1. Introduction
Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2), first detected in Wuhan, China, in Winter 2019, was identified as the causative agent for the COVID-19 pandemic and is responsible for more than 773 million confirmed infections and close to 7 million deaths worldwide, as of December 2023 [
1]. The first isolation of the novel agent, which took place on 7 January 2020, and sharing of its genetic sequence with the public by the Chinese authorities [
2] led to the rapid development of numerous specific PCR-based diagnostic assays.
With respect to the scientific nomenclature, the World Health Organization (WHO) has developed criteria to classify SARS-CoV-2 variants based on their potential for increased transmissibility, virulence, clinical disease presentation, and the effectiveness of public health measures or available diagnostics, vaccines, and therapeutics [
3]. The WHO uses a Greek nomenclature and differentiates between Variants Under Monitoring (VUMs), Variants of Interest (VOIs), and Variants of Concern (VOCs).
On 14 December 2020, the United Kingdom reported the first SARS-CoV-2 VOC, the Alpha variant, lineage B.1.1.7, and estimated its emergence back to September 2020 [
4]. Among other notable genetic alterations that were found to increase viral binding affinity with angiotensin-converting-enzyme 2 receptor (N501Y) [
5] or facilitate epithelial cell entry (P681H) [
6], deleterious mutations at positions H69/V70 were the initial clue to the new lineage [
7]. These deletions affected a widely used polymerase chain reaction assay, the TaqPathTM COVID-19 CE-IVD RT-PCR Kit (ThermoFisher, Luzern, Switzerland), preventing the amplification of the S-gene target, resulting in S-gene target failure (SGTF). While the TaqPath Kit targets three different SARS-CoV-2 genes, the Open Reading Frame 1ab (ORF1ab), the nucleocapsid (N), and the spike (S) gene, a positive result is given with the faultless amplification of two out of three target sequences. After the discovery of the SGTF signature, positive samples presenting without spike target signals then quickly became synonymous with the Alpha variant and were subsequently used as a proxy for its presence as the worldwide prevalence increased [
7,
8,
9,
10,
11,
12,
13].
With the emergence of the B.1.617.2 lineage, designated as Delta by the WHO [
14] and lacking the ΔH69/V70, triple-gene positives started to increase again in frequency after a long period of Alpha dominance, where more than 90% of samples presented with the SGTF signature [
15,
16]. While there were still a few SGTF-negative lineages in circulation at that time, an increase in triple-gene positives could nevertheless be attributed to the Delta variant, and the absence of the SGTF was subsequently used as a proxy for its proportion among the different lineages [
15,
16].
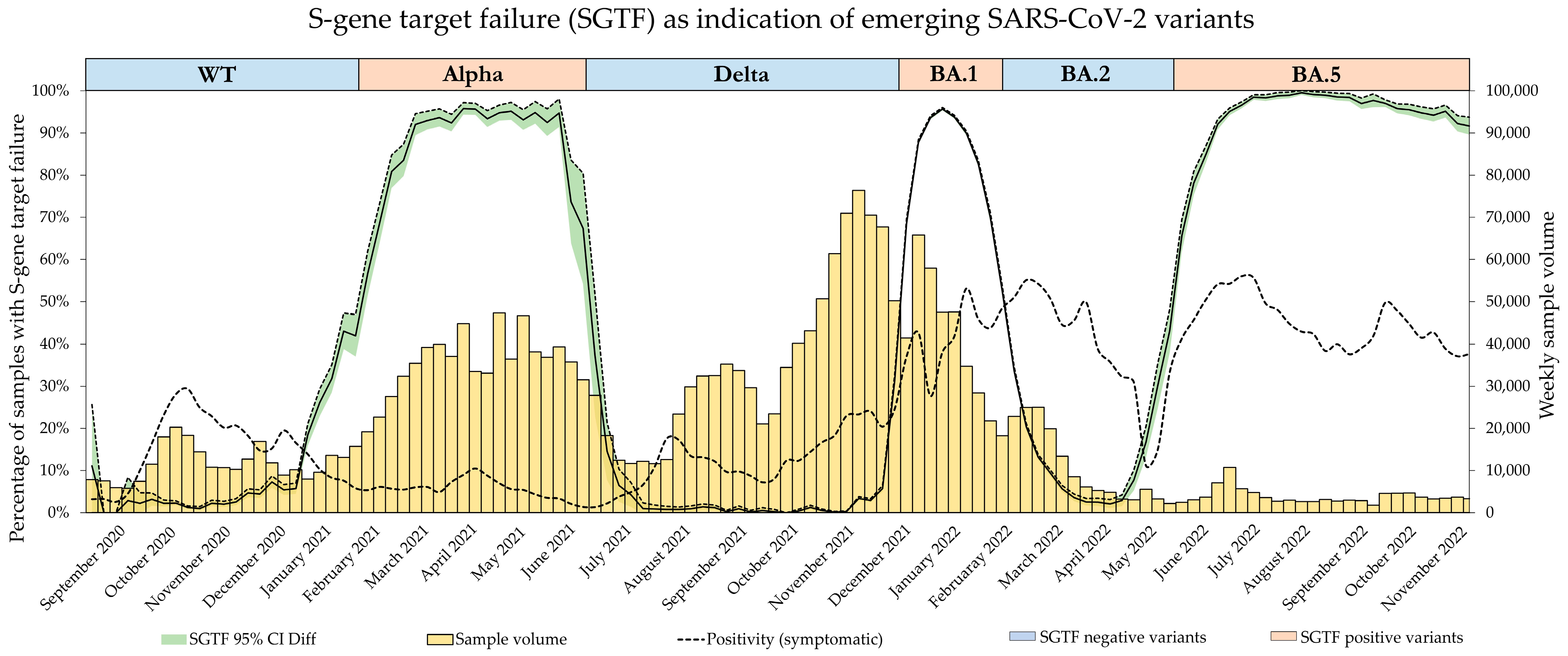
The emergence of the Omicron variants BA.1, BA.2, and BA.4/BA.5 then followed this alternating pattern of S-gene amplification presence and absence.
While the SGTF signature was extensively used as a surrogate for different VOCs during the pandemic, systematic studies on its accuracy and effectiveness as a surveillance tool are scarce. In this study, we therefore investigated the diagnostic accuracy of the absence and presence of the SGTF signature as a proxy for dominant variants throughout the pandemic in six regions of Switzerland and the principality of Liechtenstein.
2. Materials and Methods
2.1. Setting
Throughout the pandemic, Dr. Risch laboratories served all six regions of Switzerland as well as the principality of Liechtenstein with SARS-CoV-2 PCR testing. Referred samples originated from every canton and were mostly nasopharyngeal swabs or saliva samples. Alongside other testing methods, routine PCR testing was performed using the TaqPath COVID-19 CE-IVD RT-PCR Kit by ThermoFisher Scientific, Lucerne, Switzerland (TaqPath). All positive samples, starting from calendar week 37 of 2020 up to calendar week 47 of 2022, tested with the TaqPath Kit were included in the study, spanning a period encompassing the SARS-CoV-2 variant waves of B.1.1.7 (Alpha), B.1.617.2 (Delta), as well as Omicron variants BA.1, BA.2, and BA.4/5. Omicron variants BA.4 and BA.5 were summarized due to their concurrent presence and identical SGTF pattern.
2.2. SARS-CoV-2 RT-PCR
The TaqPath Kit was used in a high-throughput manner, and tests were performed according to the manufacturer’s instructions with either the QuantstudioTM 5 or the QuantstudioTM 7 qPCR System (Amplitude Solution) using the MagMax Viral/Pathogen Nucleic Acid Isolation Kit (ThermoFisher Scientific, Lucerne, Switzerland). Only samples tested using the TaqPath Kit were included in this study. This kit targets the genes for ORF1ab, the nucleocapsid protein (N-gene), as well as the spike protein (S-gene). As per the manufacturer’s instructions, samples were reported as positive when the detection of at least two out of these three target genes was achieved with a cycle threshold (Ct) value less than 37.
2.3. S-Gene Target Failure (SGTF) Definition
As a diagnostic criterium, complete S-gene target failure (cSGTF) was defined as the absence of S-gene target amplification in the presence of the faultless detection of ORF1ab- and N-gene amplicons. A partial SGTF (pSGTF) was then specified to present a diminished amplification of the S-gene (higher Ct-value) compared to the coherent detection of ORF1ab- and N-gene amplicons. For this, the S-gene shift was calculated for each SGTF-negative sample as follows:
Samples exhibiting a significant S-gene shift were specified as pSGTF, and an empirical approach was used to differentiate between unspecific pSGTF and “real” pSGTF to establish a reasonable cutoff value for sensitivity and specificity calculations.
2.4. Confirmation of Variants of Concern (VOCs)
Only samples with a Ct value < 30 were considered for analysis. This cutoff was defined as such because (i) samples with Ct > 30 have the potential to produce unspecific target failures in any of the three target amplifications (see
Supplementary Material Figure S1), and (ii) whole-genome sequencing with adequate coverage is rarely achievable in samples with Ct > 30. Variant confirmation was then performed with mutation-specific PCR (VirSNiP, TIB MOLBIOL, Berlin, Germany) or whole-genome sequencing (WGS). WGS was performed using a GridIon nanopore sequencer (Oxford Nanopore Technologies, Oxford, UK) for approximately 24 h and according to the manufacturer’s instructions using the ARTIC and Midnight protocols [
17]. Amplification was performed with the most recent primer versions to address potential sequence dropouts due to the evolution of the viral genome. Analysis was based on the ARTIC pipeline. All genomic sequences were analyzed using the Pangolin COVID-19 Lineage Assigner (version v4.3, pangolin-data version v1.20) [
18] based on the Pangolin nomenclature [
19]. The VirSNiP Assays used are listed in
Table 1.
Since all dominant SARS-CoV-2 variants in this time span showed an alternating pattern of SGTF presence and absence, variant confirmation was performed when the frequency of either one started to increase again after the steady-state period of the previously dominant variant (e.g., with the Alpha variant being dominant during the beginning of 2021, stable frequencies of SGTF at a high level were observed. The emergence of the Delta variant, however, increased the number of samples without SGTF, which triggered variant confirmation by decreasing the frequency of SGTF). Frequency surveillance was performed on a weekly basis.
2.5. Logistic Growth Rates and Sigmoid’s Midpoint
For a comparison of the different variant waves, a logistic model was fitted to the data corresponding to the introduction of each variant. In this case, Sigmoid’s midpoint (t
0) describes the time value (
x-axis) where 50% of the circulating virus belongs to the variant of interest, while the logistic growth rate (α) describes the slope at this point. The parameters α and t
0 were estimated according to Chen et al. [
21] with help of the nonlinear regression function in MedCalc
® statistical software v20.027 according to the equation for logistic regression:
where α is the logistic growth rate, and t
0 is the sigmoid’s midpoint.
A logistic model was fitted to the frequency of SGTF per week for each SGTF-positive variant (i.e., Alpha, Omicron BA.1, and Omicron BA.4/BA.5). Inversely, 1-SGTF was used as a basis for a logistic model fitted to SGTF-negative variants (i.e., Delta and Omicron BA.2). With the help of the above logistic regression formula, α and t
0 were estimated. The models were calculated over the periods reported in
Table 2.
2.6. Data Collection and Analysis
According to Article 2 of the Swiss Federal Act on Research involving Human Beings, an analysis on anonymized biological material and anonymized health data does not qualify as research in a strict sense of the law, and approval of a cantonal ethics commission as well as informed consent can thus be waived. Only data pertaining to sampling date and time, as well as the sampling place (canton) corresponding to the sampled materials, were used for this analysis, meaning our study was conducted without the use of any personal information of patients.
Descriptive statistics were obtained using Microsoft Excel v2312(Microsoft, Seattle, WA, USA), while Medcalc (Mariakerke, Belgium) was used for computations.
2.7. Comparison with GISAID
As the main goal of this study was to evaluate the effectiveness of the SGTF as a tool for estimating the proportion of different SARS-CoV-2 variants in real time, we compared our data to data from the Global Initiative on Sharing All Influenza Data (GISAID), as accumulated sequencing results on GISAID are most likely the best approximation to real-world variant proportion.
For this, CoV-Spectrum, enabled by data from GISAID, was accessed on the 27 February 2023. We only searched for data from Switzerland. The results for B.1.1.529.4* and B.1.1.529.5* were combined, as both exhibit the SGTF and were collectively responsible for the corresponding wave. The search terms and corresponding time periods used are presented in
Table 3.
Subsequently, data for proportion from the “sequences over time” and the “international comparison” tab were downloaded and used for comparison.
4. Discussion
We extensively investigated the accuracy and effectiveness of the SGTF signature and its absence as a surrogate marker for the emergence of the Alpha and Delta variants, as well as the Omicron variants BA.1, BA.2, and BA.4/BA.5. We determined the sensitivity and specificity of complete SGTF to be 93.2% [92.7%, 93.7%] and 99.3% [99.2%, 99.5%], respectively. We have also shown that classifying SGTF-negative samples with S-gene shift (pSGTF) as complete SGTF increased sensitivity and defined the best cutoff value at pSGTF > 3.8 Ct-values. This has already been reported by Borges V et al. [
26], but no clear cutoff value had been established previously. By establishing this cutoff value, accuracy, sensitivity, and specificity were improved to 98.5% [98.3%, 98.7%], 98.2% [97.9% to 98.4%], and 98.9% [98.6% to 99.1%], respectively.
Many publications have evaluated different approaches to determining the accuracy and applicability of the SGTF in variant detection [
27,
28,
29,
30,
31,
32,
33]. In our study, the sensitivity and specificity values based on WGS VOC determination were in line with previously reported values [
34]; our calculations based on variant-specific PCR, however, were consistently lower than for WGS, indicating a reduced accuracy in lineage classification.
Additionally, weekly logistic growth rates and Sigmoid’s midpoint were estimated for each variant wave based on SGTF data and did not significantly differ to estimates based on comprehensive data from GISAID for the Alpha, Delta, BA.2, and BA.4/BA.5 variants. However, the estimates for Omicron variant BA.1 showed clear deviations for growth rate, while the estimates for Sigmoid’s midpoint were significantly different. This deviation is visible in
Figure 5, as the BA.1 estimate based on SGTF data lags the GISAID estimate by 1 week during calendar weeks 49–51 of 2021. Although the samples analyzed in this study originated from all over Switzerland, the distribution was not equal among all regions and cantons, and it is possible that the very fast emergence of the Omicron BA.1 variant, coupled with potentially low testing coverage in certain regions, might have introduced bias in our data during this time. Generally, the variant proportion estimates based on SGTF or GISAID agreed well, and considering two weeks delay for sequencing results, the upside of using such a surrogate marker during a fast-paced pandemic seems obvious. This, however, is owed to the specific dynamics we witnessed during the SARS-CoV-2 pandemic, as dominant variants emerged quickly, while non-dominant variants had a very low prevalence in general. The highest proportion of non-dominant variants was observed during the Alpha period, where proportion estimates for SGTF and GISAID diverge the most. However, this did not have a significant impact on the sigmoid function and, therefore, the variant dynamics estimates.
The greatest sample volume was experienced during the dominance of the Omicron BA.1 variant. Due to its short period of dominance, the Delta, Omicron BA.1, and Omicron BA.2 variants were present simultaneously at its peak. Since the SGTF-positive samples almost certainly belonged to the Omicron BA.1 variant, we extensively confirmed the SGTF-negative samples to distinguish between the vanishing Delta variant and the newly emerging Omicron BA.2 variant. The unprecedented number of samples has prompted us to favor mutation-specific PCR for VOC confirmation as it is less time-consuming than WGS. With the increase in samples, however, there was also an increase in samples presenting with pSGTF, and through the extensive VOC confirmation of the SGTF-negative samples, we introduced bias into our data. This is clearly noticeable in
Table S4, where sensitivity and specificity for Omicron BA.2 were 100.0% [99.8%, 100.0%] and 5.5% [3.6%, 8.1%], respectively, for SNP confirmation, while WGS confirmation was in line with that of the other variants. This low specificity can therefore be explained by the extensive variant confirmation of the SGTF-negative samples during the period with the highest sample volume, leading to a relative increase in confirmed samples with S-gene shift. The increase in specificity to 70.9% [66.4%, 75.1%] for pSGTF > 3.8 supports this observation and shows the importance of distinguishing between true triple positives and pSGTF. Importantly, WGS did not reveal any additional mutations that might be causative for pSGTF. However, pSGTF was found to occur more frequently in high-viral-load samples, suggesting low frequent primer/probe mismatch if high numbers of S-gene amplicons are in the reaction. This observation is in agreement with previous reports from Portugal [
26]. Further research is needed to precisely determine the molecular mechanism behind pSGTF.
The reasons behind the alternating pattern of SGTF and non-SGTF dominant variants remain inadequately understood, and no real evidence for as to why this switching was favored by viral evolution has been presented. In 2021, ΔH69/V70 in the spike glycoprotein was found to have arisen independently at least 13 times [
35]. In vitro studies have demonstrated that this deletion does increase infectivity by higher levels of spike incorporation into virions and that it mediates faster fusion than the wild type [
36]. It does not, however, significantly reduce the sensitivity of neutralizing antibodies from recovered individuals and is probably not an immune escape mechanism [
37]. Recently, it was also reported that the SGTF can help identify the development of sub-lineages within a patient with persistent SARS-CoV-2 infection [
38].
Our study has some limitations. First, we only investigated samples with relatively high viral load (Ct < 30); therefore, our study may be biased towards these types of samples. Second, while the TaqPath Kit was the main method used for SARS-CoV-2 PCR testing, testing was also performed with multiple other platforms; therefore, not all potential SGTF samples were included in the study. Third, the TaqPath Kit is a commercially available PCR assay that does not grant flexibility for primer design or primer binding regions or knowledge thereof. Therefore, only assumptions about the primer/probe binding sites as well as the location of ΔH69/V70 on these sites are possible. Based on our data, however, it seems that, at least for the S-gene, the test design allows for a certain amount of mismatch so that detectable PCR signals can still be produced with high numbers of S-gene amplicons in the reaction.
Importantly, this study also demonstrates that multiple target assays are critical in PCR diagnostics. Diagnostic targets may be under an evolutionary pressure if a lot of cases occur, and pattern observation can therefore help in observing diagnostic driven evolution.
In conclusion, in this paper, we have reported the SGTF signature as a surrogate marker with high predictability for SARS-CoV-2 variants that acquired ΔH69/V70. Since SGTF surveillance relies on a diagnostic RT-PCR test, it allows for faster turnaround times with higher throughput while being less expensive than genomic sequencing. It is limited by the fact that no mutations other than ΔH69/V70 are being assessed, and specific lineage classification by WGS is therefore still required. It also relies on a continued alternating pattern of SGTF-positive and SGTF-negative dominant variants. Despite its limitations, the SGTF signature allowed us to make exceptionally accurate and fast predictions about the changing dynamics of the SARS-CoV-2 pandemic. Continued monitoring in combination with genomic sequencing will likely prove useful in the future.


 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}