Abstract
Sulfidogenic bacteria cause numerous issues in the oil industry since they produce sulfide, corroding steel equipment, reducing oil quality, and worsening the environmental conditions in oil fields. The purpose of this work was to isolate and taxonomically identify the sulfidogenic bacteria responsible for the corrosion of steel equipment at the Karazhanbas oil field (Kazakhstan). In this study, we characterized five sulfidogenic strains of the genera Pseudodesulfovibrio, Oleidesulfovibrio, and Acetobacterium isolated from the formation water of the Karazhanbas oil field (Kazakhstan). Sulfate-reducing strain 9FUST revealed 98.9% similarity of the 16S rRNA gene sequence with the closely related strain ‘Pseudodesulfovibrio methanolicus’ 5S69T and was studied in detail to enhance the taxonomic resolution. Strain 9FUST grew optimally at 23–28 °C, pH 6.5, and 0–2% (w/v) NaCl. The strain used lactate, pyruvate, methanol, ethanol, fructose, ribose, and H2/CO2 (in the presence of acetate) as carbon and energy sources for sulfate reduction. Iso-C17:1 ω11, C15:0, iso-C15:0, and C16:0 were the predominant fatty acids. The genome is 4.20 Mbp with a G + C content of 64.0%. The average nucleotide identity and digital DNA–DNA hybridization values with Pseudodesulfovibrio spp. genomes were 72.5–91.6% (<95%) and 18.5–45.0% (<70%), respectively, and supported our conclusion that 9FUST (=VKM B-3654T = KCTC 25498T) belonged to a novel Pseudodesulfovibrio species, for which the name Pseudodesulfovibrio karagichevae sp. nov. is proposed. Pangenome analysis of sixteen Pseudodesulfovibrio species and functional annotation analysis of identified genes revealed complete modules of enzymes of the main metabolic pathways, characteristic of bacteria of this genus, and unique genes highlighting the adaptations of strain 9FUST in carbohydrate metabolism, nutrient uptake, and environmental stress response. Isolation of these strains expands our understanding of the diversity of sulfidogens in oil reservoirs and can be used to test the effectiveness of biocides used in an oil field.
1. Introduction
Corrosion of steel equipment is an important problem in the water supply system, as well as in the oil and gas industry [1,2]. For example, in 2015, global losses due to corrosion amounted to about 2.5 trillion dollars [3]. The main agents of microbially (microbiologically) influenced corrosion (MIC) of steel equipment are sulfate-reducing prokaryotes (SRP): bacteria (SRB) and archaea (SRA), which reduce sulfate of the reservoir water to form sulfide [4,5,6]. In petroleum reservoirs, the substrates for SRP are organic acids and alcohols dissolved in reservoir water, petroleum hydrocarbons, as well as molecular hydrogen/CO2 [7,8,9]. In the absence of sulfates, SRPs are able to ferment organic substrates or grow syntrophically using methanogens as biological electron acceptors. Hydrogen sulfide is a toxic gas; it dissolves in reservoir water and oil and causes environmental problems during oil extraction, transportation, and refining. The mechanisms of microbially influenced corrosion are discussed in the review [6]. High rates of sulfate reduction in oil fields with sulfate-containing formation water, as well as in those flooded with seawater, were registered by radiotracer techniques [10,11]. Along with SRP, corrosion is also caused by microorganisms that reduce other oxidized sulfur compounds (thiosulfate-, sulfite-, and sulfur-reducing), iron-reducing, iron-oxidizing, and organic acid-producing fermentative bacteria and methanogenic archaea [12,13,14]. Hydrogenotrophic homoacetogens and methanogens consume cathodic hydrogen from Fe with the formation of acetate and methane, respectively, and contribute to the corrosion process.
This study is part of the work aimed at identifying the potential producers of hydrogen sulfide and corrosion agents at the Karazhanbas oil field (Kazakhstan) using molecular and microbiological approaches [15]. The oil-bearing horizons of the Karazhanbas field are located at shallow depths (250–500 m) and vary in temperature from 25 to 45 °C. A feature of the Karazhanbas field is the presence of high-viscosity sulfurous oil. Although sulfate was not registered in formation and injection water at the time of sampling, earlier it was detected at low concentrations (10–69 mg·L−1) in formation water, and corrosion of oil-processing equipment and sulfide release into the produced fluids were observed at the oilfield [16].
In a previous study [15], using a molecular approach based on metabarcoding of V3–V4 regions of the 16S rRNA genes, methanogenic archaea of the genera Methanococcus, Methanobacterium, and Methanothrix; syntrophic bacteria of the genus Smithella; as well as bacteria of the genera Arcobacter, Acetobacterium, Marinobacter, Thiomicrospira, and Thermovirga were detected in formation water. The share of sulfate-reducing bacteria in water samples was low (<1% total). Mesophilic bacteria of the genera Pseudodesulfovibrio, Desulfovibrio, Desulfosarcina, Desulfoglaeba, and Desulfotignum, and thermophilic bacteria of the genera Thermodesulfobacterium, Thermodesulfovibrio, and Defluviitoga were detected by the molecular approach in formation water and sulfidogenic enrichments. Prokaryotes revealed in formation water samples by this approach were potentially able to carry out the key processes (sulfate reduction, methane formation, and degradation of hydrocarbons).
The purpose of this work was the isolation and identification of pure cultures of sulfidogenic bacteria from the formation water of the Karazhanbas oil field (Kazakhstan) and the characterization of new isolates, as well as sequencing and analysis of the strain 9FUST genome to clarify its potential functional role in the subsurface environment. In this study, using different electron donors and acceptors, five strains of sulfate-reducing bacteria and an acetogenic strain were isolated and identified. The results of phenotypic studies, analysis of the genome, and unique genes allowed us to assign the sulfate-reducing strain 9FUST to the genus Pseudodesulfovibrio and to provide a description of a new species, Pseudodesulfovibrio karagichevae sp. nov. The isolated strains can be used to select effective biocides for injection into the oil reservoir
2. Materials and Methods
2.1. Source of Enrichment and Pure Cultures
Sulfidogenic bacteria were isolated from formation water collected in June 2019 at the Karazhanbas oil field (Buzachi Peninsula, Mangystau Province, Kazakhstan). The water sample was obtained from the production well 6069 (43°22′33.3″ N, 52°59′27.1″ E), exploiting the Lower Cretaceous layer G, located at 350 m below sea level and having a temperature of 25 °C. Formation water belonged to the calcium-chloride type and had a total salinity (of 32,959.2 mg·L−1) and pH 6.6; while sulfate was not registered in this water sample at the moment of investigation, sulfide was detected at a concentration of 62.9 mg·L−1 (Table S1). The G horizon is composed mainly of loose sands and weakly cemented sandstones with interlayers of red-brown clays; reservoir porosity is 35%, and permeability is 125 mD. The oil of this deposit has a density of 939–944 kg·m−3 and high viscosity (160–660 mPa·s at 50 °C). It contains high levels of sulfur (1.6–2.2 mass %), resins (24 mass %), asphaltenes (24.9–29.1 mass %), and paraffin (0.7–1.4 mass %). There is a significant content of trace elements in oil, in particular, vanadium pentoxide −308.6 g·t−1. The average gas content is 8.9–9.8 m3 per 1 t oil [16]. Well, 6069 is located in an area flooded with production water re-injected into the reservoir (PWRI) after oil separation. Injection water contained both sulfate (14 mg·L−1) and sulfide (25 mg·L−1). The physicochemical and microbiological characteristics of the oil field were described previously [15].
2.2. Isolation, Media Composition and Phenotypic Characterization
Five strains of sulfate-reducing bacteria (9FUST (=VKM B-3654T = KCTC 25,498T), 9ES, 09, DNS2, 09S) and one strain of sulfur-reducing bacteria (9FOS) were isolated from the Karazhanbas oil field. For enrichment, isolation, and cultivation of sulfate-reducing bacteria, the following basal mineral medium was used (per liter distilled water): 0.2 g KH2PO4, 0.25 g NH4Cl, 2.0 g NaCl, 0.4 g MgCl2·6H2O, 0.5 g KCl, 0.1 g CaCl2·2H2O, and 0.5 g Na2S·9H2O. The medium was amended with 0.2 g of yeast extract, 1 mL of 0.1% (w/v) Mohr’s salt solution (FeSO4·(NH4)2SO4·6H2O), and 1 mL each of vitamin [17] and microelements solutions [18]. The medium was reduced with cysteine-HCl; the pH of the medium was adjusted to pH 6.5 with the addition of HCl (10%) or NaOH (10%) at 25 °C.
The medium was prepared anaerobically under a stream of Ar, dispensed into Hungate tubes [19], sealed with butyl rubber stoppers and plastic caps, and autoclaved at 121 °C for 60 min. Sulfate-reducing enrichments were obtained in a basal medium supplemented with 3.5 g sodium lactate and 2 g sodium sulfate per liter. Pure cultures were isolated by the method of successive transfers of grown cultures from the highest growth-positive dilutions (10−7–10−8) on a fresh medium of the same composition. The cultures were incubated at a temperature of 23–25 °C. Replacement of lactate with other substrates [ethanol (2 mL·L−1), formate (2 g·L−1), fumarate (2 g·L−1), H2/CO2 (4:1, v/v)] and sulfate with elemental sulfur (2.5 g·L−1) allowed us to isolate a number of other sulfidogenic bacteria. The purity of the cultures was checked by phase-contrast microscopy of wet biomass and by 16S rRNA sequencing of the total liquid culture from the tube. Growth was monitored by optical density (OD600) measurements and by sulfide production, which was determined using the colorimetric method [20].
Strain 9FUST was studied simultaneously with the type strain 5S69T of the new species ‘Pseudodesulfovibrio methanolicus’, which was used for comparison [21]. Cell morphology, physiology, DNA isolation, 16S rRNA gene sequencing, composition of cellular fatty acids, phospholipids, and isoprenoid quinones were analyzed as described previously [21].
2.3. Genome Sequencing and Annotation
The genomic DNA of strain 9FUST was isolated using the DNeasy PowerSoils Kit (Qiagen, Düsseldorf, Germany). The library for Illumina sequencing was constructed using the NEBNext Ultra II DNA Library Prep Kit (New England Biolabs, Ipswich, MA, USA). Illumina MiSeq run output was 3,031,658 paired-end reads (2 × 300 nt, ~874 Mbp in total). FLASH v.1.2.11 [22] was used to merge paired-end reads into longer sequences, and low-quality read fragments were removed with Sickle v.1.33 (https://github.com/najoshi/sickle, accessed on 28 January 2023) [23]. The processed reads were assembled into contigs by SPAdes v. 3.15.4 [24] in isolate mode. The NCBI Prokaryotic Genome Annotation Pipeline (PGAP), executed as a part of the submission process to the NCBI GenBank database, produced genome annotation. All software was run using default settings.
2.4. Genomic and Phylogenetic Analyses
Pairwise average nucleotide identity (ANI) and digital DNA-DNA hybridization (dDDH) values were calculated using FastANI v1.3 [25] and the Genome-to-Genome Distance Calculator v3.0 [26], respectively. Phylogenomic analysis of Pseudodesulfovibrio genomes utilized a concatenated alignment of 120 single-copy phylogenetic marker genes obtained via GTDB-Tk v1.4.0 [27]. A maximum-likelihood (MLH) tree of the 16S rRNA gene sequences was constructed using a GTR + F + I + G4 model, while the phylogenomic tree was built using an LG + F + I + G4 model as recommended by ModelFinder [28] in IQ-TREE [29]. Branching supports were evaluated with 10,000 ultrafast bootstraps [30]. Maximum parsimony (MP) and neighbor-joining (NJ) trees were reconstructed using MPBoot [31] and MEGA11 [32], respectively. Pangenomic analysis was conducted using the bioinformatics pipeline suggested previously [33] and Anvi’o v8.0 [34]. Genomes were organized based on the distribution of gene clusters using the MCL algorithm (Euclidean distance, Ward linkage). Orthologous Groups (COG) of proteins were predicted and classified using COGclassifier v1.0.5 (https://github.com/moshi4/COGclassifier, accessed on 7 September 2022). Potential metabolic pathways were reconstructed by comparing Pseudodesulfovibrio genomes using the BlastKOALA tool [35] of KEGG v3.0 [36].
2.5. Nucleotide Sequence Accession Numbers
The GenBank accession numbers for the 16S rRNA gene sequences of six strains are PQ453521–PQ453525 and PQ474646. The genome sequence of strain 9FUST has been deposited at GenBank under the accession number GCF_041414475.1.
3. Results and Discussion
This article describes six strains of anaerobic sulfidogenic bacteria isolated from the Karazhanbas oil field (Kazakhstan). The strains were identified by 16S rRNA gene analysis. To clarify the taxonomic position and the potential functional role in the underground habitat, the morphology, physiology, and chemotaxonomic features of the strain 9FUST were studied in detail, and its genome was sequenced and annotated, which made it possible to describe the new species Pseudodesulfovibrio karagichevae sp. nov.
3.1. Isolation and Identification of Pure Cultures from the Oil Field
Using media with different electron donors and acceptors, six pure cultures of mesophilic obligately anaerobic sulfidogenic bacteria were isolated from reservoir water samples taken at the Karazhanbas oil field. To isolate the strains, the mineral medium base was used, to which formate, acetate, ethanol, fumarate, or H2/CO2 were added as electron and carbon donors; sulfate, elemental sulfur, and fumarate served as electron acceptors.
The isolated strains (9FUST, 9ES, 09, DNS2, 09S, and 9FOS) reduced sulfate and/or elemental sulfur with sulfide formation. In addition to the substrates listed in Table 1, all five sulfate-reducing strains (9FUST, 9ES, 09, DNS2, and 09S) grew on a medium with H2/CO2 supplemented with acetate, forming sulfide, and strain 9FOS was a homoacetogen, growing on a mixture of H2/CO2 in the absence of an electron acceptor, forming acetic acid.

Table 1.
Taxonomic affiliation of sulfidogenic bacterial strains isolated from the Karazhanbas oil field.
A high level of the 16S rRNA gene sequence similarity (99–100%) of four strains with the GeneBank sequences allowed the strain DNS2 to be classified as Oleidesulfovibrio alaskensis (previously Desulfovibrio alaskensis [37,38] and the strain 9FOS—to the species Acetobacterium carbinolicum [39,40]. The 16S rRNA gene sequences of strains 9FUST, 9ES, 09S, and 09 had 100% similarity to each other and 98.4–98.7% similarity to the gene Pseudodesulfovibrio mercurii ND132T [41].
These results indicated that strains 9FUST, 9ES, and 09 could represent a novel species of the genus Pseudodesulfovibrio. The phenotypic features of the strain 9FUST, selected as the type strain, were investigated, and its genome was sequenced and analyzed.
3.2. Physiological and Chemotaxonomic Characterization of the Strain 9FUST
Cells of the sulfate-reducing strain 9FUST were non-spore-forming, curved rods, 0.3–0.5 µm in diameter and 1–2 µm long, motile in the exponential growth phase due to a single polar flagellum (Figure 1a,b). The cells were Gram-stain-negative and had Gram-negative cell wall structure, which was confirmed by the presence of an inner cytoplasmic membrane and an outer lipoprotein membrane on microphotographs of ultrathin cell sections (Figure 1c).
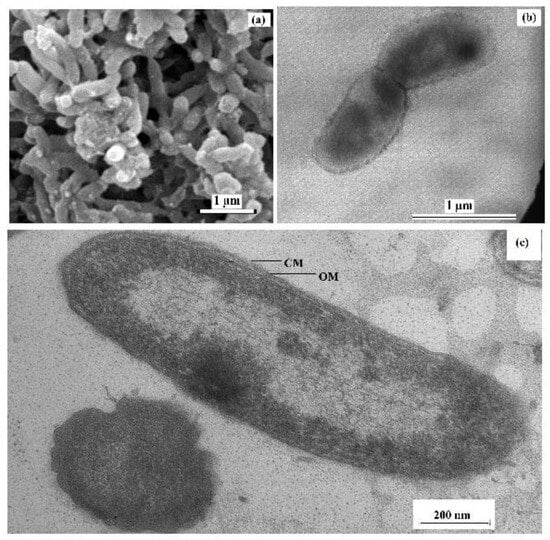
Figure 1.
Scanning electron micrograph (a) and transmission electron micrographs of negatively stained cells with a flagellum (b) and an ultrathin section of a cell of strain 9FUST showing the visible cellular membrane (CM) and outer membrane (OM) divided by the periplasmic space (c). The strain was grown in the lactate/sulfate medium for 7 days at 25 °C.
In the lactate/sulfate medium, strain 9FUST grew at the temperature range from 15 to 37 °C with an optimum at 23–28 °C (Figure S1a). No growth was observed at 10 and 42 °C. NaCl range was 0–5% (w/v) with a growth optimum at 0–2% (w/v) NaCl (Figure S1b). The pH range for growth was 4.1–8.6, with optimum pH around 6.5 (Figure S1c). However, the pH of the medium, which was 4.1–5.6 at the beginning of incubation, increased by the end of incubation to pH 6.5 (Table 2).
Table 2.
The differentiating characteristics of strain 9FUST and some Pseudodesulfovibrio species. Strains: 1, 9FUST (this study); 2, P. methanolicus 5S69T [21]; 3, P. hydrargyri BerOc1T [42]; 4, P. mercurii ND132T [41]; 5, ‘P. thermohalotolerans’ MCM B-1480T [43].
In sulfate-containing media with yeast extract (0.2 g·L−1), strain 9FUST used lactate, pyruvate, formate, fumarate, succinate, malate, methanol, ethanol, glycerol, and fructose; weak growth was observed on tryptone, ribose, and galactose, while the strain did not use benzoate, acetate, propionate, butyrate, lactose, glucose, mannose, glutamate, glycine, methylamine hydrochloride, or citrate. The strain was not capable of autotrophic growth on H2/CO2 but grew on H2/CO2 in the presence of acetate as a carbon source for constructive needs. The strain fermented pyruvate in the absence of sulfate (cultivation time 15 days at 25 °C) to form ethanol (1 mM), acetate (12 mM), CO2 (6% of the volume of the gas phase), and H2 (11%, v/v). Sulfate, sulfite, thiosulfate, elemental sulfur, and fumarate, but not nitrate, were used as terminal electron acceptors in the presence of lactate.
The predominant fatty acids in strain 9FUST were iso-C17:1 ω11 (20.8%), C15:0 (14.6%), iso-C15:0 (9.0%), C16:0 (8.5%), and C17:0 (7.7%) (Table S2). This fatty acid profile was different from that of ‘Pseudodesulfovibrio methanolicus’ 5S69T. The major polar lipids in strain 9FUST were phosphatidylethanolamine, diphosphatidylglycerol, and phosphatidylglycerol; among the minor lipids, unidentified glycolipid and aminophosholipids were detected (Figures S2 and S3). The main respiratory quinone of strain 9FUST was MK-6(H2). Physiological and biochemical characteristics that distinguish strain 9FUST from phylogenetically closely related species of the genus Pseudodesulfovibrio are summarized in Table 2.
3.3. Phylogenetic Analysis of 16S rRNA Gene Sequences
On the 16S rRNA gene phylogenetic tree, constructed using the MLH, NJ, and MP methods (Figure 2), strain 9FUST clustered within a well-supported clade, which included ‘P. methanolicus’ 5S69T, P. hydrargyri BerOc1T, P. mercurii ND132T, ‘P. thermohalotolerans’ MCM B-1480T, and P. indicus J2T. These results confirm that strain 9FUST belongs to the genus Pseudodesulfovibrio. Comparison of the 16S rRNA gene sequences of strain 9FUST revealed that the strain had similarities ranging from 92.8% to 98.9% with members of the genus Pseudodesulfovibrio (Table 3). However, it was impossible to definitively determine whether strain 9FUST represents a new species, as its 16S rRNA gene sequence is similar to ‘P. methanolicus’ 5S69T, which exceeds 98.7% [44,45], which is the typical threshold for species delineation. To resolve this, whole-genome sequencing and further comparative genomic analyses were performed to assess additional genomic features and clarify the taxonomic position of strain 9FUST.
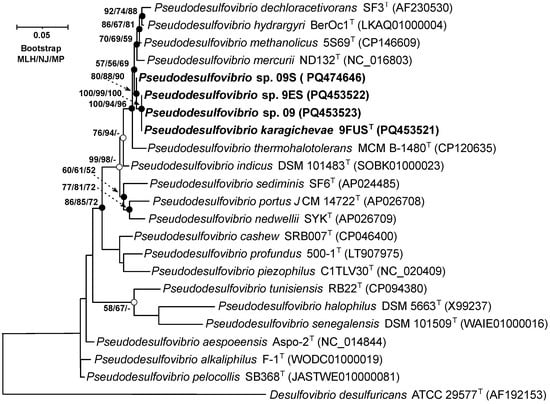
Figure 2.
Maximum-likelihood phylogenetic tree based on 16S rRNA gene sequences (1444 nucleotide sites), demonstrating the position of strain 9FUST within the genus Pseudodesulfovibrio. White circles indicate that the relevant nodes were recovered using the neighbor-joining algorithm; black circles indicate that the relevant nodes were also recovered based on the neighbor-joining and maximum-parsimony algorithms. Bootstrap values (>50%) are listed as percentages at the branching points. Bar, 0.05 substitutions per nucleotide position. The tree was rooted using Desulfovibrio desulfuricans ATCC 29577T as the outgroup. GenBank accession numbers for 16S rRNA genes are indicated in parentheses. The name of the studied strain is marked in boldface.
Table 3.
The genome-relatedness indices (%) between strain 9FUST and type strains of the genus Pseudodesulfovibrio.
3.4. Genome Features and Phylogeny
The genome of strain 9FUST was composed of 142 contigs with a total genomic length of 4,201,585 bp, an N50 value of 50.2 kb, a G + C content of 64.0%, and coverage of 132.0×. The genome comprises 3944 annotated genes, including 3851 protein-coding sequences, 23 pseudogenes, and 64 RNA genes. The genome of strain 9FUST shows ANI values ranging from 72.5% to 91.6% and dDDH values from 18.5% to 45.0% when compared with the genomes of the type strains of the genus Pseudodesulfovibrio (Table 3). These values are below the established thresholds of 95–96% for ANI and 70% for dDDH, which are generally accepted for species delineation [44,45]. This indicates that strain 9FUST represents a novel species within the genus Pseudodesulfovibrio.
To identify the phylogenetic position of strain 9FUST, a maximum-likelihood tree was constructed based on the concatenated alignment of 120 single-copy proteins comprising 37,730 amino acid positions (Figure 3). On the phylogenetic tree, strain 9FUST formed a separate branch with ‘P. methanolicus’ 5S69T, P. hydrargyri BerOc1T, P. mercurii ND132T, and ‘P. thermohalotolerans’ MCM B-1480T.
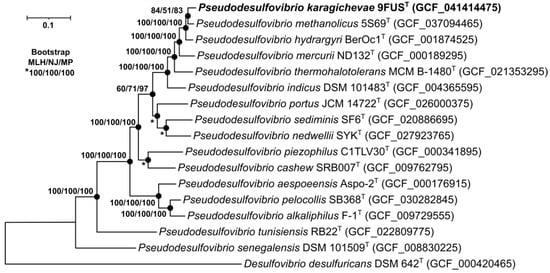
Figure 3.
The maximum-likelihood phylogenetic tree derived from concatenated 120 single-copy proteins demonstrates the position of strain 9FUST within the genus Pseudodesulfovibrio. For phylogenetic analysis, 37,730 amino acid positions were used. Black circles indicate that the corresponding nodes were also recovered based on the neighbor-joining and maximum-parsimony algorithms. Bar, 0.05 amino acid substitutions per site. Bootstrap values (>50%) are listed as percentages at the branching points. The tree was rooted using Desulfovibrio desulfuricans DSM 624T as the outgroup. Accession numbers for the genomic assemblies are indicated in parentheses. The name of the studied strain is marked in boldface.
A comprehensive pangenomic analysis was performed on sixteen Pseudodesulfovibrio species, encompassing a total of 55,478 genes, which were grouped into 12,348 gene clusters (GCs). Among these, 1472 core gene clusters were consistently found across all analyzed genomes, including 1311 single-copy genes (SCGs) (Figure 4). The phylogenetic tree, reconstructed from these SCGs and covering 429,648 amino acid positions, exhibited a topology highly consistent with the phylogenomic tree derived from the concatenated analysis of 120 single-copy proteins. This robust congruence further supports the phylogenetic placement of strain 9FUST within the genus Pseudodesulfovibrio.
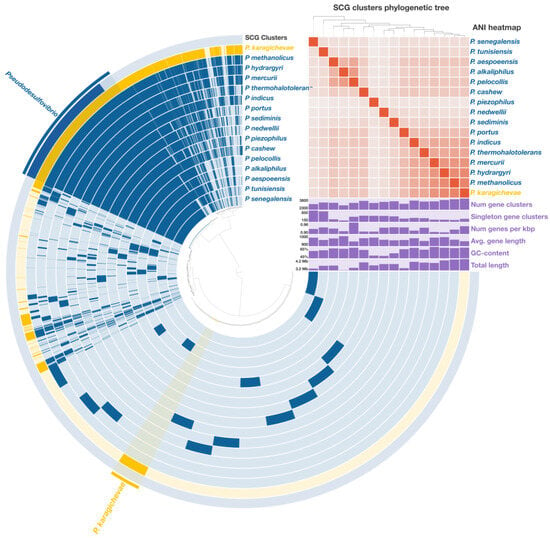
Figure 4.
Pangenome analysis of sixteen Pseudodesulfovibrio species calculated with Anvi’o v. 8.0. The central dendrogram illustrates the relationships between 12,348 gene clusters (55,478 genes) across the analyzed genomes. Dark circular regions indicate the presence of specific genes in each genome. A phylogenetic tree was reconstructed based on single-copy genes, providing insights into the evolutionary relationships among the species. ANI heatmap illustrates similarity values ranging from 70% to 100%.
3.5. Genome Insights and Unique Genes of Strain 9FUST
The functional annotation analysis of identified genes in the 9FUST genome using the COG database revealed a total of 82.89% (3192 of 3851) sequences classified into the COG functional categories and assigned to 23 functional groups (Figure 5). Among the groups, signal transduction mechanisms (10.1%), which allow cells to translate signals from the extracellular environment into changes within the cell, amino acid transport and metabolism (10.1%), and energy production and conversion (9.7%) were the most abundant.
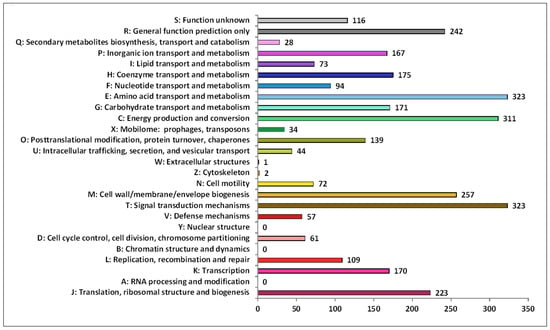
Figure 5.
Functional classification in Clusters of Orthologous Groups of proteins (COG). The X-axis indicates the number of genes in functional categories.
According to KEGG analysis of the genome of strain 9FUST, its genome contains the genes encoding complete modules of enzymes of the main metabolic pathways, characteristic of bacteria of the genus Pseudodesulfovibrio. Using the BlastKOALA tool of KEGG, the main metabolic pathways of strain 9FUST were shown to coincide with those described earlier for the closely related ‘P. methanolicus’ 5S69T [21]. Physiological and genomic characteristics of strain 9FUST were very similar to those of the type strain ‘Pseudodesulfovibrio methanolicus’ 5S69T, for which a detailed genomic analysis has recently been performed [21]. Both strains, as well as other bacteria of the genus Pseudodesulfovibrio, contained genes responsible for dissimilatory reduction in sulfate to sulfide; biosynthesis of ATP, amino acids, lipopolysaccharides, terpenoids, and menaquinones; metabolism of purines, pyrimidines, fatty acids, and hydrogen utilization. The genes coding complete modules of central carbohydrate metabolism, including the Embden–Meyerhof pathway, the Pentose phosphate pathway, the Leloir pathway of galactose degradation, and others, were revealed. In order not to duplicate the description of genes common to the genus and both new strains, 5S69T and 9FUST, an analysis of unique genes inherent only to the studied strain 9FUST was performed.
In this analysis, individual enzymes were identified that were specific to the metabolism of the studied strain and distinguished it from other members of the genus Pseudodesulfovibrio and, first of all, from the strain ‘P. methanolicus’ 5S69T.
The genome of strain 9FUST possessed 340 GCs unique to other Pseudodesulfovibrio species, 43 of which had a predicted function according to the KEGG database. This unique gene set highlights the specialized adaptations of strain 9FUST, particularly in carbohydrate metabolism, nutrient uptake, and environmental stress response. Among the distinct features, genes encoding the key enzymes for carbohydrate processing, such as galU (AB6M95_09405, 04270) coding UTP-glucose-1-phosphate uridylyltransferase (EC: 2.7.7.9), suggest enhanced capabilities for polysaccharide biosynthesis and utilization of diverse carbon sources and ability to survive in stressful environments [64].
Among the unique genes in the genome of strain 9FUST, the ccoN1 gene (encoding the main cytochrome oxidase subunit cbb3-type subunit I (EC: 7.1.1.9)) was annotated, although the auxiliary genes of the ccoNOQP operon, encoding the remaining cbb3 oxidase subunits, were absent in the genome. This enzyme catalyzes aerobic respiration, but it can also function under anaerobic conditions. This has been shown for Pseudomonas aeruginosa PAO1, which performs denitrification [65]. The ccoN1 gene has also been annotated in other sulfate reducers. The product of the ccoN1 gene of strain 9FUST has 70.1% similarity of amino acid sequences with a similar product of the Paucidesulfovibrio longus strain DSM 6739T, which was also isolated from an oil-producing well. However, what function this product can perform in the metabolism of these strains remains unclear.
The unique hddA (AB6M95_09420) and hddC (AB6M95_09405) genes coding for the biosynthesis of nucleotide-activated glycero-manno-heptose precursors of bacterial glycoproteins and cell surface polysaccharides were revealed [66].
The genome 9FUST also harbors unique genes involved in DNA repair and stress response, such as dcm (AB6M95_04415), encoding DNA (cytosine-5)-methyltransferase (EC: 2.1.1.37) [67], and uvrD (AB6M95_08355), encoding ATP-dependent DNA helicase (K03657) [68]. The strain has notable genomic elements related to horizontal gene transfer and genome maintenance, as evidenced by the presence of integrase/recombinase genes xerC (AB6M95_00800) and xerD (AB6M95_15490), which may contribute to genetic plasticity and adaptability [69].
Unlike the ‘P. methanolicus’ 5S69T, the strain 9FUST, was able to grow on ribose. Ribose is incorporated into the pentose phosphate cycle by the enzyme ribokinase (EC: 2.7.1.15), which catalyzes D-ribose phosphorylation to D-ribose 5-phosphate.
The ability to use ribose as a substrate of strain 9FUST is confirmed by the annotation of the rbsK gene (AB6M95_08240) encoding ribokinase in its genome (Figure S4).
This gene is part of the ribose operon rbsBACKR (AB6M95_08235-08260), together with the unique genes identified in the pangenomic analysis of the ribose ATP-binding cassette transport system: rbsA (ribose transport system ATP-binding protein RbsA, EC: 7.5.2.7), rbsB (ribose transport system substrate-binding protein RbsA), and rbsC (ribose transport system permease protein RbsC), as well as the rbsR gene encoding DNA-binding transcriptional repressor of ribose metabolism (Figure 6). The ribose transport system proteins RbsA, RbsB, and RbsC work together to import ribose efficiently, particularly in nutrient-limited environments, by leveraging the energy from ATP hydrolysis for active transport across the membrane [70]. In addition to the genome of strain 9FUST, the ribose metabolism operon is annotated only in the genomes of P. profundus 500T, P. nedwellii SYKT, and P. sediminis SF6T. The use of ribose as a substrate is a rare property for bacteria of the genus Pseudodesulfovibrio that is not related to the source of their allocation.
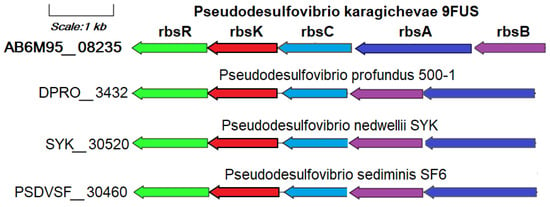
Figure 6.
The genes presumably encoding ribose metabolism in the genome of the strain 9FUST. Abbreviations: rbsK, ribokinase; rbsA, ribose transport system ATP-binding protein; rbsB, ribose transport system substrate-binding protein; rbsC, ribose transport system permease protein; rbsR, DNA-binding transcriptional repressor of ribose metabolism.
Strain 9FUST, similar to P. methanolicus 5S69T, was able to grow on fructose as a sole carbon source. According to the BlastCOALA tool, this ability of strain 9FUST is presumably provided by the fructose catabolism gene scrK (V8V93_00875), encoding fructokinase (EC: 2.7.1.4), which catalyzes phosphorylation of D-fructose to D-fructose 6-phosphate. This enzyme has been studied mainly for eukaryotes, but it also functions in bacteria. In particular, for Bifidobacterium longum DSM 20219T, it was shown that the enzyme is necessary for the assimilation of fructose [71]. Subsequently, D-fructose 6-phosphate is incorporated into glycolysis via D-fructose 1,6-bisphosphate. However, in strain 9FUST, another fructose assimilation pathway presumably additionally functions, involving the enzyme 1-phosphofructokinase (EC: 2.7.1.56), the one most widely used by bacteria, animals, and plants (Figure S5). The action of this enzyme is provided by the fructose operon genes annotated in the genome of strain 9FUST, including fruK (AB6M95_07015), encoding 1-phosphofructokinase (EC: 2.7.1.56); fruA (AB6M95_07020), encoding the fructose PTS system EIIBC or EIIC component (EC: 2.7.1.202); and fruB (AB6M95_07010), encoding a multiphosphoryl transfer protein (phosphoenolpyruvate–protein phosphotransferase; EC: 2.7.3.9). Among all members of the genus Pseudodesulfovibrio, duplicated genetic structures potentially capable of fructose assimilation, except for the genome of strain 9FUST, have been annotated only in the genomes of P. hydrargyri BerOc1T and P. sediminis SF6T.
The ability of strain 9FUST to grow on glycerol is confirmed by the annotation in its genome of a complete cluster of glycerol metabolism genes glpK-hdrBC-glpBARSTPQUV (AB6M95_08170-08225), homologous to the one previously described for P. methanolicus 5S69T, a strain also capable of glycerol utilization [21]. However, glycerol utilization by strain 9FUST may also probably follow another pathway due to the presence of the adh gene (AB6M95_01280) coding alcohol dehydrogenase (NADP+) (EC: 1.1.1.2); this gene has not been annotated in the genome of P. methanolicus 5S69T. This enzyme may catalyze the oxidation of alcohols, including glycerol, which is oxidized to D-glyceraldehyde; the latter is subsequently catabolized via the intermediate metabolites D-glycerate and 2-phospho-D-glycerate with involvement in glycolysis (Figure S6).
According to the BlastKOALA data, strain 9FUST, similar to most Pseudodesulfovibrio species, is capable of dinitrogen fixation with the formation of ammonium. The genome of strain 9FUST contains the nifVHDKBENBA operon (AB6M95_00645-00710), encoding the molybdenum–iron nitrogenase and auxiliary genes and orthologous to the one previously described for P. methanolicus strain 5S69T [21]. However, the genome of strain 9FUST also contains the anfHDKGOR gene cluster (AB6M95_01005-01040), coding an alternative iron–iron nitrogenase. This enzyme and its structural genes have originally been described for Azotobacter vinelandii [72]. The iron–iron nitrogenase of Rhodobacter capsulatus was recently shown to reduce not only N2 to NH4 but also CO2 to methane and formate; it was less selective to dinitrogen than the Mo- and V-nitrogenases [73].
Among members of the genus Pseudodesulfovibrio, a homologous cluster of alternative nitrogenase genes was found only in the genome of P. hydrargyri BerOc1T (Figure 7). Among the three known nitrogenase types, the Fe-only nitrogenase is considered the simplest since its function depends on fewer gene products compared to the orthologous but more complex Mo- and V-nitrogenases. Its investigation holds, therefore, a biotechnological promise [74].
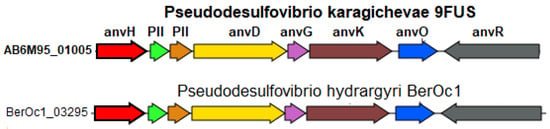
Figure 7.
Organization of gene clusters presumably encoding the alternative nitrogen fixation enzyme in the genome of strain 9FUST. Abbreviations: anvH, nitrogenase (iron–iron) reductase and maturation protein; PII, Nitrogen regulatory protein P-II, nitrogen-fixation associated; anvD, Nitrogenase (iron–iron) alpha chain; anvG, nitrogenase (iron–iron) delta chain; anvK, nitrogenase (iron–iron) beta chain; anvO, unknown; anvR, nitrogenase (iron–iron) transcriptional regulator.
Duplication of substrate utilization pathways in strain 9FUST probably provides for its metabolic lability, improving its adaptation to its environment.
3.6. Ecological Implications
The genus Pseudodesulfovibrio was described in 2016 [24], and it currently comprises 13 validly published species and 5 species not validly published under the International Code of Nomenclature of Bacteria (ICNP) [https://lpsn.dsmz.de/genus/pseudodesulfovibrio (accessed on 15 October 2024)]. This genus belongs to the family Desulfovibrionaceae, order Desulfovibrionales, class Deltaproteobacteria of the phylum Pseudomonadota [24,38]. Members of this genus were isolated from deep-sea hydrothermal areas, brackish lake sediments, terrestrial mud volcanoes, oil refinery water, and production water of petroleum reservoirs [15,21].
According to the BLAST analysis of the GenBank database on a tree including the 16S rRNA gene of the strain 9FUST and 99 other closely related sequences, the strain 9FUST forms a single cluster with several bacteria of the genera Pseudodesulfovibrio and Desulfovibrio (Figure S7). The sequences of Pseudodesulfovibrio sp. strain 09 and 9ES were isolated from the same habitat as strain 9FUST, and Desulfovibrio sp. HQM3 isolated from mudflat turned out to be the closest. The more distant sequences of this cluster belonged to Pseudodesulfovibrio sp. 09S isolated from the same habitat, as well as Desulfovibrio sp. Z1 isolated from seawater and Desulfovibrio caledoniensis SEBR 7250 isolated from oil field brines. Thus, SRBs that are taxonomically close to the strain under study (9FUST) are found not only in oil reservoirs but also in other habitats.
Sulfate-reducing strains of the genus Pseudodesulfovibrio (9FUST, 9ES, 09, DNS2, and 09S) and Oleidesulfovibrio alaskensis DNS2 were isolated at the temperature, pH, and salinity of their environment (Karazhanbas oil field) and can be considered as an indigenous microbiota to the oil reservoir. They are able to use lower alcohols (methanol, ethanol) and some sugars and grow on H2/CO2 in the presence of acetate. The latter can be formed from H2/CO2 by homoacetogenic bacteria Acetobacterium carbinolicum 9FOS, isolated from the same formation. When sulfates enter the reservoir with injected water, sulfate reducers can receive energy by their reduction to sulfide. However, at the time of the study, sulfate was not registered in reservoir water, which led to a low representation of sulfate reducers in the microbial community [15] and the predominance of methanogens as the final destructors of the organic matter of oil. Under these conditions, sulfate reducers probably grow as chemoorganoheterotrophs, fermenting carbonaceous and proteinaceous substrates, including necromass (i.e., dead biomass), and contribute to carbon turnover. Genomic analysis of strain 9FUST confirms that amino acid metabolism and carbohydrate metabolism are the major catabolic pathways in Pseudodesulfovibrio 9FUST. Apart from sulfate reducers, acetogenic and methanogenic prokaryotes occurring in the reservoir may also contribute to the corrosion of the steel equipment. The corrosive activity of these prokaryotic groups has been demonstrated previously [12,13,14].
Strain 9FUST was not found to possess the gene encoding alkylsuccinate synthase (Ass) responsible for the anaerobic degradation of alkanes and the genes encoding anaerobic degradation of the aromatic compounds present in petroleum (benzoate, phenol, catechol, and meta- and ortho-cleavage). However, in the 9FUST genome were present many genes encoding the pathways related to anaerobic respiration of oxidized sulfur compounds, important for the development of microbial corrosion. The sulfide formed by sulfate-reducing bacteria can chemically interact with metals, causing extensive steel corrosion.
There is extensive literature on the negative effects of bacteria on steel equipment in the oil and gas industry [2,11,12,13,75,76].
The production of an extracellular polymer matrix and the formation of biofilms, the presence of flagella and pili can contribute to the growth of the studied strain and other SRB on the surface of metals and to pitting corrosion of steel. The presence of pili in strain 9FUST is confirmed by the annotation of the pilA and cpaABCEF genes (AB6M95_10525-10550) encoding the formation of pili.
It was noted that the concentration of organic substrates affects the rate of corrosion [77,78,79]. In media with a high content of an organic substrate, sulfate-reducing bacteria preferentially use it and increase the biomass, decreasing the corrosion rate and pitting depth. These results indicate that the corrosion rate has no direct correlation with the number of SRBs [79]. In the absence of organic compounds, these bacteria are capable of obtaining energy using elemental iron as the electron donor for sulfate reduction [80].
Thakur and co-workers analyzed 63 genomes of bacteria of the genus Desulfovibrio and identified the genes and proteins participating in the biocorrosion process by sulfate-reducing bacteria [81]. The genes determining the metal ion binding and sulfur metabolism, hysB and hydA, and sat and dsr, respectively, were found in all 63 genomes. These genes were also present in the genome of strain 9FUST and other bacteria of the genus Pseudodesulfovibrio, which includes the species formerly assigned to Desulfovibrio.
The most important property of the strain 9FUST, which determines its potential participation in biocorrosion, is its ability to grow on molecular hydrogen. In the 9FUST genome were present genes echABCDEF (AB6M95_12990-13015) and hynAB (AB6M95_09170-09175; 045-85-04590) encoding hydrogenases, characteristic of other bacteria of the genus Pseudodesulfovibrio. However, the cooMKLXUH operon (AB6M95_00505-00550) is annotated in the genome of strain 9FUST, presumably encoding a rare for this genus membrane Coo-carbon monoxide-induced [Ni-Fe] hydrogenase. A set of hydrogenases is assumed based on the analysis of the genome of strain 9FUST, possibly participating in removing hydrogen from the metal through electron transfer to sulfate, which contributes to electrochemical corrosion and pitting formation.
2026 marks the 100th anniversary of the appearance of papers by E.S. Bastin [82] and T.L. Ginzburg-Karagicheva [83] dealing with sulfate-reducing bacteria in oil fields. These authors are considered the founders of petroleum microbiology as a science. The bacterium Desulfovibrio bastinii (at present Maridesulfovibrio bastinii) is named after E.S. Bastin [84]. To pay tribute to Tatiana L. Ginzburg-Karagicheva’s contribution to petroleum microbiology, we named a new bacterium in her honor–Pseudodesulfovibrio karagichevae sp. nov.
4. Conclusions
Metagenomic studies have revealed the distribution of Pseudodesulfovibrio members in petroleum reservoirs [21,37,43,56], including the Karazhanbas oil field located in Kazakhstan [15]. In the present work, five sulfate-reducing strains identified as members of the genera Pseudodesulfovibrio and Oleidesulfovibrio were isolated from the Karazhanbas oil field. Four strains with high 16S rRNA gene similarity between each other probably belonged to a new species of the genus Pseudodesulfovibrio. An in-depth study of strain 9FUST, which was chosen as the type strain, revealed the set of phenotypic features distinguishing this strain from its closest relatives, ‘P. methanolicus’, P. hydrargyri, P. mercurii, and ‘P. thermohalotolerans’. These phenotypic features include the ability to grow without NaCl and to use ribose, methanol, ethanol, malate, and succinate, but not lactose, as electron donors. Values of overall genomic relatedness indices (OGRI) of strain 9FUST, ANI, and dDDH, with genomes of Pseudodesulfovibrio species, were below 91.6% and 45.0%, respectively. Based on the distinct phylogenetic position of the strain 9FUST on concatenated proteins and 16S rRNA gene trees, this strain was assigned to a new species of the genus Pseudodesulfovibrio, for which the name Pseudodesulfovibrio karagichevae sp. nov. is proposed (=VKM B-3654T = KCTC 25498T). The description of the species Pseudodesulfovibrio karagichevae sp. nov. is given in the protologue (Table 4).
Table 4.
Protologue description of Pseudodesulfovibrio karagichevae sp. nov.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/microorganisms12122552/s1, Figure S1: Growth profiles of strain 9FUST in the lactate-sulfate medium at various temperatures (a), NaCl concentrations (g·L−1) (b), and pH (c) after 14 days of incubation; Figure S2: Thin layer chromatograms of polar lipids from the strains 9FUST and ‘Pseudodesulfovibrio methanolicus’ 5S69T (b, [21]). The components were visualized by staining with 5% sulfuric acid in ethanol and heating at 180 °C for 15 min. Abbreviations: PE, phosphatidylethanolamines; DPG, diphosphatidylglycerols; PG, phosphatidylglycerols; GL, glycolipids; PL, phospholipids; APL, aminophosholipids; LPE, lysophosphatidylethanolamines; GPL, glycophospholipids; Figure S3: Identification of polar lipids from the strains 9FUST (A) and 5S69T (B, [21]). The components were visualized by molybdenum blue (a), α-naphthol (b), ninhydrin (c), and Dragendorff’ reagent (d). Abbreviations as in Figure S2; Figure S4: KEGG-map of the “Pentose phosphate pathway” based on the genome analysis of the strain 9FUST. The presumptive pathway of ribose assumption is highlighted in red. The enzymes annotated in the genome are highlighted in green as in Figures S5 and S6; Figure S5: KEGG-map of the “Fructose and mannose metabolism” pathway based on the genome analysis of strain 9FUST. The presumptive pathway of fructose assumption via fructokinase is highlighted in blue, and via 1-phosphofructokinase is highlighted in red; Figure S6: KEGG-map of the “Glycerolipid metabolism” pathway based on the genome analysis of strain 9FUST. The presumptive pathway of glycerol assumption is highlighted in red; Figure S7: Phylogenetic tree of 16S rRNA gene sequences of the 99 bacterial strains closest to the strain 9FUST according to a BLAST analysis based on GenBank. The cluster of the closest strains is marked with a red asterisk; Table S1: Physicochemical characteristics of reservoir water* from the production well 6069 of the Karazhanbas oil field; Table S2: Cellular fatty acid composition of strain 9FUST and the type strain of ‘Pseudodesulfovibrio methanolicus’ 5S69T.
Author Contributions
Conceptualization, S.K.B. and T.N.N.; data curation, D.S.G., T.P.T. and A.V.M.; funding acquisition, A.V.M. and N.S.Z.; investigation, S.K.B., S.R.S., D.S.S., A.B.P., A.N.A., V.M.T., A.V.M. and N.S.Z.; supervision, T.N.N.; writing—original draft, D.S.G., T.P.T. and T.N.N.; writing—review and editing, T.P.T. and T.N.N. All authors have read and agreed to the published version of the manuscript.
Funding
This research was partly funded by the Russian Science Foundation (grant 21-64-00019).
Data Availability Statement
The whole-genome shotgun project of strain 9FUST has been deposited at DDBJ/EMBL/GenBank under the accession GCF_041414475.1, and it is the first version described in this paper.
Acknowledgments
We thank Aharon Oren (the Hebrew University of Jerusalem) for nomenclature advice. The authors express their gratitude to Nataliya G. Loiko from the Research Center of Biotechnology, the Russian Academy of Science, for scanning electron micrographs performed on the UNIQEM Collection Core Facility.
Conflicts of Interest
Author Denis S. Grouzdev was employed by the company SciBear OU. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Magot, M.; Ollivier, B.; Patel, B.K.C. Microbiology of petroleum reservoirs. Antonie Leeuwenhoek 2000, 77, 103–116. [Google Scholar] [CrossRef]
- Gieg, L.M.; Jack, T.R.; Foght, J.M. Biological souring and mitigation in oil reservoirs. Appl. Microbiol. Biotechnol. 2011, 92, 263–282. [Google Scholar] [CrossRef] [PubMed]
- Koch, G.; Varney, J.; Thompson, N.; Moghissi, O.; Gould, M.; Payer, J. International Measures of Prevention, Application, and Economics of Corrosion Technologies Study. Houston. 2016. Available online: http://impact.nace.org/economic-impact.aspx (accessed on 15 October 2024).
- Liang, R.; Grizzle, R.S.; Duncan, K.E.; McInerney, M.J.; Suflita, J.M. Roles of thermophilic thiosulfate-reducing bacteria and methanogenic archaea in the biocorrosion of oil pipelines. Front. Microbiol. 2014, 5, 89. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Zhang, B.L.; Mbadinga, S.M.; Liu, J.F.; Gu, J.D.; Mu, B.Z. Functional genes (dsr) approach reveals similar sulphidogenic prokaryotes diversity but different structure in saline waters from corroding high temperature petroleum reservoirs. Appl. Microbiol. Biotechnol. 2014, 98, 1871–1882. [Google Scholar] [CrossRef] [PubMed]
- Knisz, J.; Eckert, R.; Gieg, L.M.; Koerdt, A.; Lee, J.S.; Silva, E.R.; Skovhus, T.L.; An Stepec, B.A.; Wade, S.A. Microbiologically influenced corrosion—more than just microorganisms. FEMS Microbiol. Rev. 2023, 47, fuad041. [Google Scholar] [CrossRef] [PubMed]
- Widdel, F.; Musat, F.; Knittel, K.; Galushko, A. Anaerobic degradation of hydrocarbons with sulphate as electron donor. In Sulphate-Reducing Bacteria. Environmental and Engineered Systems; Barton, L.L., Hamilton, W.A., Eds.; Cambridge University Press: Cambridge, UK, 2007; pp. 265–303. [Google Scholar]
- Plugge, C.M.; Zhang, W.; Scholten, J.C.; Stams, A.J.M. Metabolic flexibility of sulfate-reducing bacteria. Front. Microbiol. 2011, 2, 81. [Google Scholar] [CrossRef]
- Davidova, I.A.; Marks, C.R.; Suflita, J.M. Anaerobic hydrocarbon-degrading Deltaproteobacteria. In Taxonomy, Genomics and Ecophysiology of Hydrocarbon-Degrading Microbes. Handbook of Hydrocarbon and Lipid Microbiology; McGenity, T.J., Ed.; Springer: Berlin/Heidelberg, Germany, 2018; pp. 1–38. [Google Scholar] [CrossRef]
- Nazina, T.N.; Shestakova, N.M.; Ivoilov, V.S.; Kostrukova, N.K.; Belyaev, S.S.; Ivanov, M.V. Radiotracer assay of microbial processes in petroleum reservoirs. Adv. Biotech. Microbiol. 2017, 2, 555591. [Google Scholar] [CrossRef]
- Duncan, K.E.; Gieg, L.M.; Parisi, V.A.; Tanner, R.S.; Tringe, S.G.; Bristow, J.; Suflita, J.M. Biocorrosive thermophilic microbial communities in Alaskan North Slope oil facilities. Environ. Sci. Technol. 2009, 43, 7977–7984. [Google Scholar] [CrossRef]
- Davidova, I.A.; Duncan, K.E.; Perez-Ibarra, B.M.; Suflita, J.M. Involvement of thermophilic archaea in the biocorrosion of oil pipelines. Environ. Microbiol. 2012, 14, 1762–1771. [Google Scholar] [CrossRef]
- Mand, J.; Park, H.S.; Okoro, C.; Lomans, B.P.; Smith, S.; Chiejina, L.; Voordouw, G. Microbial methane production associated with carbon steel corrosion in a Nigerian oil field. Front. Microbiol. 2016, 6, 1538. [Google Scholar] [CrossRef]
- Liang, R.; Davidova, I.A.; Marks, C.R.; Stamps, B.W.; Harriman, B.H.; Stevenson, B.S.; Duncan, K.E.; Suflita, J.M. Metabolic capability of a predominant Halanaerobium sp. in hydraulically fractured gas wells and its implication in pipeline corrosion. Front. Microbiol. 2016, 7, 988. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, D.S.; Semenova, E.M.; Grouzdev, D.S.; Ershov, A.P.; Bidzhieva, S.K.; Ivanova, A.E.; Babich, T.L.; Sissenbayeva, M.R.; Bisenova, M.A.; Nazina, T.N. Microbial diversity and potential sulfide producers in the Karazhanbas oilfield (Kazakhstan). Microbiology 2020, 89, 459–469. [Google Scholar] [CrossRef]
- Murzagaliev, R.S. Geological Model of the Karazhanbas High-Viscosity Oil Deposit and Modern Biotechnologies for Its Recovery. 2009. Available online: https://new-disser.ru/_avtoreferats/01004423876.pdf (accessed on 6 December 2024).
- Wolin, E.A.; Wolin, M.J.; Wolfe, R.S. Formation of methane by bacterial extracts. J. Biol. Chem. 1963, 238, 2882–2888. [Google Scholar] [CrossRef] [PubMed]
- Pfennig, N.; Lippert, K.D. Über das Vitamin B12-Bedürfnis phototropher Schwefelbakterien. Archiv. Mikrobiol. 1966, 55, 245–256. [Google Scholar] [CrossRef]
- Hungate, R.E. A roll tube method for the cultivation of strict anaerobes. In Methods in Microbiology; Norris, J.L., Ribbons, D.W., Eds.; Academic Press: New York, NY, USA, 1969; Volume 3, pp. 117–132. [Google Scholar]
- Trüper, H.G.; Schlegel, H.G. Sulfur metabolism in Thiorhodaceae. I. Quantitative measurements on growing cells of Chromatium okenii. Antonie Leeuwenhoek 1964, 30, 321–323. [Google Scholar] [CrossRef]
- Bidzhieva, S.K.; Tourova, T.P.; Kadnikov, V.V.; Samigullina, S.R.; Sokolova, D.S.; Poltaraus, A.B.; Avtukh, A.N.; Tereshina, V.M.; Beletsky, A.V.; Mardanov, A.V.; et al. Phenotypic and genomic characterization of a sulfate-reducing bacterium Pseudodesulfovibrio methanolicus sp. nov. isolated from a petroleum reservoir in Russia. Biology 2024, 13, 800. [Google Scholar] [CrossRef]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- A Windowed Adaptive Trimming Tool for FASTQ Files Using Quality. Available online: https://github.com/najoshi/sickle (accessed on 28 January 2023).
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Jain, C.; Rodriguez-R, L.M.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Carbasse, J.S.; Peinado-Olarte, R.L.; Göker, M. TYGS and LPSN: A database tandem for fast and reliable genome-based classification and nomenclature of prokaryotes. Nucleic Acids Res. 2022, 50, D801–D807. [Google Scholar] [CrossRef]
- Chaumeil, P.-A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk: A toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 2020, 36, 1925–1927. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Hoang, D.T.; Vinh, L.S.; Flouri, T.; Stamatakis, A.; Von Haeseler, A.; Minh, B.Q. MPBoot: Fast phylogenetic maximum parsimony tree inference and bootstrap approximation. BMC Evol. Biol. 2018, 18, 11. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Delmont, T.O.; Eren, A.M. Linking pangenomes and metagenomes: The Prochlorococcus metapangenome. PeerJ 2018, 6, e4320. [Google Scholar] [CrossRef]
- Eren, A.M.; Esen, Ö.C.; Quince, C.; Vineis, J.H.; Morrison, H.G.; Sogin, M.L.; Delmont, T.O. Anvi’o: An advanced analysis and visualization platform for ‘omics data. PeerJ 2015, 3, e1319. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Feio, M.J.; Zinkevich, V.; Beech, I.B.; Llobet-Brossa, E.; Eaton, P.; Schmitt, J.; Guezennec, J. Desulfovibrio alaskensis sp. nov.; a sulphate-reducing bacterium from a soured oil reservoir. Int. J. Syst. Evol. Microbiol. 2004, 54, 1747–1752. [Google Scholar] [CrossRef] [PubMed]
- Waite, D.W.; Chuvochina, M.; Pelikan, C.; Parks, D.H.; Yilmaz, P.; Wagner, M.; Loy, A.; Naganuma, T.; Nakai, R.; Whitman, W.B.; et al. Proposal to reclassify the proteobacterial classes Deltaproteobacteria and Oligoflexia, and the phylum Thermodesulfobacteria into four phyla reflecting major functional capabilities. Int. J. Syst. Evol. Microbiol. 2020, 70, 5972–6016. [Google Scholar] [CrossRef] [PubMed]
- Eichler, B.; Schink, B. Oxidation of primary aliphatic alcohols by Acetobacterium carbinolicum sp. nov., a homoacetogenic anaerobe. Arch. Microbiol. 1984, 140, 147–152. [Google Scholar] [CrossRef]
- Paarup, M.; Friedrich, M.; Tindall, B.; Finster, K. Characterization of the psychrotolerant acetogen strain SyrA5 and the emended description of the species Acetobacterium carbinolicum. Antonie Leeuwenhoek 2006, 89, 55–69. [Google Scholar] [CrossRef]
- Gilmour, C.C.; Soren, A.B.; Gionfriddo, C.M.; Podar, M.; Wall, J.D.; Brown, S.D.; Michener, J.K.; Urriza, M.S.G.; Elias, D.A. Pseudodesulfovibrio mercurii sp. nov., a mercury-methylating bacterium isolated from sediment. Int. J. Syst. Evol. Microbiol. 2019, 71, 004697. [Google Scholar] [CrossRef]
- Ranchou-Peyruse, M.; Goni-Urriza, M.; Guignard, M.; Goas, M.; Ranchou-Peyruse, A.; Guyoneaud, R. Pseudodesulfovibrio hydrargyri sp. nov.; a mercury-methylating bacterium isolated from a brackish sediment. Int. J. Syst. Evol. Microbiol. 2018, 68, 1461–1466. [Google Scholar] [CrossRef]
- Gaikwad, S.L.; Pore, S.D.; Dhakephalkar, P.K.; Dagar, S.S.; Soni, R.; Kaur, M.P.; Rawat, H.N. Pseudodesulfovibrio thermohalotolerans sp. nov.; a novel obligately anaerobic, halotolerant, thermotolerant, and sulfate-reducing bacterium isolated from a western offshore hydrocarbon reservoir in India. Anaerobe 2023, 83, 102780. [Google Scholar] [CrossRef]
- Chun, J.; Oren, A.; Ventosa, A.; Christensen, H.; Arahal, D.R.; Da Costa, M.S.; Rooney, A.P.; Yi, H.; Xu, X.-W.; De Meyer, S.; et al. Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int. J. Syst. Evol. Microbiol. 2018, 68, 461–466. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.-P.; Göker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 2013, 14, 60. [Google Scholar] [CrossRef]
- Brown, S.D.; Gilmour, C.C.; Kucken, A.M.; Wall, J.D.; Elias, D.A.; Brandt, C.C.; Podar, M.; Chertkov, O.; Held, B.; Bruce, D.C.; et al. Genome sequence of the mercury-methylating strain Desulfovibrio desulfuricans ND132. J. Bacteriol. 2011, 193, 2078–2079. [Google Scholar] [CrossRef]
- Cao, J.; Gayet, N.; Zeng, X.; Shao, Z.; Jebbar, M.; Alain, K. Pseudodesulfovibrio indicus gen. nov.; sp. nov.; a piezophilic sulfate-reducing bacterium from the Indian Ocean and reclassification of four species of the genus Desulfovibrio. Int. J. Syst. Evol. Microbiol. 2016, 66, 3904–3911. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, D.; Ueki, A.; Amaishi, A.; Ueki, K. Desulfovibrio portus sp. nov.; a novel sulfate-reducing bacterium in the class Deltaproteobacteria isolated from an estuarine sediment. J. Gen. Appl. Microbiol. 2009, 55, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, T.; Kondo, R. Genome sequence of the sulfate-reducing bacterium Pseudodesulfovibrio portus JCM 14722T. Microbiol. Resour. Announc. 2022, 11, e00947-22. [Google Scholar] [CrossRef] [PubMed]
- Slobodkina, G.; Merkel, A.; Novikov, A.; Slobodkin, A. Pseudodesulfovibrio pelocollis sp. nov. a sulfate-reducing bacterium isolated from a terrestrial mud volcano. Curr. Microbiol. 2024, 81, 120. [Google Scholar] [CrossRef]
- Motamedi, M.; Pedersen, K. Desulfovibrio aespoeensis sp. nov.; a mesophilic sulfate-reducing bacterium from deep groundwater at Äspö hard rock laboratory, Sweden. Int. J. Syst. Bacteriol. 1998, 48, 311–315. [Google Scholar] [CrossRef]
- Pedersen, K.; Bengtsson, A.; Edlund, J.; Rabe, L.; Hazen, T.; Chakraborty, R.; Goodwin, L.; Shapiro, N. Complete genome sequence of the subsurface, mesophilic sulfate-reducing bacterium Desulfovibrio aespoeensis Aspo-2. Genome Announc. 2014, 2, e00509-14. [Google Scholar] [CrossRef]
- Zheng, R.; Wu, S.; Sun, C. Pseudodesulfovibrio cashew sp. nov.; a novel deep-sea sulfate-reducing bacterium, linking heavy metal resistance and sulfur cycle. Microorganisms 2021, 9, 429. [Google Scholar] [CrossRef]
- Frolova, A.A.; Merkel, A.Y.; Kuchierskaya, A.A.; Bonch-Osmolovskaya, E.A.; Slobodkin, A.I. Pseudodesulfovibrio alkaliphilus, sp. nov.; an alkaliphilic sulfate-reducing bacterium isolated from a terrestrial mud volcano. Antonie Leeuwenhoek 2021, 114, 1387–1397. [Google Scholar] [CrossRef]
- Takahashi, A.; Kojima, H.; Watanabe, M.; Fukui, M. Pseudodesulfovibrio sediminis sp. nov.; a mesophilic and neutrophilic sulfate-reducing bacterium isolated from sediment of a brackish lake. Arch. Microbiol. 2022, 204, 307. [Google Scholar] [CrossRef]
- Ben Ali Gam, Z.; Oueslati, R.; Abdelkafi, S.; Casalot, L.; Tholozan, J.L.; Labat, M. Desulfovibrio tunisiensis sp. nov.; a novel weakly halotolerant, sulfate-reducing bacterium isolated from exhaust water of a Tunisian oil refinery. Int. J. Syst. Evol. Microbiol. 2009, 59, 1059–1063. [Google Scholar] [CrossRef]
- Park, M.-J.; Kim, Y.J.; Park, M.; Yu, J.; Namirimu, T.; Roh, Y.-R.; Kwon, K.K. Establishment of genome based criteria for classification of the family Desulfovibrionaceae and proposal of two novel genera, Alkalidesulfovibrio gen. nov. and Salidesulfovibrio gen. nov. Front. Microbiol. 2022, 13, 738205. [Google Scholar] [CrossRef] [PubMed]
- Thioye, A.; Gam, Z.B.A.; Mbengue, M.; Cayol, J.-L.; Joseph-Bartoli, M.; Touré-Kane, C.; Labat, M. Desulfovibrio senegalensis sp. nov.; a mesophilic sulfate reducer isolated from marine sediment. Int. J. Syst. Evol. Microbiol. 2017, 67, 3162–3166. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Yang, J.-A.; Lim, J.K.; Park, M.-J.; Yang, S.-H.; Lee, H.S.; Kang, S.G.; Lee, J.-H.; Kwon, K.K. Paradesulfovibrio onnuriensis gen. nov.; sp. nov.; a chemolithoautotrophic sulfate-reducing bacterium isolated from the Onnuri vent field of the Indian Ocean and reclassification of Desulfovibrio senegalensis as Paradesulfovibrio senegalensis comb. nov. J. Microbiol. 2020, 58, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Kondo, R. Pseudodesulfovibrio nedwellii sp. nov., a mesophilic sulphate-reducing bacterium isolated from a xenic culture of an anaerobic heterolobosean protist. Int. J. Syst. Evol. Microbiol. 2023, 73, 005826. [Google Scholar] [CrossRef] [PubMed]
- Kondo, R.; Kataoka, T. Whole-genome sequence of the sulfate-reducing bacterial strain SYK, isolated from a xenic culture of an anaerobic protist. Microbiol. Resour. Announc. 2023, 12, e01257-22. [Google Scholar] [CrossRef]
- Khelaifia, S.; Fardeau, M.-L.; Pradel, N.; Aussignargues, C.; Garel, M.; Tamburini, C.; Cayol, J.-L.; Gaudron, S.; Gaill, F.; Ollivier, B. Desulfovibrio piezophilus sp. nov., a piezophilic, sulfate-reducing bacterium isolated from wood falls in the Mediterranean Sea. Int. J. Syst. Evol. Microbiol. 2011, 61, 2706–2711. [Google Scholar] [CrossRef]
- Pradel, N.; Ji, B.; Gimenez, G.; Talla, E.; Lenoble, P.; Garel, M.; Tamburini, C.; Fourquet, P.; Lebrun, R.; Bertin, P.; et al. The first genomic and proteomic characterization of a deep-sea sulfate reducer: Insights into the piezophilic lifestyle of Desulfovibrio piezophilus. PLoS ONE 2013, 8, e55130. [Google Scholar] [CrossRef]
- Zeng, Z.; Zeng, X.; Guo, Y.; Wu, Z.; Cai, Z.; Pan, D. Determining the role of UTP-glucose-1-phosphate uridylyltransferase (GalU) in improving the resistance of Lactobacillus acidophilus NCFM to freeze-drying. Foods 2022, 11, 1719. [Google Scholar] [CrossRef]
- Hamada, M.; Toyofuku, M.; Miyano, T.; Nomura, N. cbb3-type cytochrome c oxidases, aerobic respiratory enzymes, impact the anaerobic life of Pseudomonas aeruginosa PAO1. J. Bacteriol. 2014, 196, 3881–3889. [Google Scholar] [CrossRef]
- Valvano, M.A.; Messner, P.; Kosma, P. Novel pathways for biosynthesis of nucleotide-activated glycero-manno-heptose precursors of bacterial glycoproteins and cell surface polysaccharides. Microbiology. 2002, 148, 1979–1989. [Google Scholar] [CrossRef]
- Militello, K.T.; Simon, R.D.; Mandarano, A.H.; DiNatale, A.; Hennick, S.M.; Lazatin, J.C.; Cantatore, S. 5-azacytidine induces transcriptome changes in Escherichia coli via DNA methylation-dependent and DNA methylation-independent mechanisms. BMC Microbiol. 2016, 16, 130. [Google Scholar] [CrossRef] [PubMed]
- Florés, M.; Sanchez, N.; Michel, B. A fork-clearing role for UvrD. Mol. Microbiol. 2005, 57, 1664–1675. [Google Scholar] [CrossRef] [PubMed]
- Paquola, A.C.M.; Asif, H.; Pereira, C.A.D.B.; Feltes, B.C.; Bonatto, D.; Lima, W.C.; Menck, C.F.M. Horizontal gene transfer building prokaryote genomes: Genes related to exchange between cell and environment are frequently transferred. J. Mol. Evol. 2018, 86, 190–203. [Google Scholar] [CrossRef] [PubMed]
- Clifton, M.C.; Simon, M.J.; Erramilli, S.K.; Zhang, H.; Zaitseva, J.; Hermodson, M.A.; Stauffacher, C.V. In Vitro Reassembly of the Ribose ATP-binding Cassette Transporter Reveals a Distinct Set of Transport Complexes. J. Biol. Chem. 2015, 290, 5555–5565. [Google Scholar] [CrossRef] [PubMed]
- Caescu, C.I.; Vidal, O.; Krzewinski, F.; Artenie, V.; Bouquelet, S. Bifidobacterium longum requires a fructokinase (Frk; ATP:D-fructose 6-phosphotransferase, EC 2.7.1.4) for fructose catabolism. J. Bacteriol. 2004, 186, 6515–6525. [Google Scholar] [CrossRef]
- Joerger, R.D.; Jacobson, M.R.; Premakumar, R.; Wolfinger, E.D.; Bishop, P.E. Nucleotide sequence and mutational analysis of the structural genes (anfHDGKOR) for the second alternative nitrogenase from Azotobacter vinelandii. J. Bacteriol. 1989, 171, 1075–1086. [Google Scholar] [CrossRef]
- Oehlmann, N.N.; Schmidt, F.V.; Herzog, M.; Goldman, A.L.; Rebelein, J.G. The iron nitrogenase reduces carbon dioxide to formate and methane under physiological conditions: A route to feedstock chemicals. Sci Adv. 2024, 10, eado7729. [Google Scholar] [CrossRef]
- López-Torrejón, G.; Burén, S.; Veldhuizen, M.; Rubio, L.M. Biosynthesis of cofactor-activatable iron-only nitrogenase in Saccharomyces cerevisiae. Microb. Biotechnol. 2021, 14, 1073–1083. [Google Scholar] [CrossRef]
- Wei, B.; Xu, J.; Sun, C.; Cheng, Y.F. Internal microbiologically influenced corrosion of natural gas pipelines: A critical review. J. Nat. Gas Sci. Eng. 2022, 102, 104581. [Google Scholar] [CrossRef]
- Lv, X.; Wang, C.; Liu, J.; Sand, W.; Nabuk, E., II; Zhang, Y.; Xu, A.; Duan, J.; Zhang, R. The microbiologically influenced corrosion and protection of pipelines: A detailed review. Materials 2024, 17, 4996. [Google Scholar] [CrossRef]
- Chen, Y.; Tang, Q.; Senko, J.M.; Cheng, G.; Newby, B.M.Z.; Castaneda, H.; Ju, L.K. Long-term survival of Desulfovibrio vulgaris on carbon steel and associated pitting corrosion. Corros. Sci. 2015, 90, 89–100. [Google Scholar] [CrossRef]
- Dou, W.; Liu, J.; Cai, W.; Wang, D.; Jia, R.; Chen, S.; Gu, T. Electrochemical investigation of increased carbon steel corrosion via extracellular electron transfer by a sulfate reducing bacterium under carbon source starvation. Corros. Sci. 2019, 150, 258–267. [Google Scholar] [CrossRef]
- Sun, M.; Wang, X.; Cui, W. Corrosion of sulfate-reducing bacteria on L245 steel under different carbon source conditions. Microorganisms 2024, 12, 1826. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Gu, T. Carbon source starvation triggered more aggressive corrosion against carbon steel by the Desulfovibrio vulgaris biofilm. Int. Biodeterior. Biodegrad. 2014, 91, 74–81. [Google Scholar] [CrossRef]
- Thakur, P.; Alaba, M.O.; Rauniyar, S.; Singh, R.N.; Saxena, P.; Bomgni, A.; Gnimpieba, E.Z.; Lushbough, C.; Goh, K.M.; Sani, R.K. Text-mining to identify gene sets involved in biocorrosion by sulfate-reducing bacteria: A semi-automated workflow. Microorganisms 2023, 11, 119. [Google Scholar] [CrossRef]
- Bastin, E.S. The presence of sulfate-reducing bacteria in oilfield waters. Science 1926, 63, 21–24. [Google Scholar] [CrossRef]
- Ginsburg-Karagicheva, T.L. Microbiological investigations on the sulfur salt waters off Apsheron. Azerb. Petrol. Econ. 1926, 6–7, 30–39. (In Russian) [Google Scholar]
- Magot, M.; Basso, O.; Tardy-Jacquenod, C.; Caumette, P. Desulfovibrio bastinii sp. nov. and Desulfovibrio gracilis sp. nov., moderately halophilic, sulfate-reducing bacteria isolated from deep subsurface oilfield water. Int. J. Syst. Evol. Microbiol. 2004, 54, 1693–1697. [Google Scholar] [CrossRef][Green Version]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).