
Structural Characterization of Mycobacterium tuberculosis Encapsulin in Complex with Dye-Decolorizing Peroxide

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
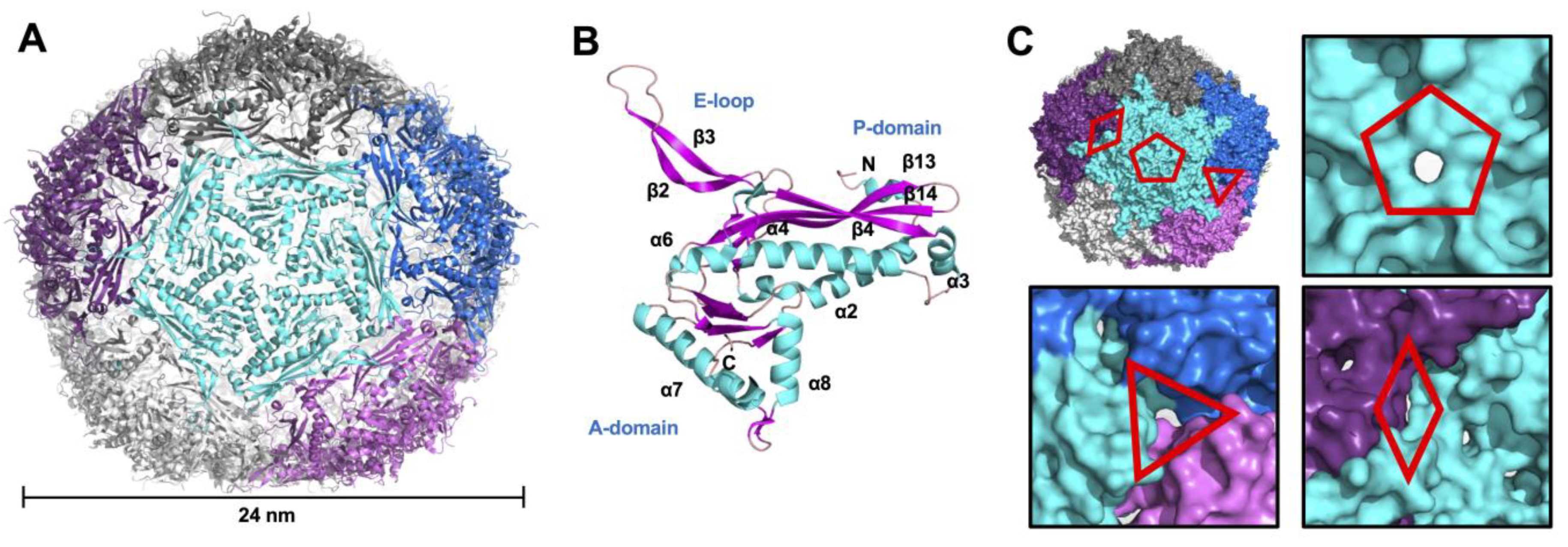
3.1. Crystallographic Structure of Mtb-Enc in Complex with DyP
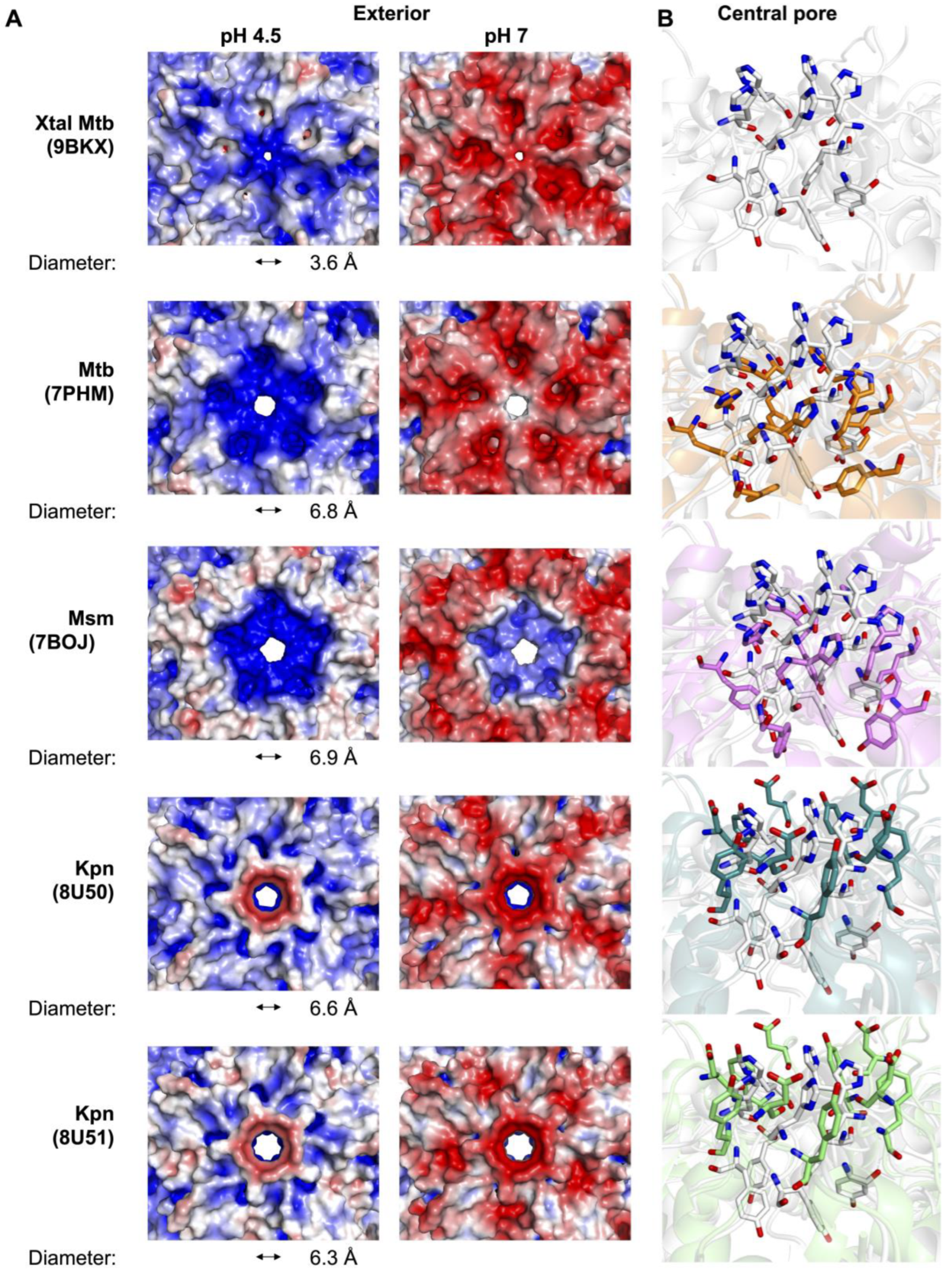
3.2. Variability of the Five-Fold Pore Across Species
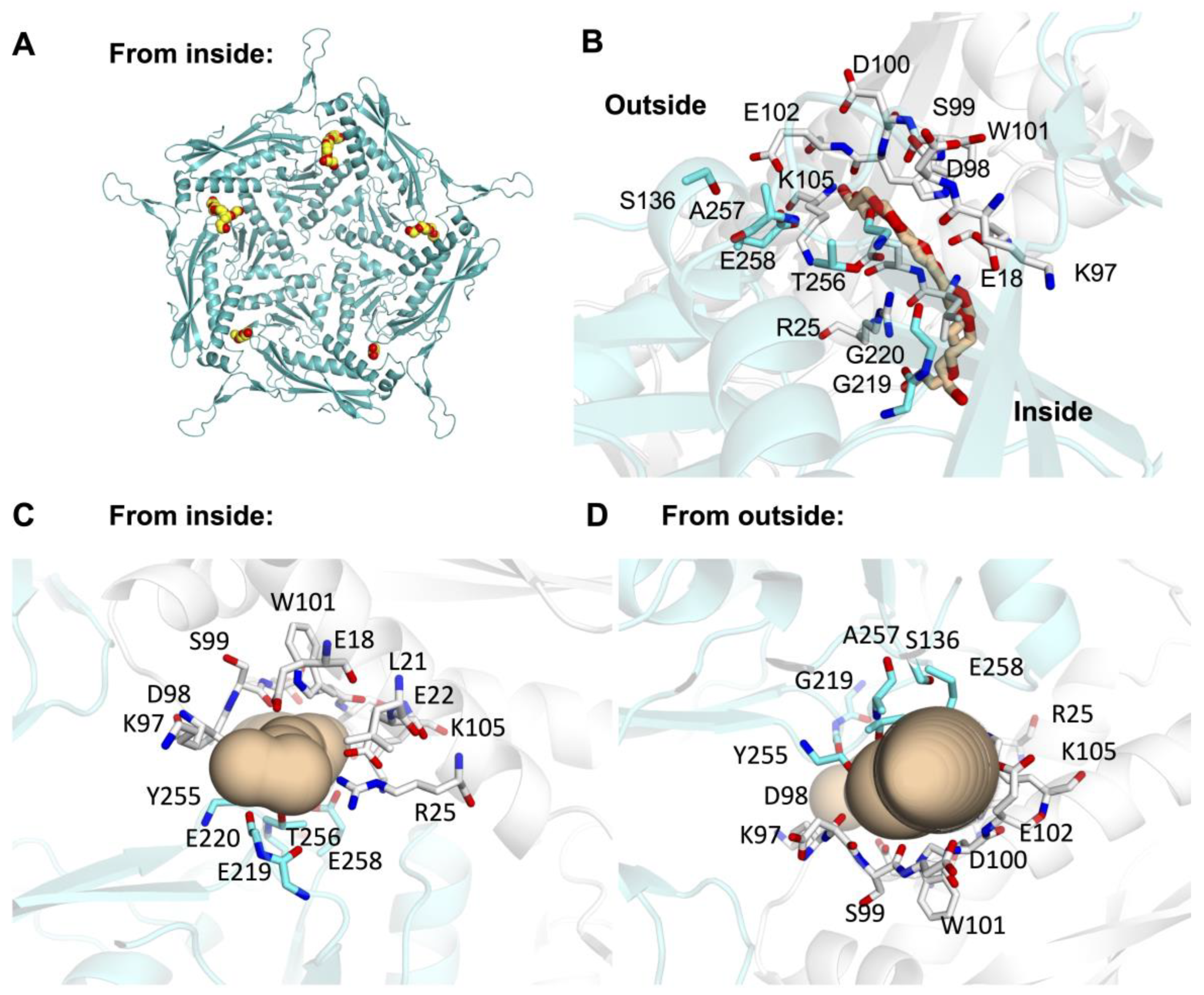
3.3. Minor Pores Surrounding the Five-Fold Central Pore May Allow Small Molecules to Enter Enc
3.4. Biologically Relevant Electrostatic Surfaces Show Differences at the Five-Fold Major Pore Across Species
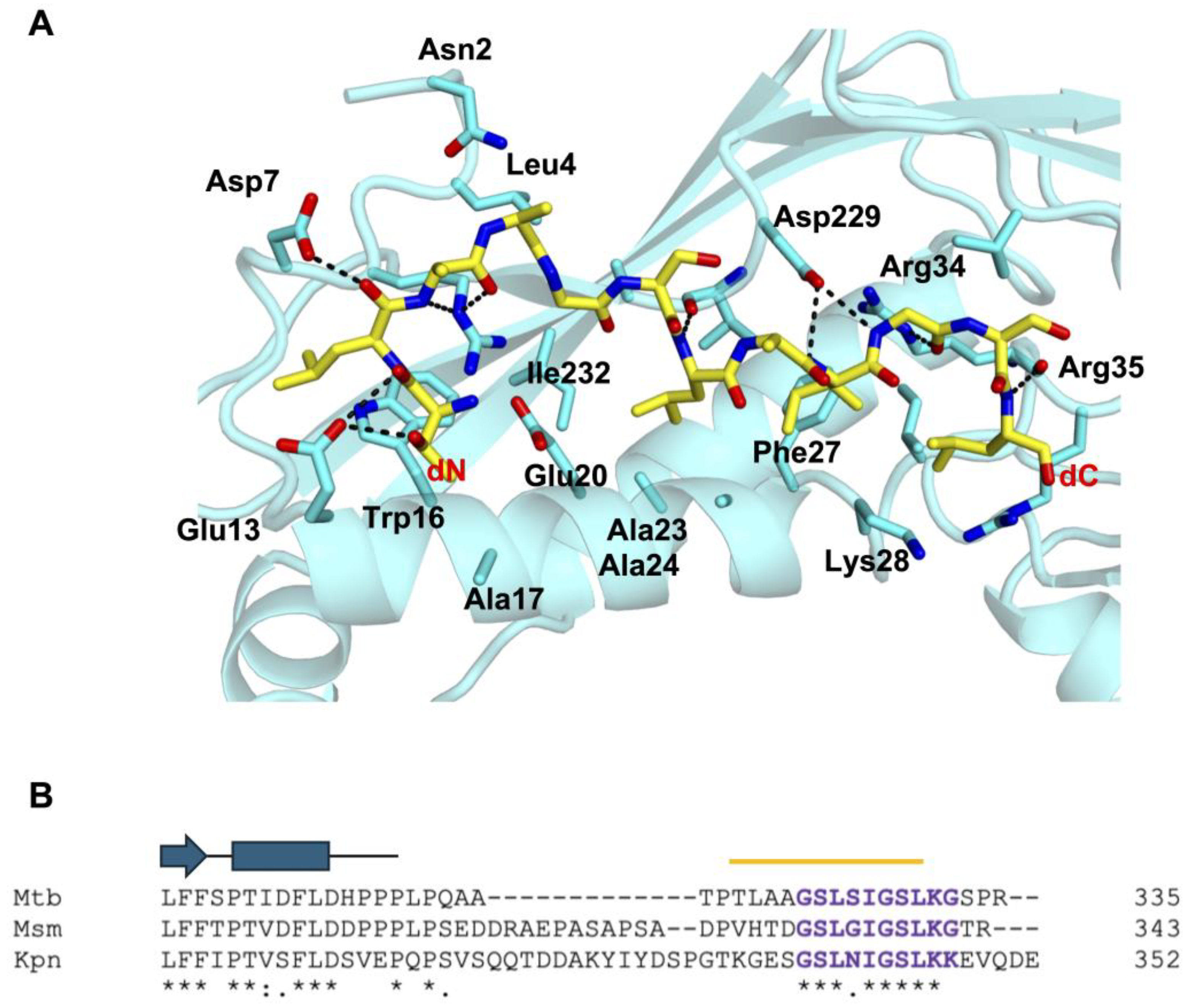
3.5. Recognition of Mtb-DyP-TP by Mtb-Enc
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- The World Health Organization. Global Tuberculosis Report 2023. Available online: https://www.who.int/teams/global-tuberculosis-programme/tb-reports/global-tuberculosis-report-2023 (accessed on 7 November 2023).
- Rojano, B.; Caminero, J.A.; Hayek, M. Curving tuberculosis: Current trends and future needs. Ann. Glob. Health 2019, 85, 5. [Google Scholar] [CrossRef] [PubMed]
- Gill, C.M.; Dolan, L.; Piggott, L.M.; McLaughlin, A.M. New developments in tuberculosis diagnosis and treatment. Breathe 2022, 18, 210149. [Google Scholar] [CrossRef]
- Sharma, K.; Ahmed, F.; Sharma, T.; Grover, A.; Agarwal, M.; Grover, S. Potential Repurposed Drug Candidates for Tuberculosis Treatment: Progress and Update of Drugs Identified in over a Decade. ACS Omega 2023, 8, 17362–17380. [Google Scholar] [CrossRef]
- Contreras, H.; Joens, M.S.; McMath, L.M.; Le, V.P.; Tullius, M.V.; Kimmey, J.M.; Bionghi, N.; Horwitz, M.A.; Fitzpatrick, J.A.J.; Goulding, C.W. Characterization of a Mycobacterium tuberculosis nanocompartment and its potential cargo proteins. J. Biol. Chem. 2014, 289, 18279–18289. [Google Scholar] [CrossRef] [PubMed]
- Lien, K.A.; Dinshaw, K.; Nichols, R.J.; Cassidy-Amstutz, C.; Knight, M.; Singh, R.; Eltis, L.D.; Savage, D.F.; Stanley, S.A. A nanocompartment system contributes to defense against oxidative stress in mycobacterium tuberculosis. eLife 2021, 10, e74358. [Google Scholar] [CrossRef]
- Eren, E.; Wang, B.; Winkler, D.C.; Watts, N.R.; Steven, A.C.; Wingfield, P.T. Structural characterization of the Myxococcus xanthus encapsulin and ferritin-like cargo system gives insight into its iron storage mechanism. Structure 2022, 30, 551–563.e4. [Google Scholar] [CrossRef]
- Giessen, T.W.; Orlando, B.J.; Verdegaal, A.A.; Chambers, M.G.; Gardener, J.; Bell, D.C.; Birrane, G.; Liao, M.; Silver, P.A. Large protein organelles form a new iron sequestration system with high storage capacity. eLife 2019, 8, e46070. [Google Scholar] [CrossRef]
- LaFrance, B.J.; Cassidy-Amstutz, C.; Nichols, R.J.; Oltrogge, L.M.; Nogales, E.; Savage, D.F. The encapsulin from Thermotoga maritima is a flavoprotein with a symmetry matched ferritin-like cargo protein. Sci. Rep. 2021, 11, 22810. [Google Scholar] [CrossRef]
- McHugh, C.A.; Fontana, J.; Nemecek, D.; Cheng, N.; Aksyuk, A.A.; Heymann, J.B.; Winkler, D.C.; Lam, A.S.; Wall, J.S.; Steven, A.C.; et al. A virus capsid-like nanocompartment that stores iron and protects bacteria from oxidative stress. EMBO J. 2014, 33, 1896–1911. [Google Scholar] [CrossRef]
- Ross, J.; McIver, Z.; Lambert, T.; Piergentili, C.; Bird, J.E.; Gallagher, K.J.; Cruickshank, F.L.; James, P.; Zarazúa-Arvizu, E.; Horsfall, L.E.; et al. Pore dynamics and asymmetric cargo loading in an encapsulin nanocompartment. Sci. Adv. 2022, 8, eabj4461. [Google Scholar] [CrossRef]
- Wiryaman, T.; Toor, N. Cryo-EM structure of a thermostable bacterial nanocompartment. IUCrJ 2021, 8, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Giessen, T.W.; Silver, P.A. Widespread distribution of encapsulin nanocompartments reveals functional diversity. Nat. Microbiol. 2017, 2, 17029. [Google Scholar] [CrossRef] [PubMed]
- Andreas, M.P.; Giessen, T.W. Large-scale computational discovery and analysis of virus-derived microbial nanocompartments. Nat. Commun. 2021, 12, 4748. [Google Scholar] [CrossRef] [PubMed]
- Benisch, R.; Andreas, M.P.; Giessen, T.W. A widespread bacterial protein compartment sequesters and stores elemental sulfur. Sci. Adv. 2024, 10, eadk9345. [Google Scholar] [CrossRef] [PubMed]
- Nichols, R.J.; Lafrance, B.; Phillips, N.R.; Radford, D.R.; Oltrogge, L.M.; Valentin-Alvarado, L.E.; Bischoff, A.J.; Nogales, E.; Savage, D.F. Discovery and characterization of a novel family of prokaryotic nanocompartments involved in sulfur metabolism. eLife 2021, 10, e59288. [Google Scholar] [CrossRef]
- Jones, J.A.; Andreas, M.P.; Giessen, T.W. Structural basis for peroxidase encapsulation inside the encapsulin from the Gram-negative pathogen Klebsiella pneumoniae. Nat. Commun. 2024, 15, 2558. [Google Scholar] [CrossRef]
- Tang, Y.; Mu, A.; Zhang, Y.; Zhou, S.; Wang, W.; Lai, Y.; Zhou, X.; Liu, F.; Yang, X.; Gong, H.; et al. Cryo-EM structure of Mycobacterium smegmatis DyP-loaded encapsulin. Proc. Natl. Acad. Sci. USA 2021, 118, e2025658118. [Google Scholar] [CrossRef]
- Sutter, M.; Boehringer, D.; Gutmann, S.; Günther, S.; Prangishvili, D.; Loessner, M.J.; Stetter, K.O.; Weber-Ban, E.; Ban, N. Structural basis of enzyme encapsulation into a bacterial nanocompartment. Nat. Struct. Mol. Biol. 2008, 15, 939–947. [Google Scholar] [CrossRef]
- Adamson, L.S.R.; Tasneem, N.; Andreas, M.P.; Close, W.; Jenner, E.N.; Szyszka, T.N.; Young, R.; Cheah, L.C.; Norman, A.; MacDermott-Opeskin, H.I.; et al. Pore structure controls stability and molecular flux in engineered protein cages. Sci. Adv. 2022, 8, eabl7346. [Google Scholar] [CrossRef]
- Lončar, N.; Rozeboom, H.J.; Franken, L.E.; Stuart, M.C.A.; Fraaije, M.W. Structure of a robust bacterial protein cage and its application as a versatile biocatalytic platform through enzyme encapsulation. Biochem. Biophys. Res. Commun. 2020, 529, 548–553. [Google Scholar] [CrossRef] [PubMed]
- Putri, R.M.; Allende-Ballestero, C.; Luque, D.; Klem, R.; Rousou, K.A.; Liu, A.; Traulsen, C.H.H.; Rurup, W.F.; Koay, M.S.T.; Castón, J.R.; et al. Structural Characterization of Native and Modified Encapsulins as Nanoplatforms for in Vitro Catalysis and Cellular Uptake. ACS Nano 2017, 11, 12796–12804. [Google Scholar] [CrossRef]
- Xiong, X.; Sun, C.; Vago, F.S.; Klose, T.; Zhu, J.; Jiang, W. Cryo-EM structure of heterologous protein complex loaded thermotoga maritima encapsulin capsid. Biomolecules 2020, 10, 1342. [Google Scholar] [CrossRef] [PubMed]
- Akita, F.; Chong, K.T.; Tanaka, H.; Yamashita, E.; Miyazaki, N.; Nakaishi, Y.; Suzuki, M.; Namba, K.; Ono, Y.; Tsukihara, T.; et al. The Crystal Structure of a Virus-like Particle from the Hyperthermophilic Archaeon Pyrococcus furiosus Provides Insight into the Evolution of Viruses. J. Mol. Biol. 2007, 368, 1469–1483. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.; Andreas, M.P.; Giessen, T.W. Structure and heterogeneity of a highly cargo-loaded encapsulin shell. J. Struct. Biol. 2023, 215, 108022. [Google Scholar] [CrossRef]
- Jones, J.A.; Andreas, M.P.; Giessen, T.W. Exploring the Extreme Acid Tolerance of a Dynamic Protein Nanocage. Biomacromolecules 2023, 24, 1388–1399. [Google Scholar] [CrossRef]
- Jones, J.A.; Benisch, R.; Giessen, T.W. Encapsulin cargo loading: Progress and potential. J. Mater. Chem. B 2023, 11, 4377–4388. [Google Scholar] [CrossRef]
- Tracey, J.C.; Coronado, M.; Giessen, T.W.; Lau, M.C.Y.; Silver, P.A.; Ward, B.B. The Discovery of Twenty-Eight New Encapsulin Sequences, Including Three in Anammox Bacteria. Sci. Rep. 2019, 9, 20122. [Google Scholar] [CrossRef]
- Giessen, T.W. Encapsulins: Microbial nanocompartments with applications in biomedicine, nanobiotechnology and materials science. Curr. Opin. Chem. Biol. 2016, 34, 1–10. [Google Scholar] [CrossRef]
- Quinton, A.R.; McDowell, H.B.; Hoiczyk, E. Chapter One—Encapsulins: Nanotechnology’s future in a shell. In Advances in Applied Microbiology; Gadd, G.M., Sariaslani, S., Eds.; Academic Press: Cambridge, MA, USA, 2023; Volume 125, pp. 1–48. [Google Scholar]
- Battye, T.G.G.; Kontogiannis, L.; Johnson, O.; Powell, H.R.; Leslie, A.G.W. iMOSFLM: A new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 271–281. [Google Scholar] [CrossRef] [PubMed]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef]
- Terwilliger, T.C.; Grosse-Kunstleve, R.W.; Afonine, P.V.; Moriarty, N.W.; Zwart, P.H.; Hung, L.W.; Read, R.J.; Adams, P.D. Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr. Sect. D Biol. Crystallogr. 2007, 64, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [PubMed]
- Afonine, P.V.; Grosse-Kunstleve, R.W.; Echols, N.; Headd, J.J.; Moriarty, N.W.; Mustyakimov, M.; Terwilliger, T.C.; Urzhumtsev, A.; Zwart, P.H.; Adams, P.D. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. Sect. D Biol. Crystallogr. 2012, 68, 352–367. [Google Scholar] [CrossRef] [PubMed]
- Liebschner, D.; Afonine, P.V.; Moriarty, N.W.; Poon, B.K.; Sobolev, O.V.; Terwilliger, T.C.; Adams, P.D. Polder maps: Improving OMIT maps by excluding bulk solvent. Acta Crystallogr. Sect. D Struct. Biol. 2017, 73, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Holm, L.; Rosenstrom, P. Dali server: Conservation mapping in 3D. Nucleic Acids Res. 2010, 38, W545–W549. [Google Scholar] [CrossRef]
- Rahmanpour, R.; Bugg, T.D.H. Assembly in vitro of Rhodococcus jostii RHA1 encapsulin and peroxidase DypB to form a nanocompartment. FEBS J. 2013, 280, 2097–2104. [Google Scholar] [CrossRef] [PubMed]
- The PyMOL Molecular Graphics System, version 2.5; Schrödinger, LLC: New York, NY, USA, 2021.
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Jurrus, E.; Engel, D.; Star, K.; Monson, K.; Brandi, J.; Felberg, L.E.; Brookes, D.H.; Wilson, L.; Chen, J.; Liles, K.; et al. Improvements to the APBS biomolecular solvation software suite. Protein Sci. 2018, 27, 112–128. [Google Scholar] [CrossRef]
- Unni, S.; Huang, Y.; Hanson, R.M.; Tobias, M.; Krishnan, S.; Li, W.W.; Nielsen, J.E.; Baker, N.A. Web Servers and Services for Electrostatics Calculations with APBS and PDB2PQR SAMIR. J. Comput. Chem. 2011, 32, 1488–1491. [Google Scholar] [CrossRef]
- Jurcik, A.; Bednar, D.; Byska, J.; Marques, S.M.; Furmanova, K.; Daniel, L.; Kokkonen, P.; Brezovsky, J.; Strnad, O.; Stourac, J.; et al. CAVER Analyst 2.0: Analysis and visualization of channels and tunnels in protein structures and molecular dynamics trajectories. Bioinformatics 2018, 34, 3586–3588. [Google Scholar] [CrossRef]
- Pravda, L.; Sehnal, D.; Toušek, D.; Navrátilová, V.; Bazgier, V.; Berka, K.; Vařeková, R.S.; Koča, J.; Otyepka, M. MOLEonline: A web-based tool for analyzing channels, tunnels and pores (2018 update). Nucleic Acids Res. 2018, 46, W368–W373. [Google Scholar] [CrossRef]
- Bienert, G.P.; Schjoerring, J.K.; Jahn, T.P. Membrane transport of hydrogen peroxide. Biochim. Biophys. Acta Biomembr. 2006, 1758, 994–1003. [Google Scholar] [CrossRef]
- Cid, H.; Bunster, M.; Canales, M.; Gazitúa, F. Hydrophobicity and structural classes in proteins. Protein Eng. Des. Sel. 1992, 5, 373–375. [Google Scholar] [CrossRef]
- Zimmerman, J.M.; Eliezer, N.; Simha, R. The characterization of amino acid sequences in proteins by statistical methods. J. Theor. Biol. 1968, 21, 170–201. [Google Scholar] [CrossRef]
- Catucci, G.; Valetti, F.; Sadeghi, S.J.; Gilardi, G. Biochemical features of dye-decolorizing peroxidases: Current impact on lignin degradation. Biotechnol. Appl. Biochem. 2020, 67, 751–759. [Google Scholar] [CrossRef]
- Sugano, Y.; Muramatsu, R.; Ichiyanagi, A.; Sato, T.; Shoda, M. DyP, a unique dye-decolorizing peroxidase, represents a novel heme peroxidase family: ASP171 replaces the distal histidine of classical peroxidases. J. Biol. Chem. 2007, 282, 36652–36658. [Google Scholar] [CrossRef] [PubMed]
- Sugano, Y.; Yoshida, T. Dyp-type peroxidases: Recent advances and perspectives. Int. J. Mol. Sci. 2021, 22, 5556. [Google Scholar] [CrossRef] [PubMed]
- Madeira, F.; Madhusoodanan, N.; Lee, J.; Eusebi, A.; Niewielska, A.; Tivey, A.R.N.; Lopez, R.; Butcher, S. The EMBL-EBI Job Dispatcher sequence analysis tools framework in 2024. Nucleic Acids Res. 2024, 52, W521–W525. [Google Scholar] [CrossRef] [PubMed]
- Kirykowicz, A.M.; Woodward, J.D. Shotgun EM of mycobacterial protein complexes during stationary phase stress. Curr. Res. Struct. Biol. 2020, 2, 204–212. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Wavelength of collection | 1 Å |
| Data collection | SSRL 7-1 |
| Space group | P 21 3 |
| Cell dimensions | |
| a, b, c (Å) | 313.5, 313.5, 313.5 |
| α, β, γ (°) | 90, 90, 90 |
| Resolution (Å) | 54.56–3.15 (3.20–3.15) 1 |
| Rmerge 2 | 0.284 (0.887) |
| I/σI | 7.4 (1.4) |
| Completeness (%) | 99.4 (99.8) |
| Redundancy | 4.5 (4.5) |
| Refinement | |
| Resolution (Å) | 49.0–3.15 (3.26–3.15) |
| Total reflections | 790,230 (38,895) |
| Unique reflections | 174,711 (8661) |
| Rwork/Rfree 3 | 20.2/23.3 (28.2/32.8) |
| Ramachandran favored (%) | 96.8 |
| Ramachandran outliers (%) | 0.2 |
| Enc subunits in ASU | 20 |
| DyP peptides in ASU | 9 |
| No. atoms | 42,837 |
| Protein | 41,266 |
| Ligands | 1179 |
| Water | 392 |
| B-factors (Å2) | |
| Protein | 41.9 |
| Ligands | 61.3 |
| Water | 26.3 |
| Root mean square deviations | |
| Bond lengths (Å) | 0.003 |
| Bond angles (°) | 0.6 |
| PDB ID | 9BKX |
| Structure | PDB ID | Bottleneck Diameter (Å) | Hydropathy [46] | Polarity [47] |
|---|---|---|---|---|
| Mtb-Enc with DyP | 9BKX | 3.4 ± 0.8 | −1.8 ± 0.4 | 26.6 ± 4.1 |
| Mtb-Enc with DyP | 8PYS | 2.8 ± 0.3 | −2.3 ± 0.3 | 30.5 ± 2.9 |
| Mtb-Enc | 7PHM | 3.4 ± 0.6 | −2.2 ± 0.3 | 29.2 ± 3.9 |
| Mtb-Enc | 8IKA | 3.5 ± 0.7 | −0.6 ± 1.0 | 15.7 ± 11.8 |
| Msm-Enc with DyP | 7BOJ | 2.2 ± 0 | −0.7 ± 0.0 | 14.5 ± 0.0 |
| Kpn-Enc | 8U50 | 4.0 ± 0.8 | −2.5 ± 0.9 | 20.0 ± 5.7 |
| Kpn-Enc with SUMO-DyP-TP | 8U51 | 4.0 ± 1.4 | −2.3 ± 1.2 | 14.8 ± 1.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cuthbert, B.J.; Chen, X.; Burley, K.; Batot, G.; Contreras, H.; Dixon, S.; Goulding, C.W. Structural Characterization of Mycobacterium tuberculosis Encapsulin in Complex with Dye-Decolorizing Peroxide. Microorganisms 2024, 12, 2465. https://doi.org/10.3390/microorganisms12122465
Cuthbert BJ, Chen X, Burley K, Batot G, Contreras H, Dixon S, Goulding CW. Structural Characterization of Mycobacterium tuberculosis Encapsulin in Complex with Dye-Decolorizing Peroxide. Microorganisms. 2024; 12(12):2465. https://doi.org/10.3390/microorganisms12122465
Chicago/Turabian StyleCuthbert, Bonnie J., Xiaorui Chen, Kalistyn Burley, Gaëlle Batot, Heidi Contreras, Shandee Dixon, and Celia W. Goulding. 2024. "Structural Characterization of Mycobacterium tuberculosis Encapsulin in Complex with Dye-Decolorizing Peroxide" Microorganisms 12, no. 12: 2465. https://doi.org/10.3390/microorganisms12122465
APA StyleCuthbert, B. J., Chen, X., Burley, K., Batot, G., Contreras, H., Dixon, S., & Goulding, C. W. (2024). Structural Characterization of Mycobacterium tuberculosis Encapsulin in Complex with Dye-Decolorizing Peroxide. Microorganisms, 12(12), 2465. https://doi.org/10.3390/microorganisms12122465