
Untranslated Regions of a Segmented Kindia Tick Virus Genome Are Highly Conserved and Contain Multiple Regulatory Elements for Viral Replication

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Tick Collection
2.2. Whole-Genome Sequencing
2.3. Analysis of Whole-Genome Sequencing
2.4. 5′ UTR–3′ UTR Sequences and Structures
3. Results
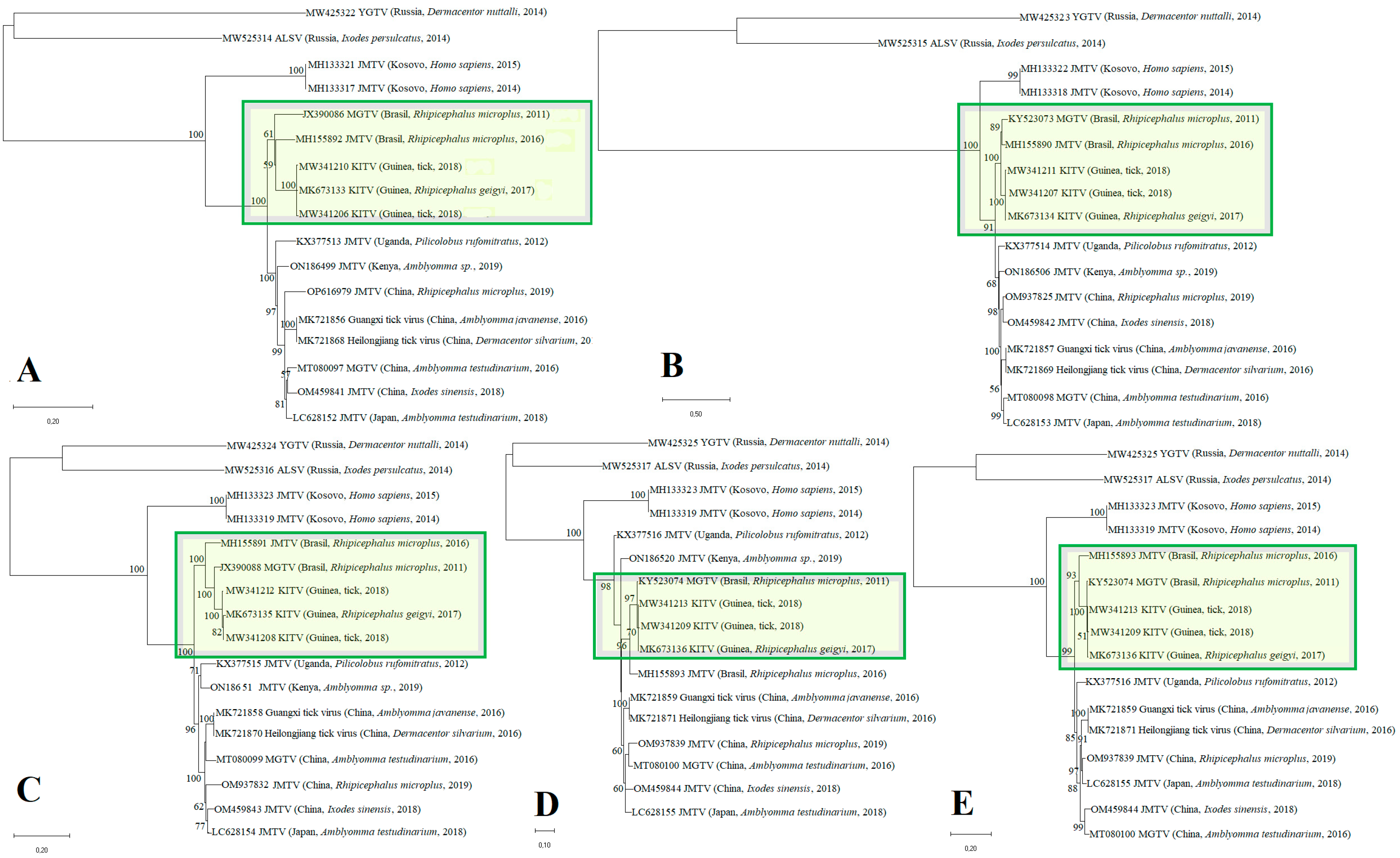
3.1. Phylogenetic Tree and Substitution Rate
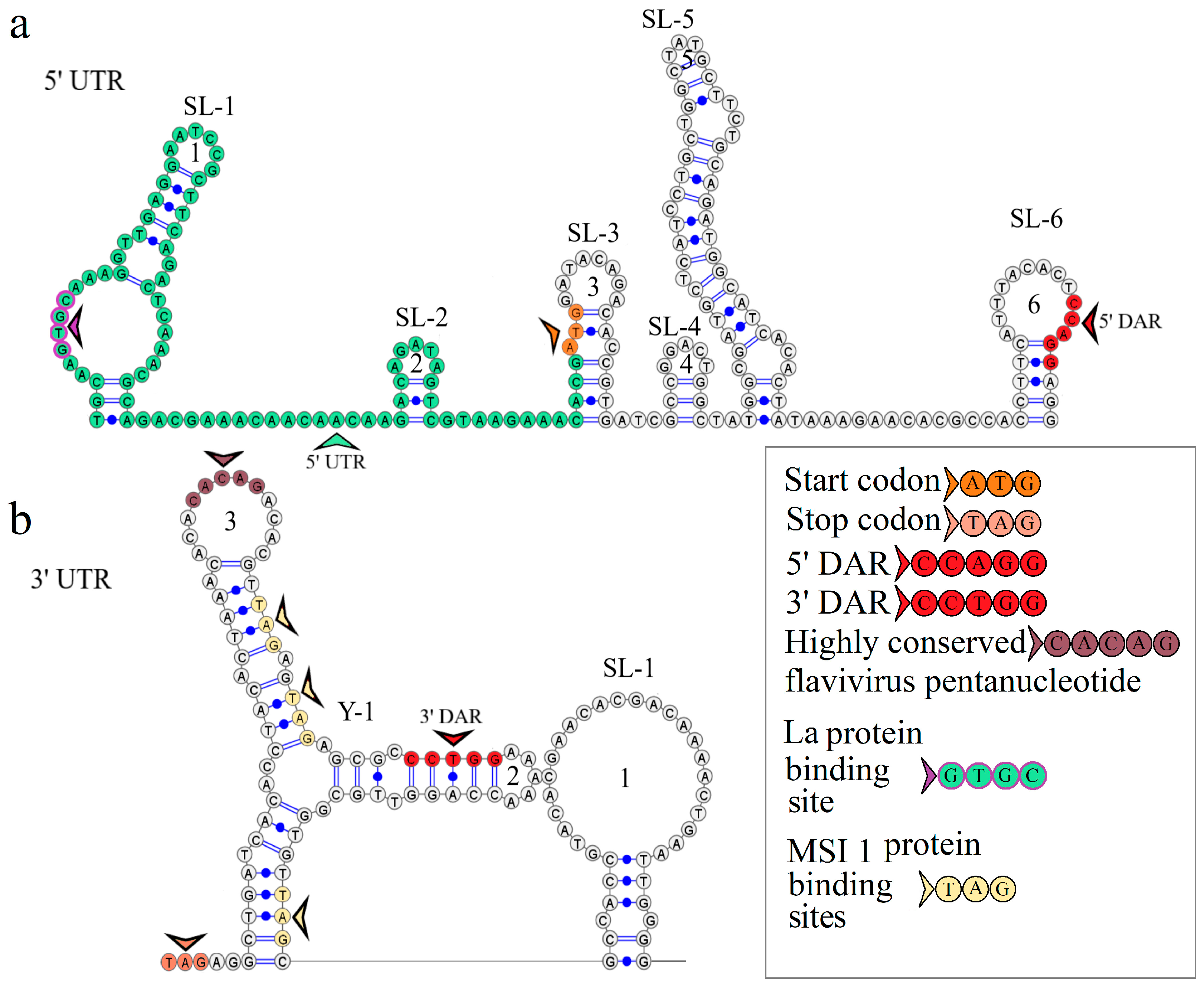
3.2. KITV 5′ and 3′ UTR Sequences
3.3. KITV Segment 1’s 5′ and 3′ UTR Structures
3.4. KITV Segment 2’s 5′ and 3′ UTR Structures
3.5. KITV Segment 3’s 5′ UTR Structure
3.6. KITV Segment 4’s 5′ and 3′ UTR Structures
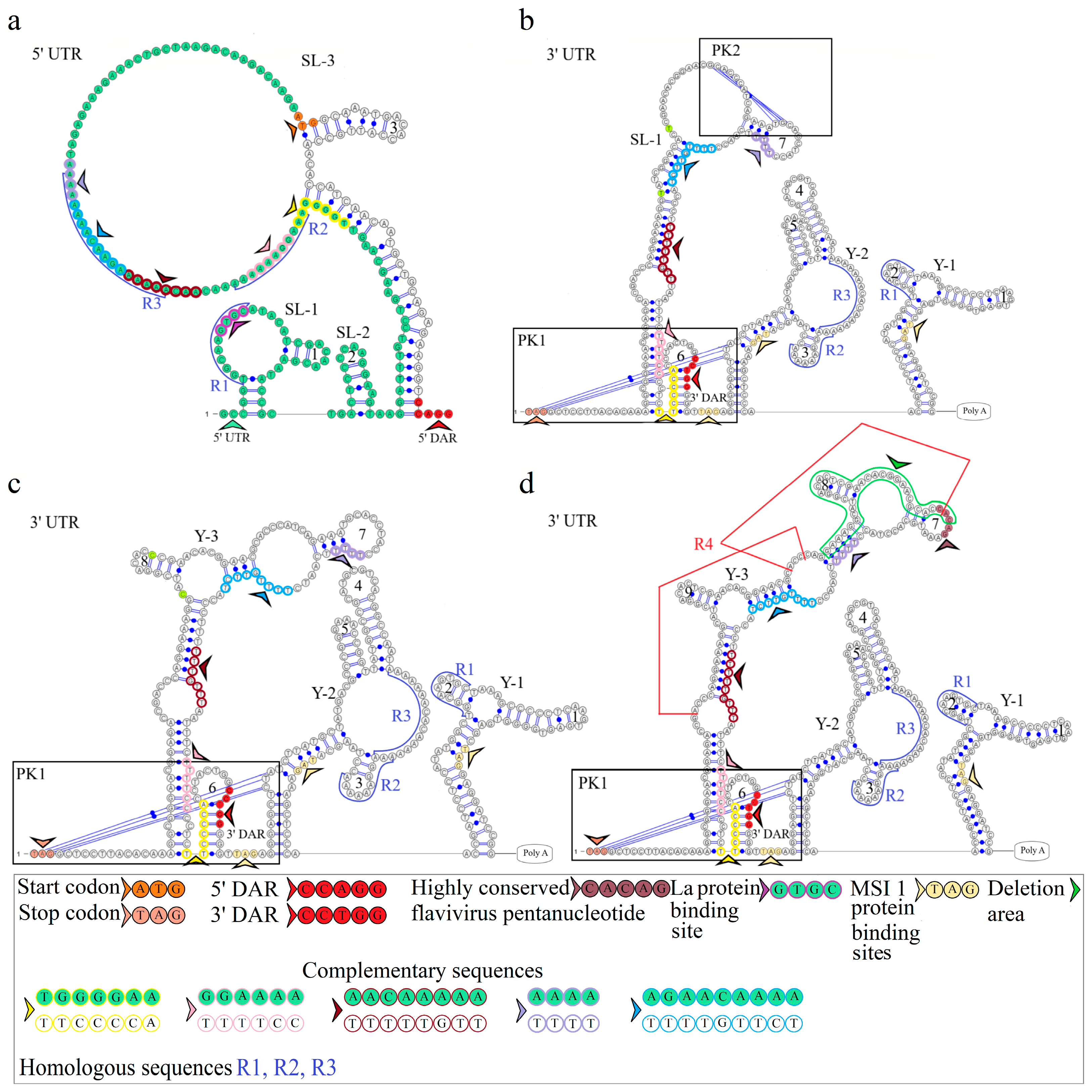
3.7. Conserved Regions of KITV 5′ and 3′ UTRs
3.7.1. Segment 1’s 5′ UTR
3.7.2. Segment 1’s 3′ UTR
3.7.3. Segment 2’s 5′ UTR
3.7.4. Segment 2’s 3′ UTR
3.7.5. Segment 3’s 5′ UTR
3.7.6. Segment 3’s 3′ UTR
3.7.7. Segment 4’s 5′ UTR
3.7.8. Segment 4’s 3′ UTR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, X.; Wang, N.; Wang, Z.; Liu, Q. The Discovery of Segmented Flaviviruses: Implications for Viral Emergence. Curr. Opin. Virol. 2020, 40, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.-C.; Shi, M.; Tian, J.-H.; Lin, X.-D.; Gao, D.-Y.; He, J.-R.; Wang, J.-B.; Li, C.-X.; Kang, Y.-J.; Yu, B.; et al. A Tick-Borne Segmented RNA Virus Contains Genome Segments Derived from Unsegmented Viral Ancestors. Proc. Natl. Acad. Sci. USA 2014, 111, 6744–6749. [Google Scholar] [CrossRef] [PubMed]
- Kuivanen, S.; Levanov, L.; Kareinen, L.; Sironen, T.; Jääskeläinen, A.J.; Plyusnin, I.; Zakham, F.; Emmerich, P.; Schmidt-Chanasit, J.; Hepojoki, J.; et al. Detection of Novel Tick-Borne Pathogen, Alongshan Virus, in Ixodes ricinus Ticks, South-Eastern Finland, 2019. Eurosurveillance 2019, 24, 1900394. [Google Scholar] [CrossRef] [PubMed]
- Ladner, J.T.; Wiley, M.R.; Beitzel, B.; Auguste, A.J.; Dupuis, A.P.; Lindquist, M.E.; Sibley, S.D.; Kota, K.P.; Fetterer, D.; Eastwood, G.; et al. A Multicomponent Animal Virus Isolated from Mosquitoes. Cell Host Microbe 2016, 20, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-D.; Wang, W.; Wang, N.-N.; Qiu, K.; Zhang, X.; Tana, G.; Liu, Q.; Zhu, X.-Q. Prevalence of the Emerging Novel Alongshan Virus Infection in Sheep and Cattle in Inner Mongolia, Northeastern China. Parasites Vectors 2019, 12, 450. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-D.; Wang, B.; Wei, F.; Han, S.-Z.; Zhang, L.; Yang, Z.-T.; Yan, Y.; Lv, X.-L.; Li, L.; Wang, S.-C.; et al. A New Segmented Virus Associated with Human Febrile Illness in China. N. Engl. J. Med. 2019, 380, 2116–2125. [Google Scholar] [CrossRef]
- Wu, Z.; Zhang, M.; Zhang, Y.; Lu, K.; Zhu, W.; Feng, S.; Qi, J.; Niu, G. Jingmen Tick Virus: An Emerging Arbovirus with a Global Threat. mSphere 2023, 8, e0028123. [Google Scholar] [CrossRef]
- Kholodilov, I.S.; Belova, O.A.; Morozkin, E.S.; Litov, A.G.; Ivannikova, A.Y.; Makenov, M.T.; Shchetinin, A.M.; Aibulatov, S.V.; Bazarova, G.K.; Bell-Sakyi, L.; et al. Geographical and Tick-Dependent Distribution of Flavi-Like Alongshan and Yanggou Tick Viruses in Russia. Viruses 2021, 13, 458. [Google Scholar] [CrossRef]
- Ternovoi, V.; Gladysheva, A.; Sementsova, A.; Zaykovskaya, A.; Volynkina, A.; Kotenev, E.; Loktev, V.; Agafonov, A. Detection of the RNA for New Multicomponent Virus in Patients with Crimean-Congo Hemorrhagic Fever in Southern Russia. Ann. Russ. Acad. Med. Sci. 2020, 75, 129–134. [Google Scholar] [CrossRef]
- Jia, N.; Liu, H.-B.; Ni, X.-B.; Bell-Sakyi, L.; Zheng, Y.-C.; Song, J.-L.; Li, J.; Jiang, B.-G.; Wang, Q.; Sun, Y.; et al. Emergence of Human Infection with Jingmen Tick Virus in China: A Retrospective Study. EBioMedicine 2019, 43, 317–324. [Google Scholar] [CrossRef]
- Gao, X.; Zhu, K.; Wojdyla, J.A.; Chen, P.; Qin, B.; Li, Z.; Wang, M.; Cui, S. Crystal Structure of the NS3-like Helicase from Alongshan Virus. IUCrJ 2020, 7, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Litov, A.G.; Okhezin, E.V.; Kholodilov, I.S.; Belova, O.A.; Karganova, G.G. Conserved Sequences in the 5′ and 3′ Untranslated Regions of Jingmenvirus Group Representatives. Viruses 2023, 15, 971. [Google Scholar] [CrossRef] [PubMed]
- Ternovoi, V.; Protopopova, E.; Shvalov, A.; Kartashov, M.; Bayandin, R.; Tregubchak, T.; Yakovlev, S.; Nikiforov, K.; Konovalova, S.; Loktev, V.; et al. Complete Coding Genome Sequence for a Novel Multicomponent Kindia Tick Virus Detected from Ticks Collected in Guinea 2020. bioRxiv 2020. [Google Scholar] [CrossRef]
- Kartashov, M.Y.; Gladysheva, A.V.; Naidenova, E.V.; Zakharov, K.S.; Shvalov, A.N.; Krivosheina, E.I.; Senichkina, A.M.; Bah, M.B.; Ternovoi, V.A.; Boumbaly, S.; et al. Molecular and Genetic Characteristics of the Multicomponent Flavi-like Kindia Tick Virus (Flaviviridae) Found in Ixodes Ticks on the Territory of the Republic of Guinea. Probl. Virol. 2023, 67, 487–495. [Google Scholar] [CrossRef]
- Gladysheva, A.A.; Gladysheva, A.V.; Ternovoi, V.A.; Loktev, V.B. Structural Motifs and Spatial Structures of Helicase (NS3) and RNA-Dependent RNA-Polymerase (NS5) of a Flavi-like Kindia Tick Virus (Unclassified Flaviviridae). Probl. Virol. 2023, 68, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.; Bouattour, A.; Camicas, J.-L.; Estrada-Peña, A.; Horak, I.; Latif, A.; Pegram, R.G.; Preston, P.M. Ticks of Domestic Animals in Africa: A Guide to Identification of Species; Bioscience Reports: Edinburgh, UK, 2003. [Google Scholar]
- Bellaousov, S.; Kayedkhordeh, M.; Peterson, R.J.; Mathews, D.H. Accelerated RNA Secondary Structure Design Using Preselected Sequences for Helices and Loops. RNA 2018, 24, 1555–1567. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, R.; Bernhart, S.H.; Höner zu Siederdissen, C.; Tafer, H.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011, 6, 26. [Google Scholar] [CrossRef]
- Markham, N.R.; Zuker, M. UNAFold: Software for nucleic acid folding and hybridization. Methods Mol. Biol. 2008, 453, 3–31. [Google Scholar] [CrossRef]
- Darty, K.; Denise, A.; Ponty, Y. VARNA: Interactive Drawing and Editing of the RNA Secondary Structure. Bioinformatics 2009, 25, 1974–1975. [Google Scholar] [CrossRef]
- Vashist, S.; Anantpadma, M.; Sharma, H.; Vrati, S. La Protein Binds the Predicted Loop Structures in the 3′ Non-Coding Region of Japanese Encephalitis Virus Genome: Role in Virus Replication. J. Gen. Virol. 2009, 90, 1343–1352. [Google Scholar] [CrossRef]
- Friebe, P.; Shi, P.-Y.; Harris, E. The 5′ and 3′ Downstream AUG Region Elements Are Required for Mosquito-Borne Flavivirus RNA Replication. J. Virol. 2011, 85, 1900–1905. [Google Scholar] [CrossRef] [PubMed]
- Souza, W.M.D.; Fumagalli, M.J.; Torres Carrasco, A.D.O.; Romeiro, M.F.; Modha, S.; Seki, M.C.; Gheller, J.M.; Daffre, S.; Nunes, M.R.T.; Murcia, P.R.; et al. Viral Diversity of Rhipicephalus microplus Parasitizing Cattle in Southern Brazil. Sci. Rep. 2018, 8, 16315. [Google Scholar] [CrossRef]
- Al-Khelaifi, F.; Yousri, N.A.; Diboun, I.; Semenova, E.A.; Kostryukova, E.S.; Kulemin, N.A.; Borisov, O.V.; Andryushchenko, L.B.; Larin, A.K.; Generozov, E.V.; et al. Genome-Wide Association Study Reveals a Novel Association Between MYBPC3 Gene Polymorphism, Endurance Athlete Status, Aerobic Capacity and Steroid Metabolism. Front. Genet. 2020, 11, 595. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-D.; Deng, C.-L.; Yuan, Z.-M.; Ye, H.-Q.; Zhang, B. Different Degrees of 5′-to-3′ DAR Interactions Modulate Zika Virus Genome Cyclization and Host-Specific Replication. J. Virol. 2020, 94, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Sommer, G.; Heise, T. Role of the RNA-Binding Protein La in Cancer Pathobiology. RNA Biol. 2021, 18, 218–236. [Google Scholar] [CrossRef]
- Kumar, A.; Ray, U.; Das, S. Human La Protein Interaction with GCAC near the Initiator AUG Enhances Hepatitis C Virus RNA Replication by Promoting Linkage between 5′ and 3′ Untranslated Regions. J. Virol. 2013, 87, 6713–6726. [Google Scholar] [CrossRef]
- Zhang, Q.-Y.; Liu, S.-Q.; Li, X.-D.; Li, J.-Q.; Zhang, Y.-N.; Deng, C.-L.; Zhang, H.-L.; Li, X.-F.; Fang, C.-X.; Yang, F.-X.; et al. Sequence Duplication in 3′ UTR Modulates Virus Replication and Virulence of Japanese Encephalitis Virus. Emerg. Microbes Infect. 2022, 11, 123–135. [Google Scholar] [CrossRef]
- Slonchak, A.; Parry, R.; Pullinger, B.; Sng, J.D.J.; Wang, X.; Buck, T.F.; Torres, F.J.; Harrison, J.J.; Colmant, A.M.G.; Hobson-Peters, J.; et al. Structural Analysis of 3′UTRs in Insect Flaviviruses Reveals Novel Determinants of SfRNA Biogenesis and Provides New Insights into Flavivirus Evolution. Nat. Commun. 2022, 13, 1279. [Google Scholar] [CrossRef]
- Schneider, A.D.B.; Wolfinger, M.T. Musashi Binding Elements in Zika and Related Flavivirus 3′UTRs: A Comparative Study in Silico. Sci. Rep. 2019, 9, 6911. [Google Scholar] [CrossRef]
- Chavali, P.L.; Stojic, L.; Meredith, L.W.; Joseph, N.; Nahorski, M.S.; Sanford, T.J.; Sweeney, T.R.; Krishna, B.A.; Hosmillo, M.; Firth, A.E.; et al. Neurodevelopmental Protein Musashi-1 Interacts with the Zika Genome and Promotes Viral Replication. Science 2017, 357, 83–88. [Google Scholar] [CrossRef]
- Darai, N.; Mahalapbutr, P.; Wolschann, P.; Lee, V.S.; Wolfinger, M.T.; Rungrotmongkol, T. Theoretical Studies on RNA Recognition by Musashi 1 RNA-Binding Protein. Sci. Rep. 2022, 12, 12137. [Google Scholar] [CrossRef] [PubMed]
- Klase, Z.A.; Khakhina, S.; Schneider, A.D.B.; Callahan, M.V.; Glasspool-Malone, J.; Malone, R. Zika Fetal Neuropathogenesis: Etiology of a Viral Syndrome. PLoS Negl. Trop. Dis. 2016, 10, e0004877. [Google Scholar] [CrossRef] [PubMed]
- Upstone, L.; Colley, R.; Harris, M.; Goonawardane, N. Functional Characterization of 5′ Untranslated Region (UTR) Secondary RNA Structures in the Replication of Tick-Borne Encephalitis Virus in Mammalian Cells. PLoS Negl. Trop. Dis. 2023, 17, e0011098. [Google Scholar] [CrossRef] [PubMed]
- Berzal-Herranz, A.; Berzal-Herranz, B.; Ramos-Lorente, S.E.; Romero-López, C. The Genomic 3′ UTR of Flaviviruses Is a Translation Initiation Enhancer. Int. J. Mol. Sci. 2022, 23, 8604. [Google Scholar] [CrossRef]
- Avila-Bonilla, R.G.; Salas-Benito, J.S. Interactions of Host MiRNAs in the Flavivirus 3′UTR Genome: From Bioinformatics Predictions to Practical Approaches. Front. Cell Infect. Microbiol. 2022, 12, 976843. [Google Scholar] [CrossRef]
- Ternovoi, V.A.; Gladysheva, A.V.; Ponomareva, E.P.; Mikryukova, T.P.; Protopopova, E.V.; Shvalov, A.N.; Konovalova, S.N.; Chausov, E.V.; Loktev, V.B. Variability in the 3′ Untranslated Regions of the Genomes of the Different Tick-Borne Encephalitis Virus Subtypes. Virus Genes 2019, 55, 448–457. [Google Scholar] [CrossRef]
- Rafalsky, V.; Averyanov, A.; Bart, B.; Minina, E.; Putilovskiy, M.; Andrianova, E.; Epstein, O. Efficacy and Safety of Ergoferon versus Oseltamivir in Adult Outpatients with Seasonal Influenza Virus Infection: A Multicenter, Open-Label, Randomized Trial. Int. J. Infect. Dis. 2016, 51, 47–55. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name of the Isolate of KITV and MGTV, Place of Isolation | Size of UTRs | |||||||
|---|---|---|---|---|---|---|---|---|
| Segment 1 | Segment 2 | Segment 3 | Segment 4 | |||||
| 5′ UTR | 3′ UTR | 5′ UTR | 3′ UTR | 5′ UTR | 3′ UTR | 5′ UTR | 3′ UTR | |
| KITV/2018/1, Guinea, Africa | 91 | 127 * | 154 | 352 | 110 | 177 * | 130 | 247 |
| KITV/2018/2, Guinea, Africa | 91 | 127 * | 154 | 352 | 100 | 143 * | 130 | 247 |
| KITV/2017/1, Guinea, Africa | 104 | 119 * | 156 | 387 | 97 | 143 * | 130 | 244 |
| MGTV/V4/11, Brazil, South America | 91 | 130 * | 51 * | 318 * | 102 | 179 * | 130 | 250 |
| MGTV Yunnan2016, China, Asia | 105 | 226 | 176 | 348 | 117 | 254 | 135 | 258 |
| Other KITV, Guinea, Africa | OP612399.1–OP612413.1 | OP612414.1–OP612428.1 | OP612443.1–OP612439.1 | OP612458.1–OP612444.1 | ||||
| 91 | 127 * | 122 * | 352 | 99 | 175 * | 130 | 247 | |
| Orthoflavivirus | ||||||||
| Name of the virus | Size of 5′ UTR | Size of 3′ UTR | ||||||
| Zika virus | 107 | 428 | ||||||
| Tick-borne encephalitis virus | 132 | 428–764 | ||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsishevskaya, A.A.; Alkhireenko, D.A.; Bayandin, R.B.; Kartashov, M.Y.; Ternovoi, V.A.; Gladysheva, A.V. Untranslated Regions of a Segmented Kindia Tick Virus Genome Are Highly Conserved and Contain Multiple Regulatory Elements for Viral Replication. Microorganisms 2024, 12, 239. https://doi.org/10.3390/microorganisms12020239
Tsishevskaya AA, Alkhireenko DA, Bayandin RB, Kartashov MY, Ternovoi VA, Gladysheva AV. Untranslated Regions of a Segmented Kindia Tick Virus Genome Are Highly Conserved and Contain Multiple Regulatory Elements for Viral Replication. Microorganisms. 2024; 12(2):239. https://doi.org/10.3390/microorganisms12020239
Chicago/Turabian StyleTsishevskaya, Anastasia A., Daria A. Alkhireenko, Roman B. Bayandin, Mikhail Yu. Kartashov, Vladimir A. Ternovoi, and Anastasia V. Gladysheva. 2024. "Untranslated Regions of a Segmented Kindia Tick Virus Genome Are Highly Conserved and Contain Multiple Regulatory Elements for Viral Replication" Microorganisms 12, no. 2: 239. https://doi.org/10.3390/microorganisms12020239
APA StyleTsishevskaya, A. A., Alkhireenko, D. A., Bayandin, R. B., Kartashov, M. Y., Ternovoi, V. A., & Gladysheva, A. V. (2024). Untranslated Regions of a Segmented Kindia Tick Virus Genome Are Highly Conserved and Contain Multiple Regulatory Elements for Viral Replication. Microorganisms, 12(2), 239. https://doi.org/10.3390/microorganisms12020239