K. Mortality event in freshwater drum Aplodinotus grunniens from Lake Ontario, Canada, associated with viral haemorrhagic septicemia virus, type IV. Dis. Aquat. Org. 2007, 76, 99–111. [Google Scholar] [CrossRef]
- Bacherach, E.; Mishra, N.; Briese, T.; Zody, M.C.; Tsofack, J.E.K.; Zamostiano, R.; Berkowitz, A.; Ng, J.; Nitido, A.; Corvelo, A.; et al. Characterization of a novel orthomyxo-like virus causing mass die-offs of tilapia. mBio 2016, 7, e00431. [Google Scholar] [CrossRef]
- Miaud, C.; Pozet, F.; Curt Grand Gaudin, N.; Martel, A.; Pasmans, F.; Labrut, S. Ranavirus causes mass die-offs of Alpine amphibians in the southwestern Alps, France. J. Wildlife Dis. 2016, 52, 242–252. [Google Scholar] [CrossRef]
- Thresher, R.E.; Allman, J.; Stremick-Thompsom, L. Impacts of an invasive virus (CyHV-3) on established invasive populations of common carp (Cyprinus carpio) in North America. Biol. Invasions 2018, 20, 1703–1718. [Google Scholar] [CrossRef]
- Grandjean, F.; Gilbert, C.; Razafimafondy, F.; Vucic, M.; Delaunay, C.; Gindre, P.; Bouchard, J.; Raimond, M.; Moumen, B. A new bunya-like virus associated with mass mortality of white-clawed crayfish in the wild. Virology 2019, 533, 115–124. [Google Scholar] [CrossRef]
- Richard, J.C.; Leis, E.; Dunn, C.D.; Agbalog, R.; Waller, D.; Knowles, S.; Putnam, J.; Goldberg, T.L. Mass mortality in freshwater mussels (Actinonaias pectorosa) in the Clinch River, USA, linked to a novel densovirus. Sci. Rep. 2020, 10, 14498. [Google Scholar] [CrossRef]
- Hooper, C.; Debnath, P.P.; Biswas, S.; van Aerle, R.; Bateman, K.S.; Basak, S.K.; Rahman, M.M.; Mohan, C.V.; Islam, H.M.R.; Ross, S.; et al. A novel RNA virus, Macrobrachium rosenbergii Golda virus (MrGV), linked to mass mortalities of the larval giant freshwater prawn in Bangladesh. Viruses 2020, 12, 1120. [Google Scholar] [CrossRef] [PubMed]
- Walker, P.; Winton, J.R. Emerging viral diseases of fish and shrimp. Vet. Res. 2010, 41, 51. [Google Scholar] [CrossRef] [PubMed]
- Johnson, P.T.J.; Paull, S.H. The ecology and emergence of diseases in fresh waters. Freshw. Biol. 2011, 56, 638–657. [Google Scholar] [CrossRef]
- Roux, S.; Adriaenssens, E.M.; Dutilh, B.E.; Koonin, E.V.; Kropinski, A.M.; Krupovic, M.; Kuhn, J.H.; Lavigne, R.; Brister, J.R.; Varsani, A.; et al. Minimum information about an uncultivated virus genome (MIUViG). Nat. Biotechnol. 2019, 37, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Krupovic, M.; Dolja, V.V. The global virome: How much diversity and how many independent origins? Environ. Microbiol. 2023, 25, 40–44. [Google Scholar] [CrossRef]
- Gorbalenya, A.E.; Krupovic, M.; Mushegian, A.; Kropinksi, A.M.; Siddell, S.G.; Varsani, A.; Adams, M.J.; Davison, A.J.; Dutilh, B.E.; Harrach, B.; et al. The new scope of virus taxonomy: Partitioning the virosphere into 15 hierarchical ranks. Nat. Microbiol. 2020, 5, 668–674. [Google Scholar] [CrossRef]
- Simmonds, P.; Adams, M.J.; Benko, M.; Breitbart, M.; Brister, J.R.; Carstens, E.B.; Davison, A.J.; Delwart, E.; Gorbalenya, A.E.; Harrach, B.; et al. Virus taxonomy in the age of metagenomics. Nat. Rev. Microbiol. 2017, 15, 161–168. [Google Scholar] [CrossRef]
- Dutilh, B.E.; Varsani, A.; Tong, Y.; Simmonds, P.; Sabanadzovic, S.; Rubino, L.; Roux, S.; Munoz, A.R.; Lood, C.; Lefkowitz, E.J.; et al. Perspective on taxonomic classification of uncultivated viruses. Curr. Opin. Virol. 2021, 51, 207–215. [Google Scholar] [CrossRef]
- Holmes, E.C.; Krammer, F.; Goodrum, F.D. Virology—The next fifty years. Cell 2024, 187, 5128–5145. [Google Scholar] [CrossRef]
- Zell, R.; Groth, M.; Selinka, L.; Selinka, H.C. Exploring the diversity of plant-associated viruses and related viruses in riverine freshwater samples collected in Berlin, Germany. Pathogens 2023, 12, 1458. [Google Scholar] [CrossRef]
- Zell, R.; Groth, M.; Selinka, L.; Selinka, H.C. Hepeliviruses in two waterbodies in Berlin, Germany. Arch. Virol. 2023, 168, 9. [Google Scholar] [CrossRef] [PubMed]
- Wyn-Jones, A.P.; Carducci, A.; Cook, N.; D’Agostino, M.D.; Divizia, M.; Fleischer, J.; Gantzer, A.; Girones, R.; Höller, C.; de Roda Husman, A.M.; et al. Surveillance of adenoviruses and noroviruses in European recreational waters. Water Res. 2011, 45, 1025–1038. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequence reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Nurk, S.; Melshko, D.; Korobeynikov, A.; Pevzner, P.A. metaSPAdes: A new versatile metagenomic assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Johnson, K.N.; Johnson, K.L.; Dasgupta, R.; Gratsch, T.; Ball, L.A. Comparisons among the larger genome segments of six nodaviruses and their encoded RNA replicases. J. Gen. Virol. 2001, 82, 1855–1866. [Google Scholar] [CrossRef]
- Sahul Hameed, A.S.; Ninawe, A.S.; Nakai, T.; Chi, S.C.; Johnson, K.L.; ICTV Report Consortium. ICTV Virus Taxonomy Profile: Nodaviridae. J. Gen. Virol. 2019, 100, 3–4. [Google Scholar] [CrossRef]
- Matthijnssens, J.; Attoui, H.; Bányai, K.; Brussaard, C.P.D.; Danthi, P.; del Vas, M.; Dermody, T.S.; Duncan, R.; Fang, Q.; Johne, R.; et al. ICTV Virus Taxonomy Profile: Sedoreoviridae 2022. J. Gen. Virol. 2022, 103, 001782. [Google Scholar] [CrossRef] [PubMed]
- Matthijnssens, J.; Attoui, H.; Bányai, K.; Brussaard, C.P.D.; Danthi, P.; del Vas, M.; Dermody, T.S.; Duncan, R.; Fang, Q.; Johne, R.; et al. ICTV Virus Taxonomy Profile: Spinareoviridae 2022. J. Gen. Virol. 2022, 103, 001781. [Google Scholar] [CrossRef] [PubMed]
- Trask, S.D.; McDonald, S.M.; Patton, J.T. Structural insights into the coupling of virion assembly and rotavirus replication. Nat. Rev. Microbiol. 2012, 10, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Schmaljohn, C.S.; Nichol, S. Bunyaviridae. In Fields Virology, 5th ed.; Knipe, D.M., Howley, P., Eds.; Lippincott, Williams and Wilkins: Philadelphia, PA, USA, 2007; pp. 1741–1789. [Google Scholar]
- Delmas, B.; Attoui, H.; Ghosh, S.; Malik, Y.S.; Mundt, E.; Vakharia, V.N.; ICTV Consortium. ICTV virus taxonomy profile: Birnaviridae. J. Gen. Virol. 2019, 100, 5–6. [Google Scholar] [CrossRef]
- Gorbalenya, A.E.; Pringle, F.M.; Zeddam, J.L.; Luke, B.T.; Cameron, C.E.; Kalmakoff, J.; Hanzlik, T.N.; Gordon, K.H.J.; Ward, V.K. The palm subdomain-based active site is internally permuted in viral RNA-dependent RNA polymerases of an ancient lineags. J. Mol. Biol. 2002, 324, 47–62. [Google Scholar] [CrossRef]
- Pan, J.; Vakharia, V.N.; Tao, Y.J. The structure of a birnavirus polymerase reveals a distinct active site topology. Proc. Natl. Acad. Sci. USA 2007, 104, 7385–7390. [Google Scholar] [CrossRef]
- Zeddam, J.L.; Gordon, K.H.J.; Lauber, C.; Felipe Alves, C.A.; Luke, B.R.; Hanzlik, T.N.; Ward, V.K.; Gorbalenya, A.E. Euprosterna elaeasa virus genome sequence and evolution of the Tetraviridae family: Emergence of bipartite genomes and conservation of the VPg signal with the dsRNA Birnaviridae family. Virology 2010, 397, 145–154. [Google Scholar] [CrossRef]
- Gorbalenya, A.E.; Enjuanes, L.; Ziebuhr, J.; Snijder, E.J. Nidovirales: Evolving the largest RNA virus genome. Virus Res. 2006, 117, 17–37. [Google Scholar] [CrossRef]
- Simmonds, P.; Becher, P.; Bukh, J.; Gould, E.A.; Meyers, G.; Monath, T.; Muerhoff, S.; Pletnev, A.; Rico-Hesse, R.; Smith, D.B.; et al. ICTV Report Consortium. ICTV Virus Taxonomy Profile: Flaviviridae. J. Gen. Virol. 2017, 98, 2–3. [Google Scholar] [CrossRef]
- Kuhn, R.J.; Zhang, W.; Rossmann, M.G.; Pletnev, S.V.; Corver, J.; Lenches, E.; Jones, C.T.; Mukhopadhyay, S.; Chipman, P.R.; Strauss, E.G.; et al. Structure of dengue virus: Implications of flavivirus organization, maturation, and fusion. Cell 2002, 108, 717–725. [Google Scholar] [CrossRef]
- Shi, M.; Lin, X.D.; Vasilakis, N.; Tian, J.H.; Li, C.X.; Chen, L.J.; Eastwood, G.; Diao, X.N.; Chen, M.H.; Chen, X.; et al. Divergent viruses discovered in arthropods and vertebrates revise the evolutionary history of the Flaviviridae and related viruses. J. Virol. 2015, 90, 659–669. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.C.; Shi, M.; Tian, J.H.; Lin, X.D.; Gao, D.Y.; He, J.R.; Wang, J.B.; Li, C.X.; Kang, Y.J.; Yu, B.; et al. A tick-borne segmented RNA virus contains genome segments derived from unsegmented viral ancestors. Proc. Natl. Acad. Sci. USA 2014, 111, 6744–6749. [Google Scholar] [CrossRef] [PubMed]
- Urayama, S.; Takaki, Y.; Nunoura, T. FLDS: A comprehensive dsRNA sequencing method for intracellular RNA virus surveillance. Microbes Environ. 2016, 31, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Vasilakis, N.; Forrester, N.L.; Palacios, G.; Nasar, F.; Savji, N.; Rossi, S.L.; Guzman, H.; Wood, T.G.; Popov, V.; Gorchakov, R.; et al. Negevirus: A proposed new taxon of insect-specific viruses with wide geographic distribution. J. Virol. 2013, 87, 2475–2488. [Google Scholar] [CrossRef] [PubMed]
- Kallies, R.; Kopp, A.; Zirkel, F.; Estrada, A.; Gillespie, T.R.; Drosten, C.; Junglen, S. Genetic characterization of Goutanap virus, a novel virus related to negeviruses, cileviruses and higreviruses. Viruses 2014, 6, 4346–4357. [Google Scholar] [CrossRef] [PubMed]
- Dietzgen, R.G.; Kondo, H.; Goodin, M.M.; Kurath, G.; Vasilakis, N. The family Rhabdoviridae: Mono- and bipartite negative-sense RNA viruses with diverse genome organization and common evolutionary origins. Virus Res. 2017, 227, 158–170. [Google Scholar] [CrossRef]
- Walker, P.; Dietzgen, R.G.; Joubert, D.A.; Blasdell, K.R. Rhabdovirus accessory genes. Virus Res. 2011, 162, 110–125. [Google Scholar] [CrossRef]
- Walker, P.; Freitas-Astúa, J.; Bejerman, N.; Blasdell, K.R.; Breyta, R.; Dietzgen, R.G.; Fooks, A.R.; Kondo, H.; Kurath, G.; Kuzmin, I.V.; et al. ICTV Virus Taxonomy Profile: Rhabdoviridae 2022. J. Gen. Virol. 2022, 103, 001689. [Google Scholar] [CrossRef]
- Li, C.X.; Shi, M.; Tian, J.H.; Lin, X.D.; Kang, Y.J.; Chen, L.J.; Qin, X.C.; Xu, J.; Holmes, E.C.; Zhang, Y.Z. Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses. eLife 2015, 4, e05378. [Google Scholar] [CrossRef]
- Di Paola, N.; Dheilly, N.M.; Junglen, S.; Paraskevopoulou, S.; Postler, T.S.; Shi, M.; Kuhn, J.H. Jingchuvirales: A new taxonomical framework for a rapidly expanding order of unusual monjiviricete viruses broadly distributed among arthropod subphyla. Appl. Environ. Microbiol. 2021, 88, e0195421. [Google Scholar] [CrossRef]
- Shwed, P.S.; Dobos, P.; Cameron, L.A.; Vakharia, V.N.; Duncan, R. Birnavirus VP1 proteins form a distinct subgroup of RNA-dependent RNA polymerases lacking a GDD motif. Virology 2002, 296, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Wolf, Y.I.; Silas, S.; Wang, Y.; Wu, S.; Bocek, M.; Kazlauskas, D.; Krupovic, M.; Fire, A.; Dolja, V.V.; Koonin, E.V. Doubling of the known set of RNA viruses by metagenomic analysis of an aquatic virome. Nat. Microbiol. 2020, 5, 1262–1270. [Google Scholar] [CrossRef] [PubMed]
- Wille, M.; Holmes, E.C. Wild birds as reservoirs for disease and abundant gamma- and deltacoronaviruses. FEMS Microbiol. Rev. 2020, 44, 631–644. [Google Scholar] [CrossRef] [PubMed]
- Vlasova, A.N.; Kenney, S.P.; Jung, K.; Wang, Q.; Saif, L.J. Deltacoronavirus evolution and transmission: Current scenario and evolutionary perspectives. Front. Vet. Sci. 2021, 7, 626785. [Google Scholar] [CrossRef]
- Saberi, A.; Gulyaeva, A.A.; Brubacher, J.L.; Newmark, P.A.; Gorbalenya, A.E. A planarian nidovirus expands the limits of RNA genome size. PLoS Pathog. 2018, 14, e1007314. [Google Scholar] [CrossRef]
- Koonin, E.V.; Dolja, V.V.; Krupovic, M. Origins and evolution of viruses of eukaryotes: The ultimate modularity. Virology 2015, 479−480, 2–25. [Google Scholar] [CrossRef]
- Dolja, V.V.; Koonin, E.V. Metagenomics reshapes the concepts of RNA virus evolution by revealing extensive horizontal virus transfer. Virus Res. 2018, 244, 36–52. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Capsid Protein | RdRp Superfamily | Examples |
|---|---|---|
| Viral coat protein S-domain (pfam00729) | RdRp1 (pfam00729) | Plasmopara halstedii virus A, Sclerophthora macrospora virus A, Beijing sediment noda-like virus1, Beihai noda-like virus 5, Ripothoz virus TC-noda-LV–11, –16 |
| RdRp3 (pfam00998) | Tombusviridae (except Luteovirus), Tombunodavirus | |
| RdRp4 (pfam02123) | Sobemovirus, Polemovirus | |
| permuted RdRp | Inari permutotetravirus, Beihai sphaeromadae virus 4, TC-permutotetra-LV–2, –7 | |
| Nodavirus capsid protein VNN (pfam11729) | RdRp1 (pfam00680) | Betanodavirus, Orsay nodavirus, Le Blanc nodavirus, Santeuil nodavirus |
| RdRp3 (pfam00998) | Wufeng shrew carmotetravirus 1 | |
| Luteovirus coat protein(pfam00894) | RdRp1 (pfam00729) | Craigies Hill virus |
| RdRp3 (pfam00998) | Luteovirus | |
| RdRp4 (pfam02123) | Enamovirus, Polerovirus | |
| Peptidase A6 (pfam01829) | RdRp1 (pfam00680) | Alphanodavirus, Ripothip virus, TC-noda-LV–92 |
| RdRp2 (pfam00978) | H-hepe-LV–5, TC-hepe-LV–20 | |
| RdRp3 (pfam00998) | TC-tombus-LV–336, –339, –340, H-tombus-LV–4, –6, –15, Shahe isopoda virus 5 | |
| RdRp4 (pfam02123) | H-solemo-LV–4, –6, –10, –15, TC-solemo-LV–11 | |
| permuted RdRp | TC-hepe-LV–22, –24 | |
| Peptidase A21 (pfam03566) | RdRp1 (pfam00680) | Sinhaliviridae, Lutzomyia nodavirus |
| RdRp2 (pfam00978) | Alphatetraviridae, Hubei hepe-like virus 2, TC-hepe-LV–30 | |
| RdRp3 (pfam00998) | Carmotetraviridae, TC-tombus-LV–87, –337, Sichuan mosquito tombus-like virus | |
| permuted RdRp | Permutotetraviridae, Permutotetraviridae sp. 174-k141_43091, Hubei permutotetra-like virus 6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zell, R.; Groth, M.; Selinka, L.; Selinka, H.-C. Metagenomic Analyses of Water Samples of Two Urban Freshwaters in Berlin, Germany, Reveal New Highly Diverse Invertebrate Viruses. Microorganisms 2024, 12, 2361. https://doi.org/10.3390/microorganisms12112361
Zell R, Groth M, Selinka L, Selinka H-C. Metagenomic Analyses of Water Samples of Two Urban Freshwaters in Berlin, Germany, Reveal New Highly Diverse Invertebrate Viruses. Microorganisms. 2024; 12(11):2361. https://doi.org/10.3390/microorganisms12112361
Chicago/Turabian StyleZell, Roland, Marco Groth, Lukas Selinka, and Hans-Christoph Selinka. 2024. "Metagenomic Analyses of Water Samples of Two Urban Freshwaters in Berlin, Germany, Reveal New Highly Diverse Invertebrate Viruses" Microorganisms 12, no. 11: 2361. https://doi.org/10.3390/microorganisms12112361
APA StyleZell, R., Groth, M., Selinka, L., & Selinka, H.-C. (2024). Metagenomic Analyses of Water Samples of Two Urban Freshwaters in Berlin, Germany, Reveal New Highly Diverse Invertebrate Viruses. Microorganisms, 12(11), 2361. https://doi.org/10.3390/microorganisms12112361