

Three Rounds of Read Correction Significantly Improve Eukaryotic Protein Detection in ONT Reads


Abstract
:1. Introduction
2. Materials and Methods
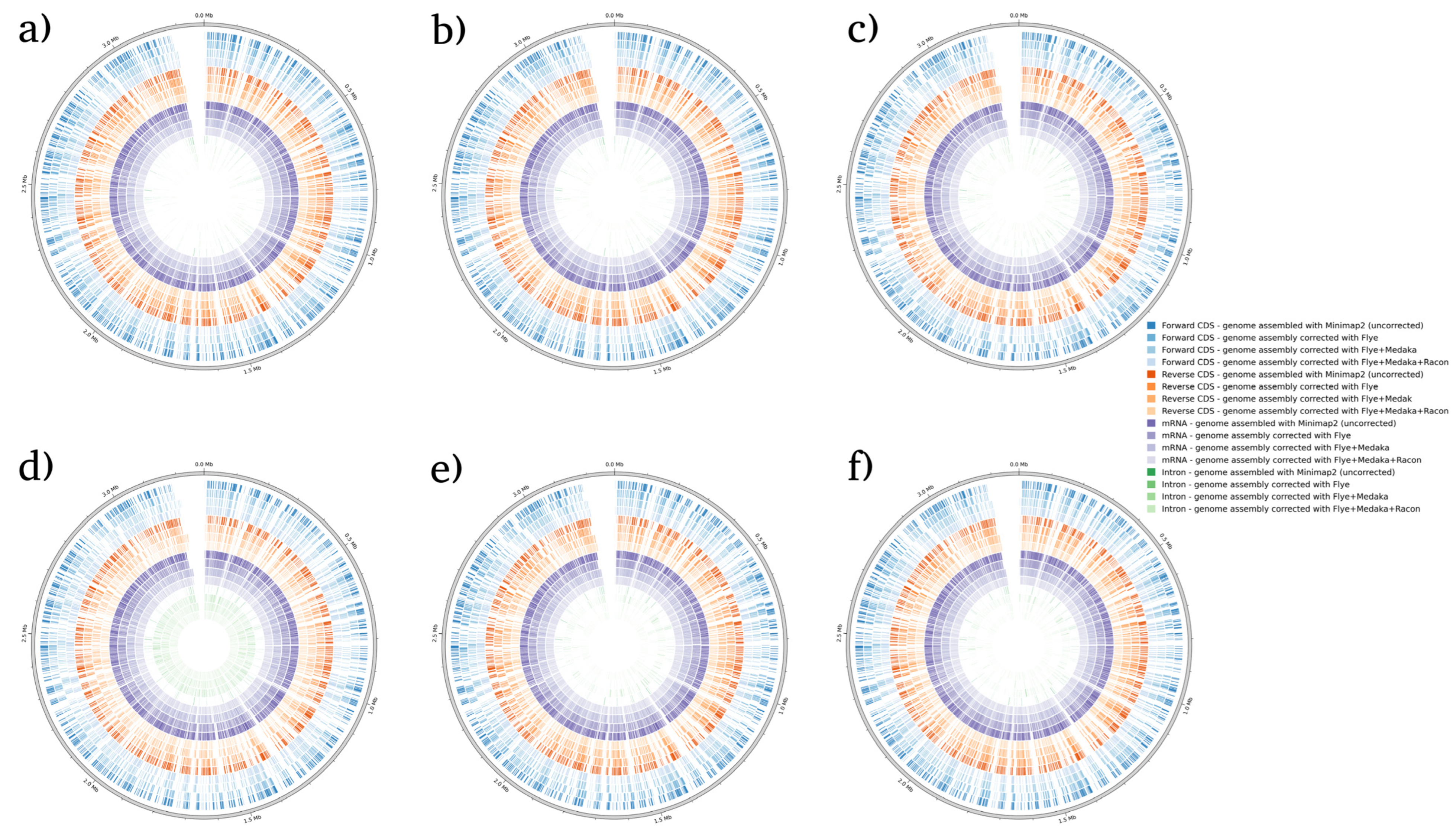
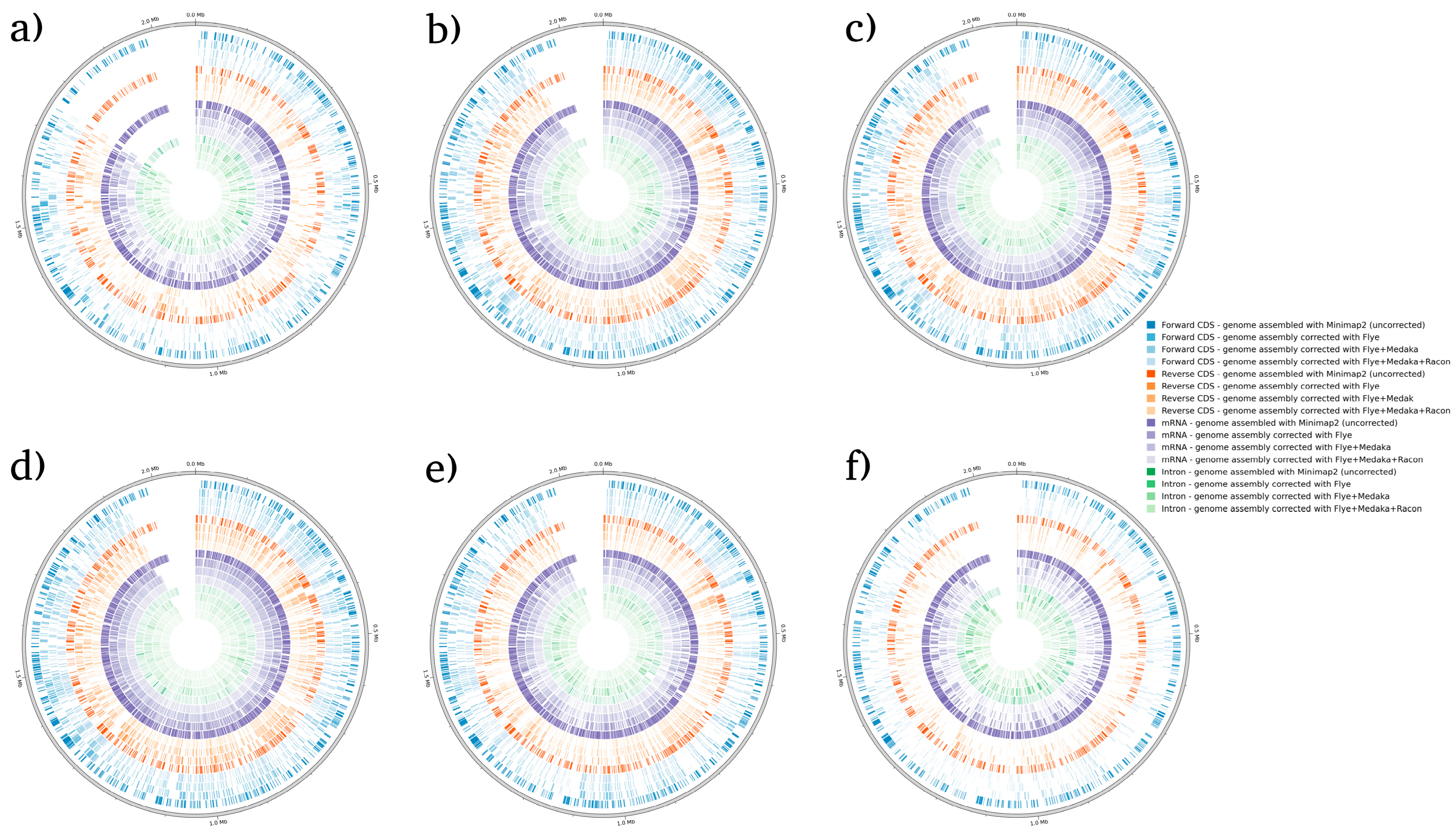
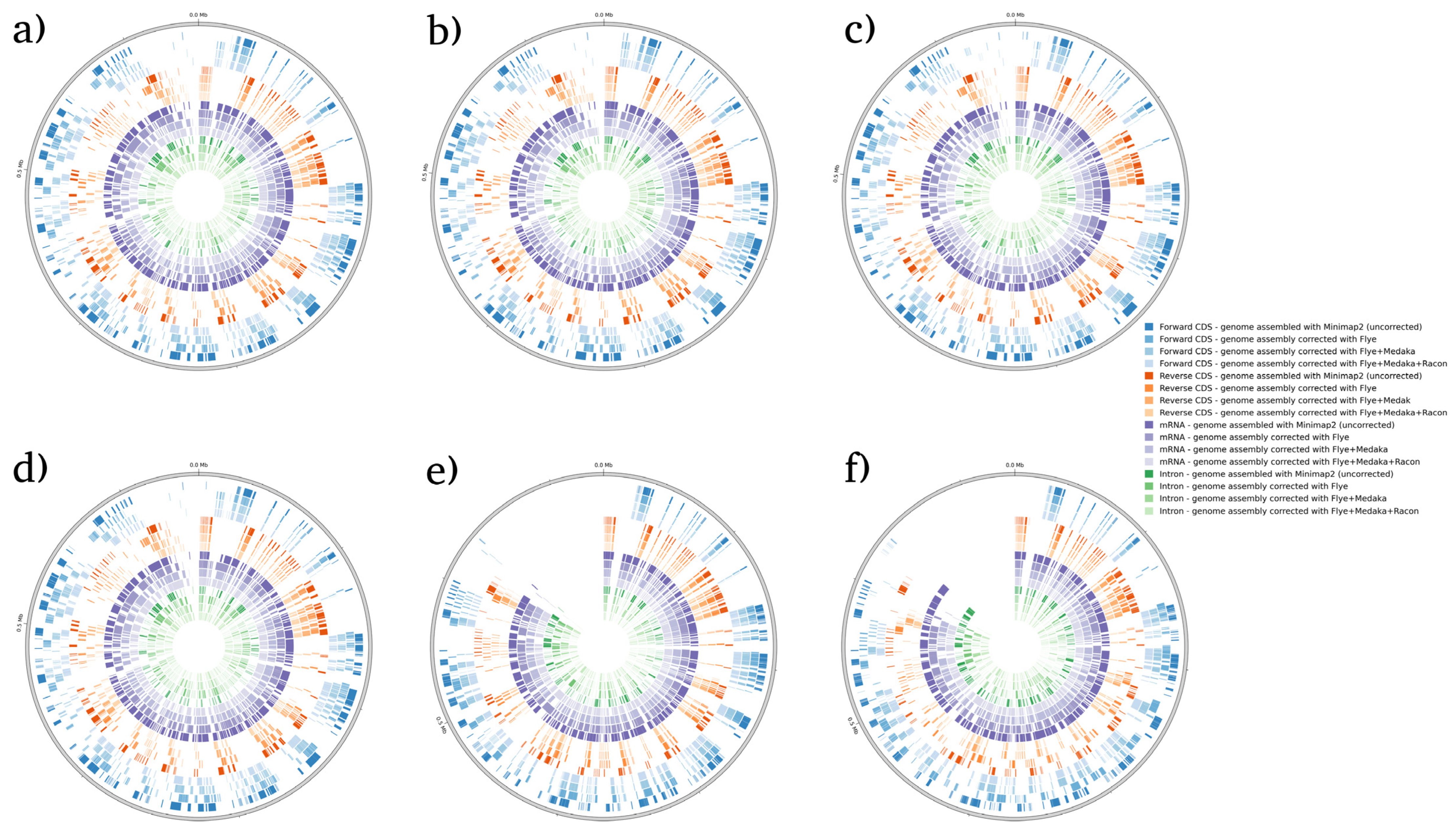
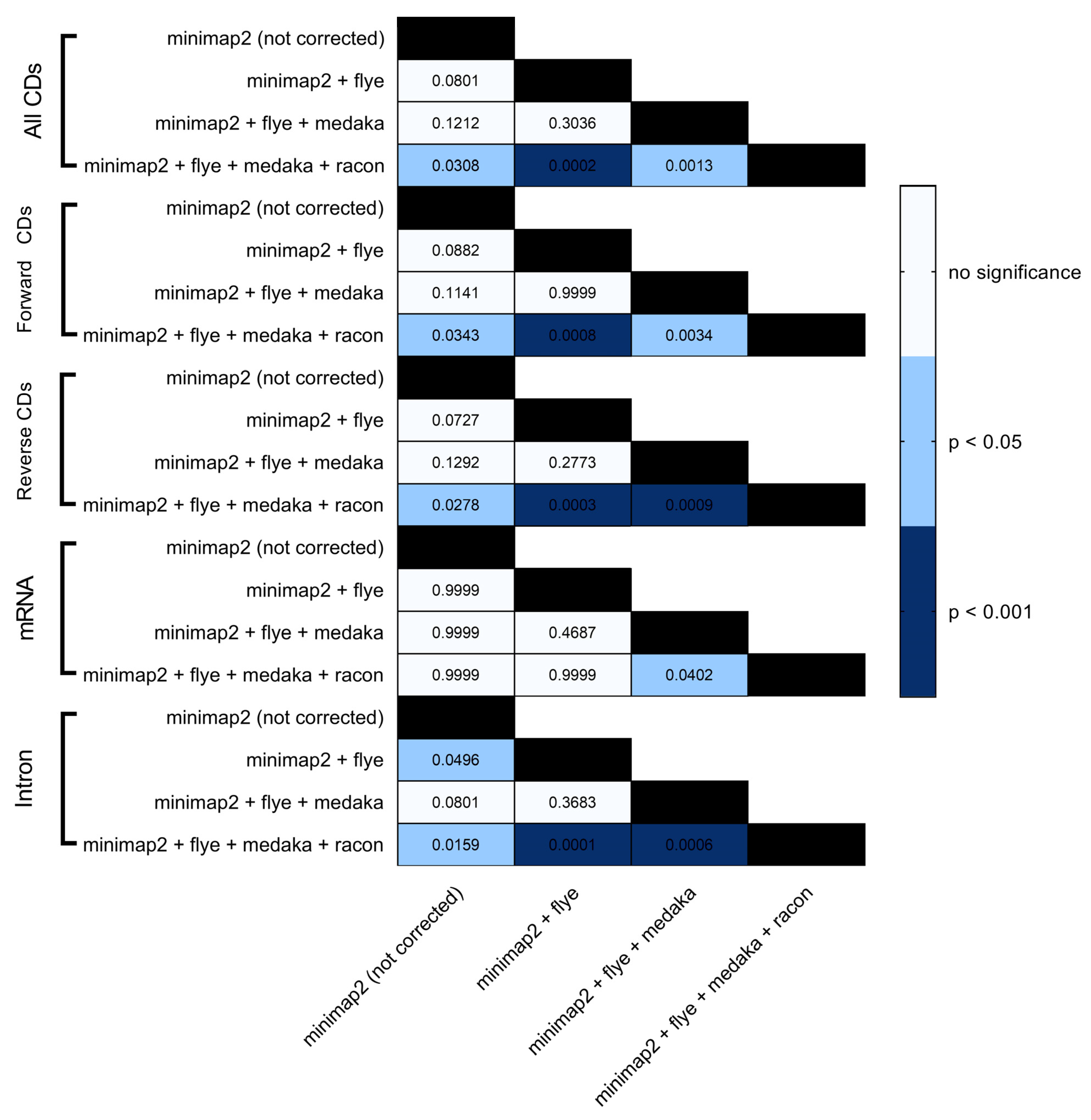
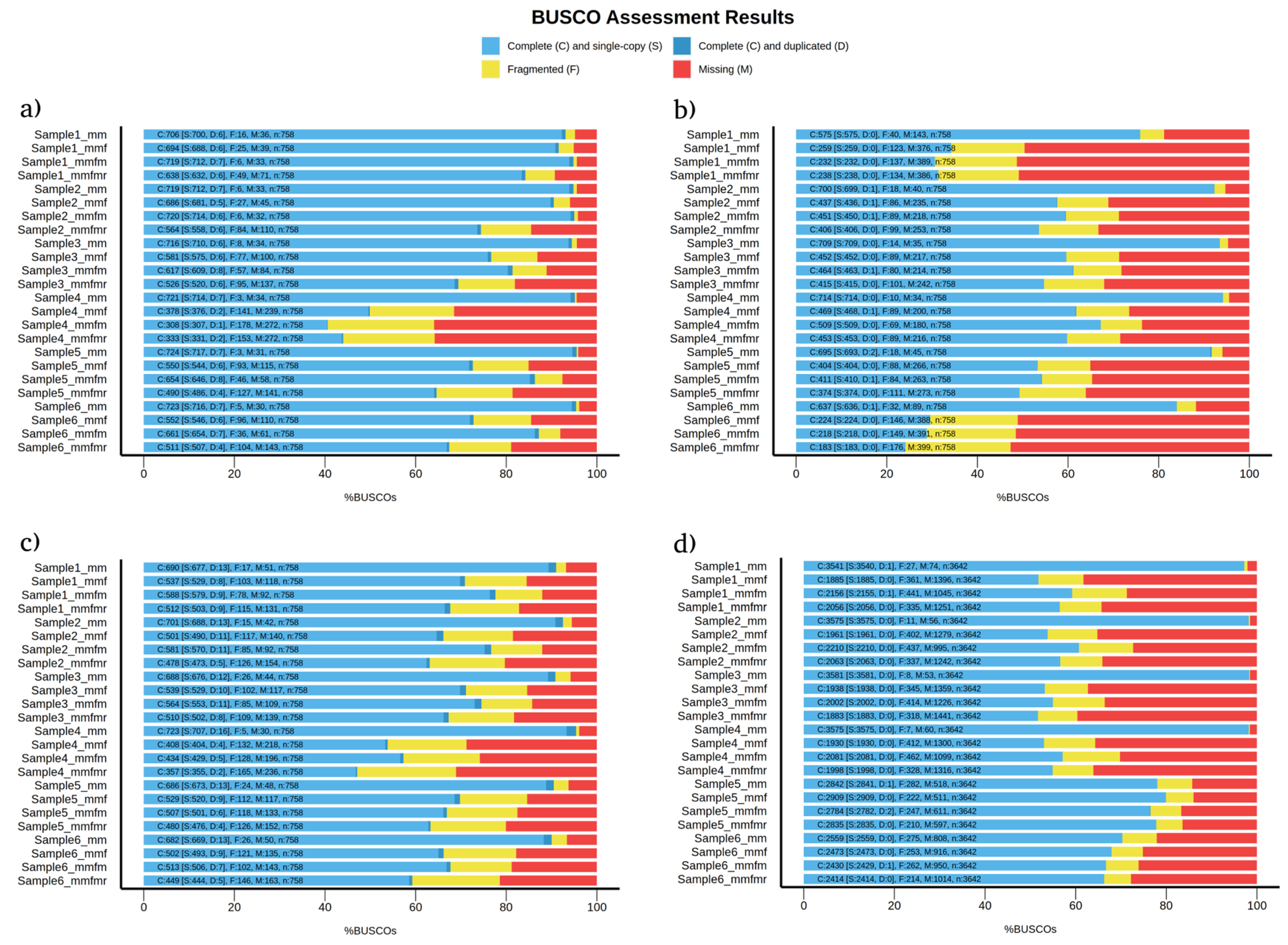
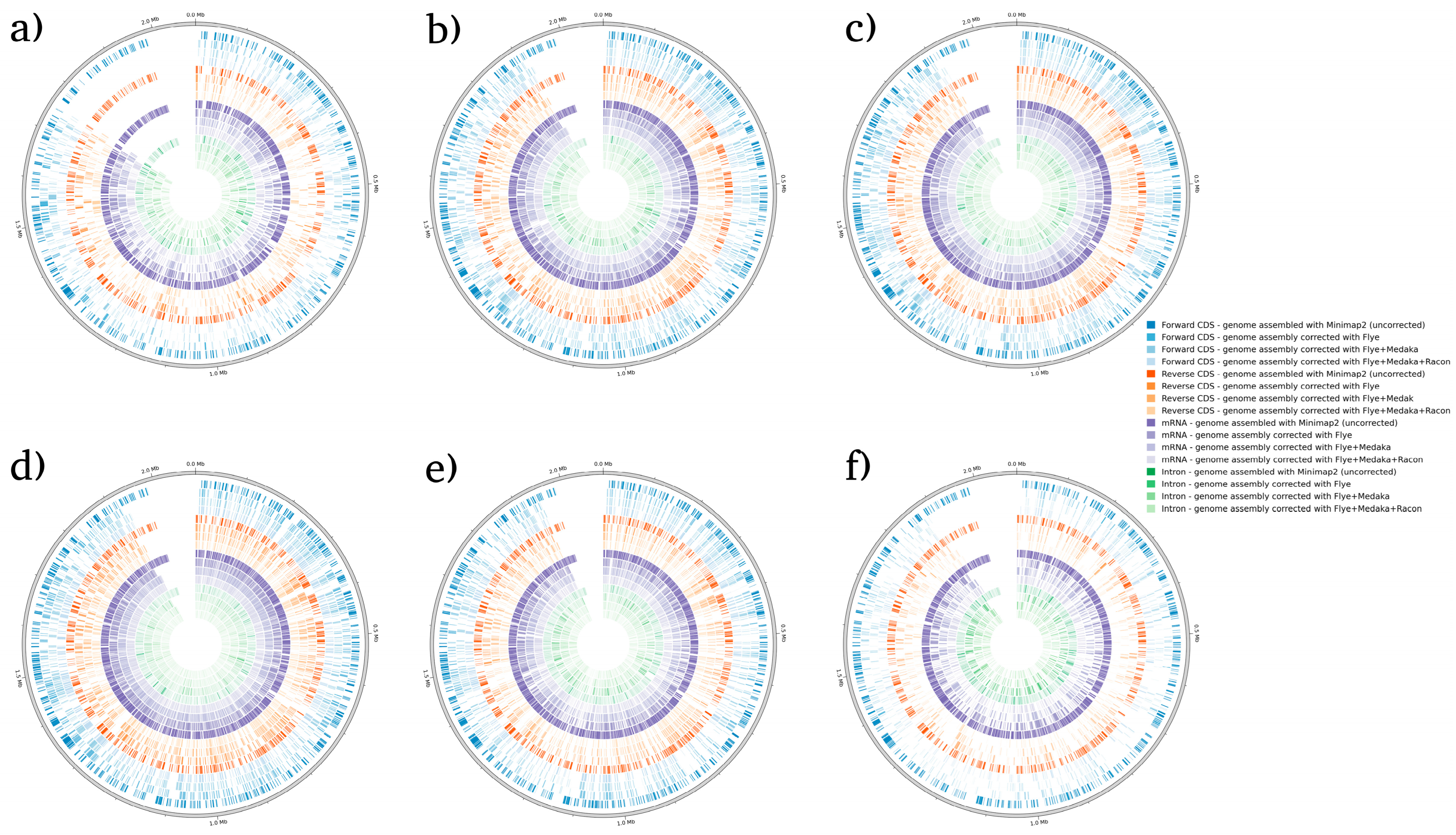
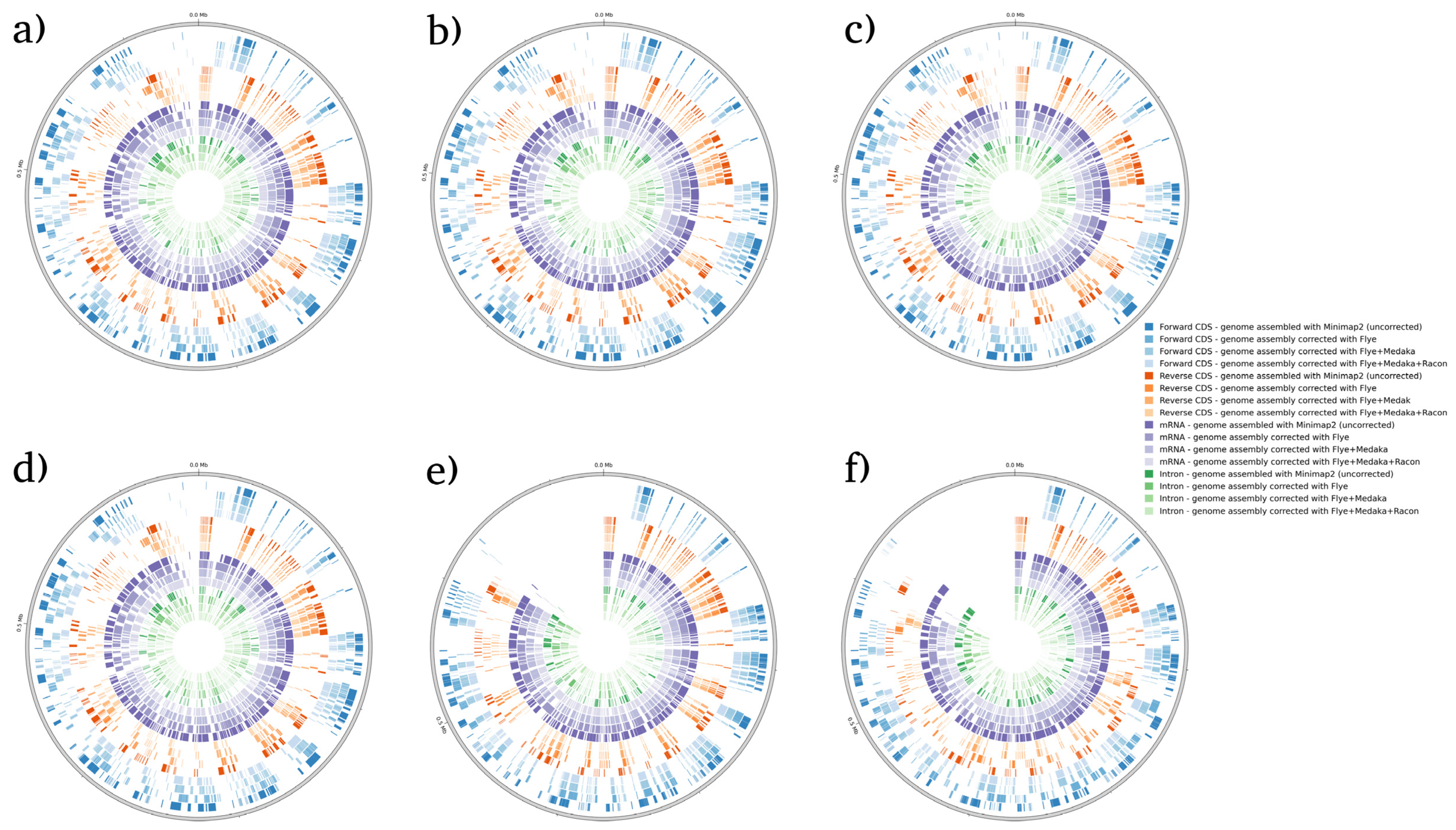
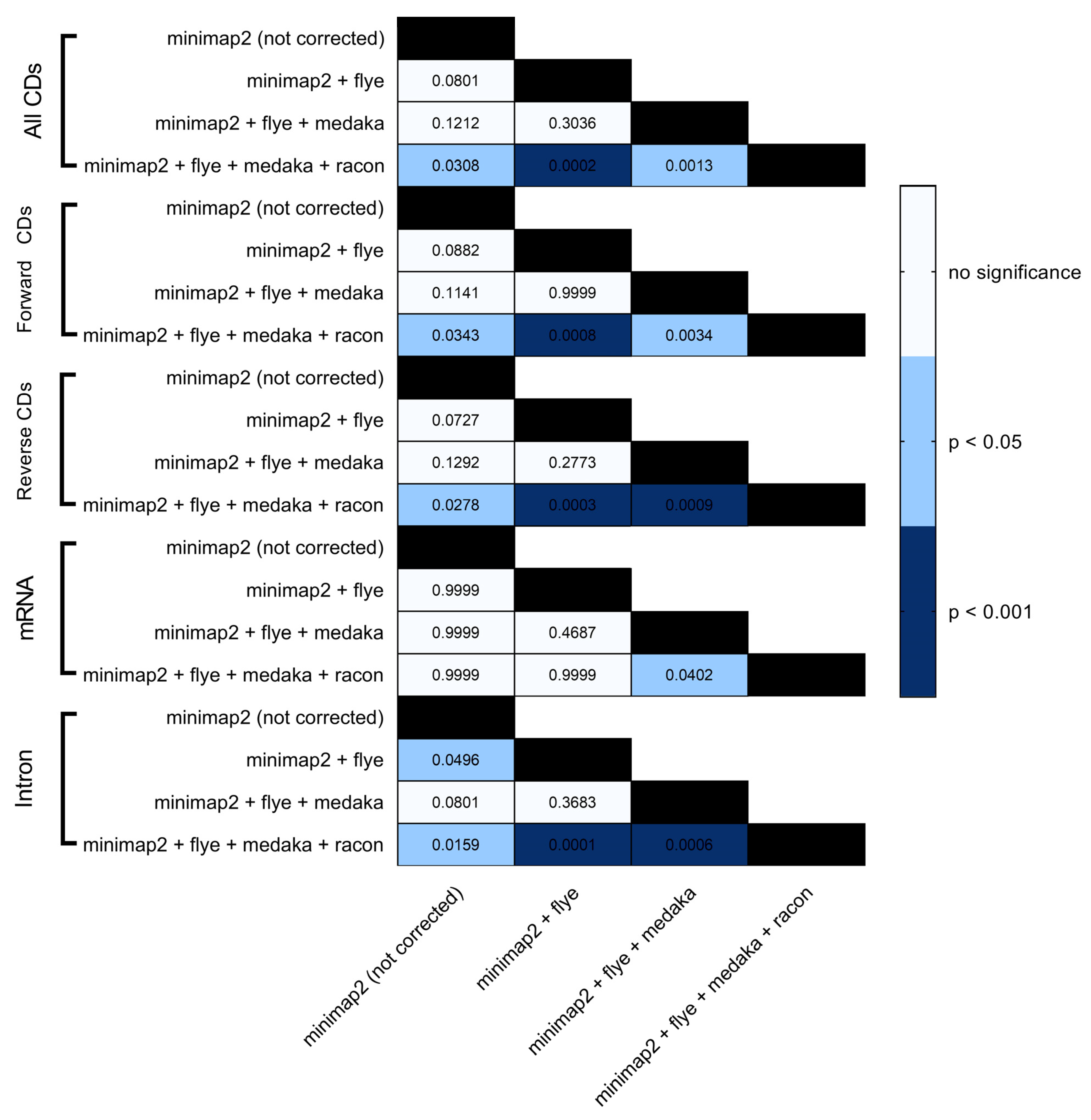
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Runtuwene, L.R.; Tuda, J.S.B.; Mongan, A.E.; Makalowski, W.; Frith, M.C.; Imwong, M.; Srisutham, S.; Thi, L.A.N.; Tuan, N.N.; Eshita, Y.; et al. Nanopore sequencing of drug-resistance-associated genes in malaria parasites, Plasmodium falciparum. Sci. Rep. 2018, 8, 8286. [Google Scholar] [CrossRef] [PubMed]
- Stevanovski, I.; Chintalaphani, S.R.; Gamaarachchi, H.; Ferguson, J.M.; Pineda, S.S.; Scriba, C.K.; Tchan, M.; Fung, V.; Ng, K.; Cortese, A.; et al. Comprehensive genetic diagnosis of tandem repeat expansion disorders with programmable targeted nanopore sequencing. Sci. Adv. 2022, 8, eabm5386. [Google Scholar] [CrossRef] [PubMed]
- Charalampous, T.; Kay, G.L.; Richardson, H.; Aydin, A.; Baldan, R.; Jeanes, C.; Rae, D.; Grundy, S.; Turner, D.J.; Wain, J.; et al. Nanopore metagenomics enables rapid clinical diagnosis of bacterial lower respiratory infection. Nat. Biotechnol. 2019, 37, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Sun, Y.; Yang, Q.; Deng, M.; Yu, Z.; Zhu, G.; Qu, J.; Liu, L.; Yang, L.; Xia, Y. A rapid bacterial pathogen and antimicrobial resistance diagnosis workflow using Oxford nanopore adaptive sequencing method. Brief. Bioinform. 2022, 23, bbac453. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Zeng, W.; Pang, B.; Luo, M.; Peng, Y.; Xu, J.; Kan, B.; Li, Z.; Lu, X. Oxford nanopore long-read sequencing enables the generation of complete bacterial and plasmid genomes without short-read sequencing. Front. Microbiol. 2023, 14, 1179966. [Google Scholar] [CrossRef] [PubMed]
- Salzberg, S.L.; Phillippy, A.M.; Zimin, A.; Puiu, D.; Magoc, T.; Koren, S.; Treangen, T.J.; Schatz, M.C.; Delcher, A.L.; Roberts, M.; et al. GAGE: A critical evaluation of genome assemblies and assembly algorithms. Genome Res. 2011, 22, 557–567. [Google Scholar] [CrossRef]
- Ashton, P.M.; Nair, S.; Dallman, T.; Rubino, S.; Rabsch, W.; Mwaigwisya, S.; Wain, J.; O’Grady, J. MinION nanopore sequencing identifies the position and structure of a bacterial antibiotic resistance island. Nat. Biotechnol. 2014, 33, 296–300. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, Y.; Bollas, A.; Wang, Y.; Au, K.F. Nanopore sequencing technology, bioinformatics and applications. Nat. Biotechnol. 2021, 39, 1348–1365. [Google Scholar] [CrossRef]
- Delahaye, C.; Delahaye, C.; Nicolas, J.; Nicolas, J. Sequencing DNA with nanopores: Troubles and biases. PLoS ONE 2021, 16, e0257521. [Google Scholar] [CrossRef]
- Sutton, J.M.; Millwood, J.D.; McCormack, A.C.; Fierst, J.L. Optimizing experimental design for genome sequencing and assembly with Oxford Nanopore Technologies. Gigabyte 2021, 2021, 1–26. [Google Scholar] [CrossRef]
- Brown, C.L.; Keenum, I.M.; Dai, D.; Zhang, L.; Vikesland, P.J.; Pruden, A. Critical evaluation of short, long, and hybrid assembly for contextual analysis of antibiotic resistance genes in complex environmental metagenomes. Sci. Rep. 2021, 11, 3753. [Google Scholar] [CrossRef] [PubMed]
- Dohm, J.C.; Peters, P.; Stralis-Pavese, N.; Himmelbauer, H. Benchmarking of long-read correction methods. NAR Genom. Bioinform. 2020, 2, lqaa037. [Google Scholar] [CrossRef] [PubMed]
- Cherukuri, Y.; Janga, S.C. Benchmarking of de novo assembly algorithms for Nanopore data reveals optimal performance of OLC approaches. BMC Genom. 2016, 17, 507. [Google Scholar] [CrossRef] [PubMed]
- Juraschek, K.; Borowiak, M.; Tausch, S.H.; Malorny, B.; Käsbohrer, A.; Otani, S.; Schwarz, S.; Meemken, D.; Deneke, C.; Hammerl, J.A. Outcome of Different Sequencing and Assembly Approaches on the Detection of Plasmids and Localization of Antimicrobial Resistance Genes in Commensal Escherichia coli. Microorganisms 2021, 9, 598. [Google Scholar] [CrossRef] [PubMed]
- Wick, R.R.; Holt, K.E. Benchmarking of long-read assemblers for prokaryote whole genome sequencing. F1000Research 2021, 8, 2138. [Google Scholar] [CrossRef] [PubMed]
- Safar, H.A.; Alatar, F.; Nasser, K.; Al-Ajmi, R.; Alfouzan, W.; Mustafa, A.S. The impact of applying various de novo assembly and correction tools on the identification of genome characterization, drug resistance, and virulence factors of clinical isolates using ONT sequencing. BMC Biotechnol. 2023, 23, 26. [Google Scholar] [CrossRef] [PubMed]
- De Coster, W.; D’Hert, S.; Schultz, D.T.; Cruts, M.; Van Broeckhoven, C. NanoPack: Visualizing and processing long-read sequencing data. Bioinformatics 2018, 34, 2666–2669. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- García-Alcalde, F.; Okonechnikov, K.; Carbonell, J.; Cruz, L.M.; Götz, S.; Tarazona, S.; Dopazo, J.; Meyer, T.F.; Conesa, A. Qualimap: Evaluating next-generation sequencing alignment data. Bioinformatics 2012, 28, 2678–2679. [Google Scholar] [CrossRef]
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef]
- Vaser, R.; Sović, I.; Nagarajan, N.; Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 2017, 27, 737–746. [Google Scholar] [CrossRef]
- Mikheenko, A.; Prjibelski, A.; Saveliev, V.; Antipov, D.; Gurevich, A. Versatile genome assembly evaluation with QUAST-LG. Bioinformatics 2018, 34, i142–i150. [Google Scholar] [CrossRef] [PubMed]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Zdobnov, E.M. BUSCO: Assessing Genomic Data Quality and Beyond. Curr. Protoc. 2021, 1, e323. [Google Scholar] [CrossRef] [PubMed]
- Stanke, M.; Schöffmann, O.; Morgenstern, B.; Waack, S. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinform. 2006, 7, 62. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2014, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Hoff, K.J.; Lange, S.; Lomsadze, A.; Borodovsky, M.; Stanke, M. BRAKER1: Unsupervised RNA-Seq-Based Genome Annotation with GeneMark-ET and AUGUSTUS. Bioinformatics 2015, 32, 767–769. [Google Scholar] [CrossRef] [PubMed]
- Hoff, K.J.; Lomsadze, A.; Borodovsky, M.; Stanke, M. Whole-genome annotation with Braker. Methods Mol. Biol. 2019, 1962, 65–95. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef]
- Chen, Z.; Erickson, D.L.; Meng, J. Benchmarking Long-Read Assemblers for Genomic Analyses of Bacterial Pathogens Using Oxford Nanopore Sequencing. Int. J. Mol. Sci. 2020, 21, 9161. [Google Scholar] [CrossRef]
- Cali, D.S.; Kim, J.S.; Ghose, S.; Alkan, C.; Mutlu, O. Nanopore sequencing technology and tools for genome assembly: Computational analysis of the current state, bottlenecks and future directions. Brief. Bioinform. 2018, 20, 1542–1559. [Google Scholar] [CrossRef]
- Lee, J.Y.; Kong, M.; Oh, J.; Lim, J.; Chung, S.H.; Kim, J.-M.; Kim, J.-S.; Kim, K.-H.; Yoo, J.-C.; Kwak, W. Comparative evaluation of Nanopore polishing tools for microbial genome assembly and polishing strategies for downstream analysis. Sci. Rep. 2021, 11, 20740. [Google Scholar] [CrossRef] [PubMed]
- Sigova, E.A.; Pushkova, E.N.; Rozhmina, T.A.; Kudryavtseva, L.P.; Zhuchenko, A.A.; Novakovskiy, R.O.; Zhernova, D.A.; Povkhova, L.V.; Turba, A.A.; Borkhert, E.V.; et al. Assembling Quality Genomes of Flax Fungal Pathogens from Oxford Nanopore Technologies Data. J. Fungi 2023, 9, 301. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, C.-G.; Yang, S.-H.; Wang, X.; Bai, F.-W.; Wang, Z. Benchmarking of long-read sequencing, assemblers and polishers for yeast genome. Brief. Bioinform. 2022, 23, bbac146. [Google Scholar] [CrossRef] [PubMed]
- Siadjeu, C.; Pucker, B.; Viehöver, P.; Albach, D.C.; Weisshaar, B. High Contiguity de novo Genome Sequence Assembly of Trifoliate Yam (Dioscorea dumetorum) Using Long Read Sequencing. Genes 2020, 11, 274. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.C.; Kim, H.; Lee, J.H.; Kim, H.-W.; Park, J.; Choi, B.-S.; Lee, S.-C.; Kim, J.H.; Lee, H.; Kim, S. Nanopore sequencing reads improve assembly and gene annotation of the Parochlus steinenii genome. Sci. Rep. 2019, 9, 5095. [Google Scholar] [CrossRef] [PubMed]
- Hereford, L.M.; Rosbash, M. Number and distribution of polyadenylated RNA sequences in yeast. Cell 1977, 10, 453–462. [Google Scholar] [CrossRef] [PubMed]
- von der Haar, T. A quantitative estimation of the global translational activity in logarithmically growing yeast cells. BMC Syst. Biol. 2008, 2, 87. [Google Scholar] [CrossRef]
- Steward, C.A.; Parker, A.P.J.; Minassian, B.A.; Sisodiya, S.M.; Frankish, A.; Harrow, J. Genome annotation for clinical genomic diagnostics: Strengths and weaknesses. Genome Med. 2017, 9, 49. [Google Scholar] [CrossRef]
- Wingfield, B.D.; Berger, D.K.; Coetzee, M.P.A.; Duong, T.A.; Martin, A.; Pham, N.Q.; Berg, N.v.D.; Wilken, P.M.; Arun-Chinnappa, K.S.; Barnes, I.; et al. IMA genome-F17. IMA Fungus 2022, 13, 19. [Google Scholar] [CrossRef]
- Goldstein, S.; Beka, L.; Graf, J.; Klassen, J.L. Evaluation of strategies for the assembly of diverse bacterial genomes using MinION long-read sequencing. BMC Genom. 2019, 20, 23. [Google Scholar] [CrossRef]
- Chen, Z.; Erickson, D.L.; Meng, J. Benchmarking hybrid assembly approaches for genomic analyses of bacterial pathogens using Illumina and Oxford Nanopore sequencing. BMC Genom. 2020, 21, 631. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chen, K.; Ren, Q.; Zhang, Y.; Liu, J.; Wang, G.; Liu, A.; Li, Y.; Liu, G.; Luo, J.; et al. Systematic Comparison of the Performances of De Novo Genome Assemblers for Oxford Nanopore Technology Reads From Piroplasm. Front. Cell. Infect. Microbiol. 2021, 11, 696669. [Google Scholar] [CrossRef] [PubMed]
- Schiavone, A.; Pugliese, N.; Samarelli, R.; Cumbo, C.; Minervini, C.F.; Albano, F.; Camarda, A. Factors Affecting the Quality of Bacterial Genomes Assemblies by Canu after Nanopore Sequencing. Appl. Sci. 2022, 12, 3110. [Google Scholar] [CrossRef]
- Deamer, D.; Akeson, M.; Branton, D. Three decades of nanopore sequencing. Nat. Biotechnol. 2016, 34, 518–524. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Li, H.; Ma, S.; Cao, J.; Liao, H.; Huang, Q.; Chen, W. The newest Oxford Nanopore R10.4.1 full-length 16S rRNA sequencing enables the accurate resolution of species-level microbial community profiling. Appl. Environ. Microbiol. 2023, 89, e0060523. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.; Liu, X.; Simeneh, Z.M.; Yang, M.; Li, R. Benchmarking of Nanopore R10.4 and R9.4.1 flow cells in single-cell whole-genome amplification and whole-genome shotgun sequencing. Comput. Struct. Biotechnol. J. 2023, 21, 2352–2364. [Google Scholar] [CrossRef] [PubMed]
- Sereika, M.; Kirkegaard, R.H.; Karst, S.M.; Michaelsen, T.Y.; Sørensen, E.A.; Wollenberg, R.D.; Albertsen, M. Oxford Nanopore R10.4 long-read sequencing enables the generation of near-finished bacterial genomes from pure cultures and metagenomes without short-read or reference polishing. Nat. Methods 2022, 19, 823–826. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, T.; Zhang, S.; Zhang, L.; Li, Q.; Lv, Q.; Kong, D.; Jiang, H.; Ren, Y.; Jiang, Y.; et al. Clinical evaluation of metagenomic next-generation sequencing in unbiased pathogen diagnosis of urinary tract infection. J. Transl. Med. 2023, 21, 762. [Google Scholar] [CrossRef]
- Panthee, S.; Hamamoto, H.; Ishijima, A.S.; Paudel, A.; Sekimizu, K. Utilization of Hybrid Assembly Approach to Determine the Genome of an Opportunistic Pathogenic Fungus, Candida albicans TIMM 1768. Genome Biol. Evol. 2018, 10, 2017–2022. [Google Scholar] [CrossRef]
- Rizzo, M.; Soisangwan, N.; Vega-Estevez, S.; Price, R.J.; Uyl, C.; Iracane, E.; Shaw, M.; Soetaert, J.; Selmecki, A.; Buscaino, A. Stress combined with loss of the Candida albicans SUMO protease Ulp2 triggers selection of aneuploidy via a two-step process. PLoS Genet. 2022, 18, e1010576. [Google Scholar] [CrossRef]
- Schotanus, K.; Heitman, J. Centromere deletion in Cryptococcus deuterogattii leads to neocentromere formation and chromosome fusions. eLife 2020, 9, e56026. [Google Scholar] [CrossRef] [PubMed]
- Farrer, R.A.; Chang, M.; Davis, M.J.; van Dorp, L.; Yang, D.-H.; Shea, T.; Sewell, T.R.; Meyer, W.; Balloux, F.; Edwards, H.M.; et al. A New Lineage of Cryptococcus gattii (VGV) Discovered in the Central Zambezian Miombo Woodlands. mBio 2019, 10, e02306–e02319. [Google Scholar] [CrossRef] [PubMed]
- Salazar, A.N.; de Vries, A.R.G.; Broek, M.v.D.; Wijsman, M.; Cortés, P.d.l.T.; Brickwedde, A.; Brouwers, N.; Daran, J.-M.G.; Abeel, T. Nanopore sequencing enables near-complete de novo assembly of Saccharomyces cerevisiae reference strain CEN.PK113-7D. FEMS Yeast Res. 2017, 17, fox074. [Google Scholar] [CrossRef] [PubMed]
- Dans, M.G.; Piirainen, H.; Nguyen, W.; Khurana, S.; Mehra, S.; Razook, Z.; Geoghegan, N.D.; Dawson, A.T.; Das, S.; Schneider, M.P.; et al. Sulfonylpiperazine compounds prevent Plasmodium falciparum invasion of red blood cells through interference with actin-1/profilin dynamics. PLoS Biol. 2023, 21, e3002066. [Google Scholar] [CrossRef]
- De Meulenaere, K.; Cuypers, W.L.; Gauglitz, J.M.; Guetens, P.; Rosanas-Urgell, A.; Laukens, K.; Cuypers, B. Selective whole-genome sequencing of Plasmodium parasites directly from blood samples by nanopore adaptive sampling. mBio 2023, e0196723. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Correction Tool | Minimap2 (Not Corrected) | Flye | Flye + Medaka | Flye + Medaka + Racon | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Total Length (bp) | Total Aligned (bp) | GC% | Total Length (bp) | Total Aligned (bp) | GC% | Total Length (bp) | Total Aligned (bp) | GC% | Total Length (bp) | Total Aligned (bp) | GC% | ||
| C. albicans | Sample 1 | 14268731 | 14255757 | 33.45 | 14272767 | 14231426 | 33.49 | 14317735 | 14250916 | 33.43 | 14319429 | 14255001 | 33.43 |
| Sample 2 | 14251618 | 14238188 | 33.46 | 14298244 | 14246769 | 33.5 | 14341847 | 14262678 | 33.43 | 14356382 | 14250132 | 33.42 | |
| Sample 3 | 14275154 | 14217242 | 33.42 | 14240646 | 14111615 | 33.51 | 14320530 | 14166519 | 33.38 | 14312009 | 14138998 | 33.4 | |
| Sample 4 | 14280549 | 14226612 | 33.4 | 14263763 | 14211021 | 33.34 | 14345200 | 14272900 | 33.2 | 14318448 | 14241382 | 33.17 | |
| Sample 5 | 14268190 | 14182812 | 33.4 | 14218801 | 14102192 | 33.48 | 14287333 | 14155066 | 33.33 | 14304631 | 14158562 | 33.29 | |
| Sample 6 | 14267575 | 14183870 | 33.41 | 14206012 | 14097160 | 33.5 | 14265963 | 14144176 | 33.37 | 14275276 | 14126308 | 33.33 | |
| median | 14268460.5 | 14221927 | 33.415 | 14252204.5 | 14161318 | 33.495 | 14319132.5 | 14208717.5 | 33.375 | 14315228.5 | 14199972 | 33.365 | |
| C. gattii | Sample 1 | 18374056 | 13963456 | 47.95 | 15618076 | 3018791 | 45.87 | 15848723 | 1127999 | 45.39 | 15649875 | 979468 | 45.62 |
| Sample 2 | 18373936 | 16738202 | 47.87 | 17275771 | 2829797 | 47.74 | 17401496 | 3193078 | 47.65 | 17335832 | 2478663 | 47.64 | |
| Sample 3 | 18373817 | 16748750 | 47.87 | 17249122 | 2811973 | 47.77 | 17403823 | 2993154 | 47.66 | 17331969 | 2314973 | 47.69 | |
| Sample 4 | 18373586 | 16911300 | 47.86 | 17292994 | 3406667 | 47.78 | 17435803 | 3916947 | 47.7 | 17395149 | 2892625 | 47.71 | |
| Sample 5 | 18371784 | 17309929 | 47.88 | 17667739 | 10488842 | 47.95 | 17746664 | 10916558 | 47.91 | 17719423 | 10110195 | 47.82 | |
| Sample 6 | 18374011 | 15590434 | 47.88 | 17093485 | 3649510 | 47.47 | 17283501 | 3129355 | 47.09 | 17341085 | 2347707 | 47.07 | |
| median | 18373876.5 | 16743476 | 47.875 | 17262446.5 | 3212729 | 47.755 | 17402659.5 | 3161216.5 | 47.655 | 17338458.5 | 2413185 | 47.665 | |
| S. cerevisiae | Sample 1 | 11900917 | 11786751 | 38.26 | 11756094 | 11627598 | 38.37 | 11762061 | 11614289 | 38.27 | 11770518 | 11610040 | 38.24 |
| Sample 2 | 11927452 | 11786979 | 38.22 | 11817583 | 11391169 | 38.31 | 11835515 | 11392663 | 38.24 | 11841970 | 11389993 | 38.23 | |
| Sample 3 | 11867150 | 11717686 | 38.28 | 11714984 | 11611725 | 38.37 | 11728646 | 11591392 | 38.3 | 11734569 | 11542743 | 38.2 | |
| Sample 4 | 12048365 | 11746218 | 38.27 | 11701491 | 11557641 | 38.31 | 11744219 | 11530244 | 38.26 | 11726032 | 11472823 | 38.12 | |
| Sample 5 | 11848014 | 11728342 | 38.26 | 11844556 | 11579727 | 38.37 | 11847283 | 11568021 | 38.25 | 11841609 | 11544386 | 38.21 | |
| Sample 6 | 11898828 | 11680204 | 38.27 | 11650537 | 11518215 | 38.35 | 11683391 | 11519687 | 38.23 | 11676382 | 11483435 | 38.13 | |
| median | 11899872.5 | 11737280 | 38.265 | 11735539 | 11568684 | 38.36 | 11753140 | 11549132.5 | 38.255 | 11752543.5 | 11513089 | 38.205 | |
| P. falciparum | Sample 1 | 23184099 | 23030452 | 19.3 | 22783133 | 22726603 | 19.63 | 23110345 | 23037187 | 19.36 | 23277887 | 23197642 | 19.16 |
| Sample 2 | 23244418 | 23191818 | 19.33 | 22846745 | 22827099 | 19.64 | 23103471 | 23077430 | 19.44 | 23251109 | 23206304 | 19.29 | |
| Sample 3 | 23278091 | 23119804 | 19.27 | 22794879 | 22740838 | 19.59 | 23071068 | 22992830 | 19.36 | 23170782 | 23115122 | 19.2 | |
| Sample 4 | 23266743 | 23186289 | 19.33 | 22843636 | 22817074 | 19.64 | 23082452 | 23052262 | 19.44 | 23222395 | 23183221 | 19.29 | |
| Sample 5 | 23167744 | 22187311 | 19.55 | 22597393 | 22360095 | 19.53 | 22902857 | 22526387 | 19.36 | 22879437 | 22148919 | 19.29 | |
| Sample 6 | 23193836 | 20645915 | 19.63 | 21278952 | 20848467 | 19.64 | 22021131 | 21099604 | 19.32 | 21995137 | 20265232 | 19.27 | |
| median | 23219127 | 23075128 | 19.33 | 22789006 | 22733720.5 | 19.635 | 23076760 | 23015008.5 | 19.36 | 23196588.5 | 23149171.5 | 19.28 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Safar, H.A.; Alatar, F.; Mustafa, A.S. Three Rounds of Read Correction Significantly Improve Eukaryotic Protein Detection in ONT Reads. Microorganisms 2024, 12, 247. https://doi.org/10.3390/microorganisms12020247
Safar HA, Alatar F, Mustafa AS. Three Rounds of Read Correction Significantly Improve Eukaryotic Protein Detection in ONT Reads. Microorganisms. 2024; 12(2):247. https://doi.org/10.3390/microorganisms12020247
Chicago/Turabian StyleSafar, Hussain A., Fatemah Alatar, and Abu Salim Mustafa. 2024. "Three Rounds of Read Correction Significantly Improve Eukaryotic Protein Detection in ONT Reads" Microorganisms 12, no. 2: 247. https://doi.org/10.3390/microorganisms12020247
APA StyleSafar, H. A., Alatar, F., & Mustafa, A. S. (2024). Three Rounds of Read Correction Significantly Improve Eukaryotic Protein Detection in ONT Reads. Microorganisms, 12(2), 247. https://doi.org/10.3390/microorganisms12020247