
Differential Microbial Composition and Interkingdom Interactions in the Intestinal Microbiota of Holstein and German Simmental × Holstein Cross F1 Calves: A Comprehensive Analysis of Bacterial and Fungal Diversity

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Experimental Procedure
2.2. Sample Collection
2.3. DNA Extraction and PCR Amplification
2.4. Bioinformatics Analysis
2.5. Statistical Analysis
2.6. Predicted Function of the Microbiome
3. Results
3.1. Birth Traits and Diarrhea Incidence in Various Cattle Breeds
3.2. Bacterial Diversity in Fecal Microbiota of Different Breeds of Calves
3.2.1. Sequencing Statistics and Bacterial Diversity
3.2.2. Taxonomic Composition and Differences in Fecal Bacterial Community
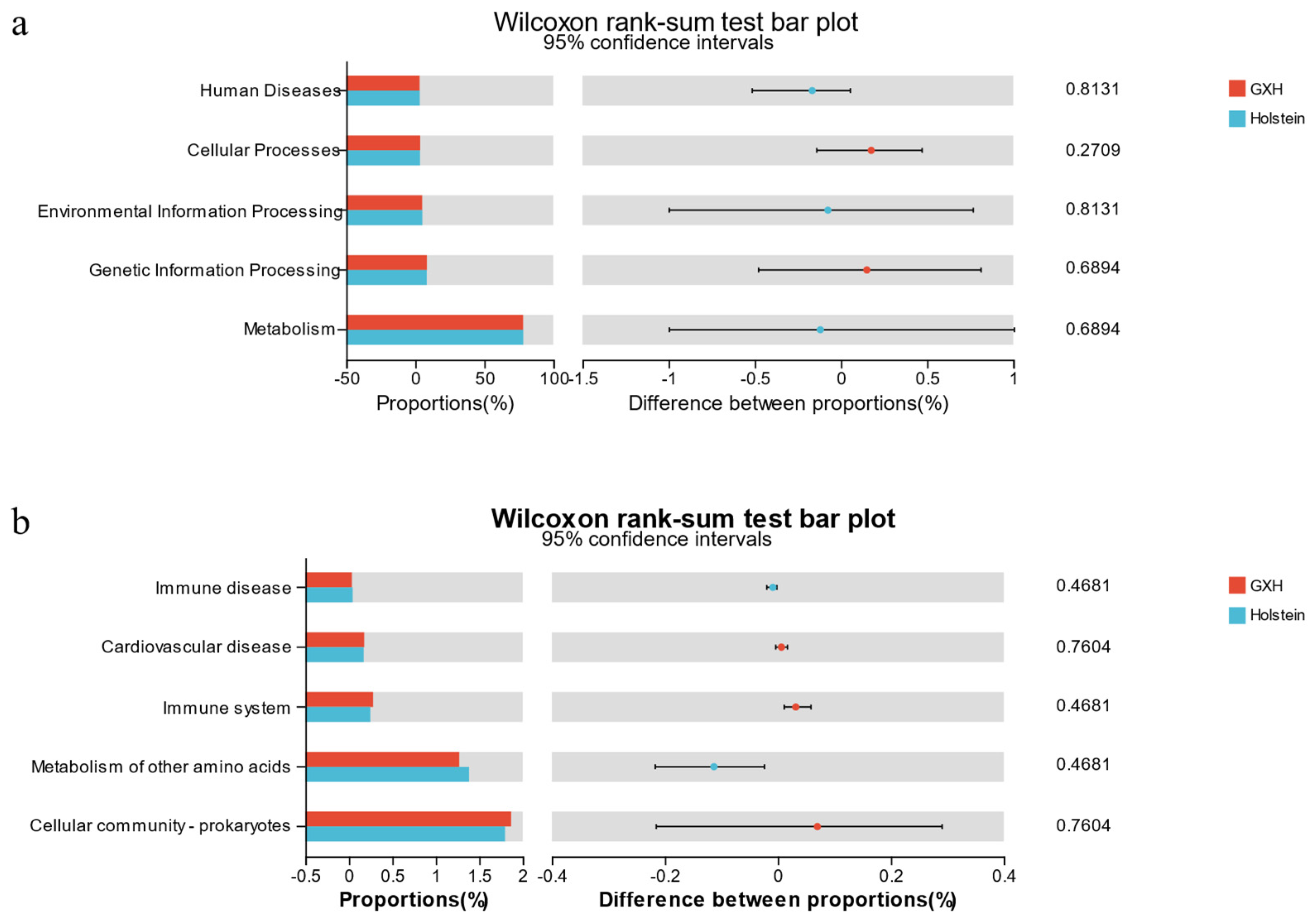
3.2.3. Functional Predictions of the Rectal Microbiota Using PICRUSt
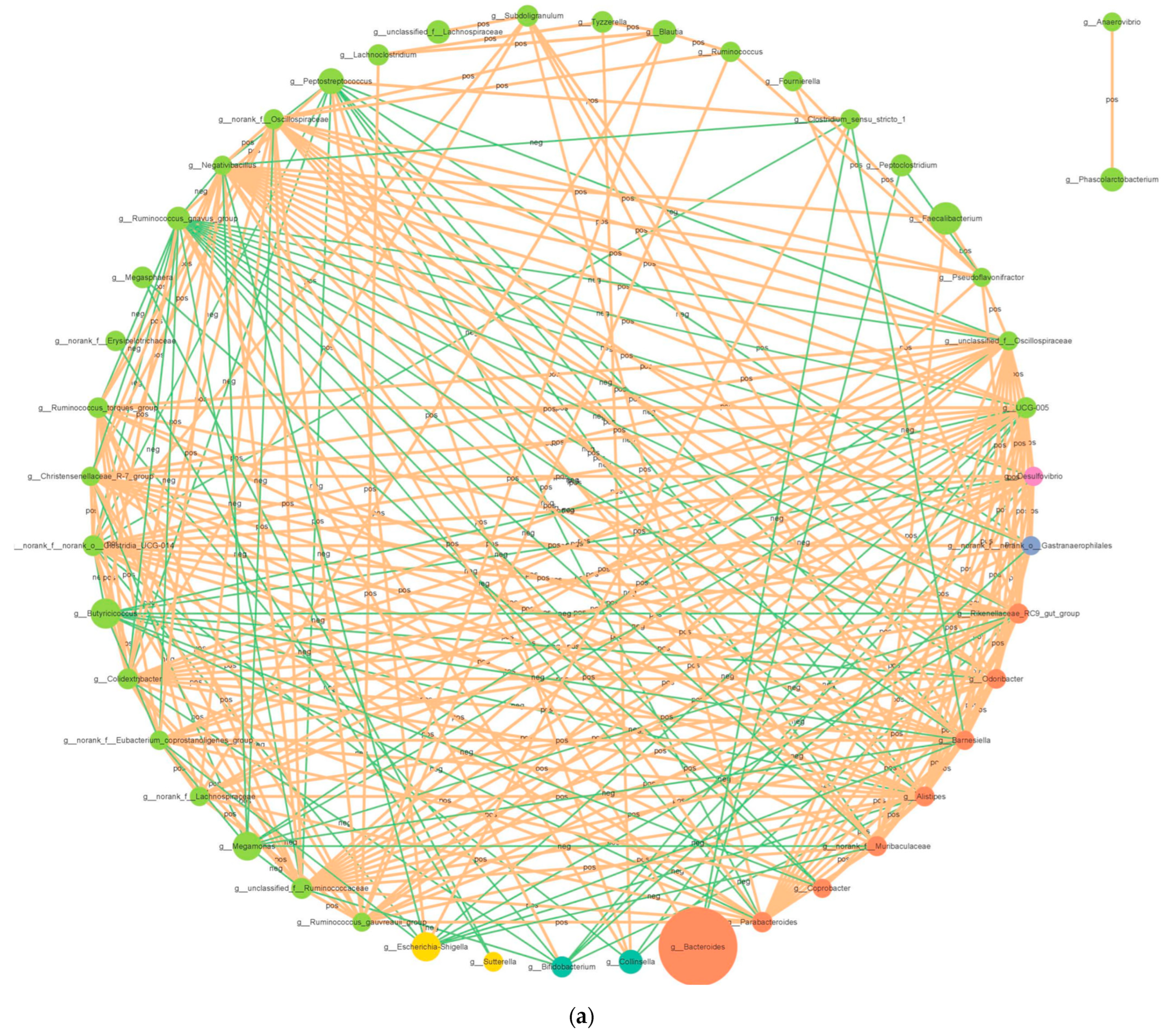
3.2.4. Correlation Network Analysis
3.3. Fungal Diversity in the Fecal Microbiota of Different Breeds of Calves
3.3.1. Sequencing Statistics and Fungal Diversity
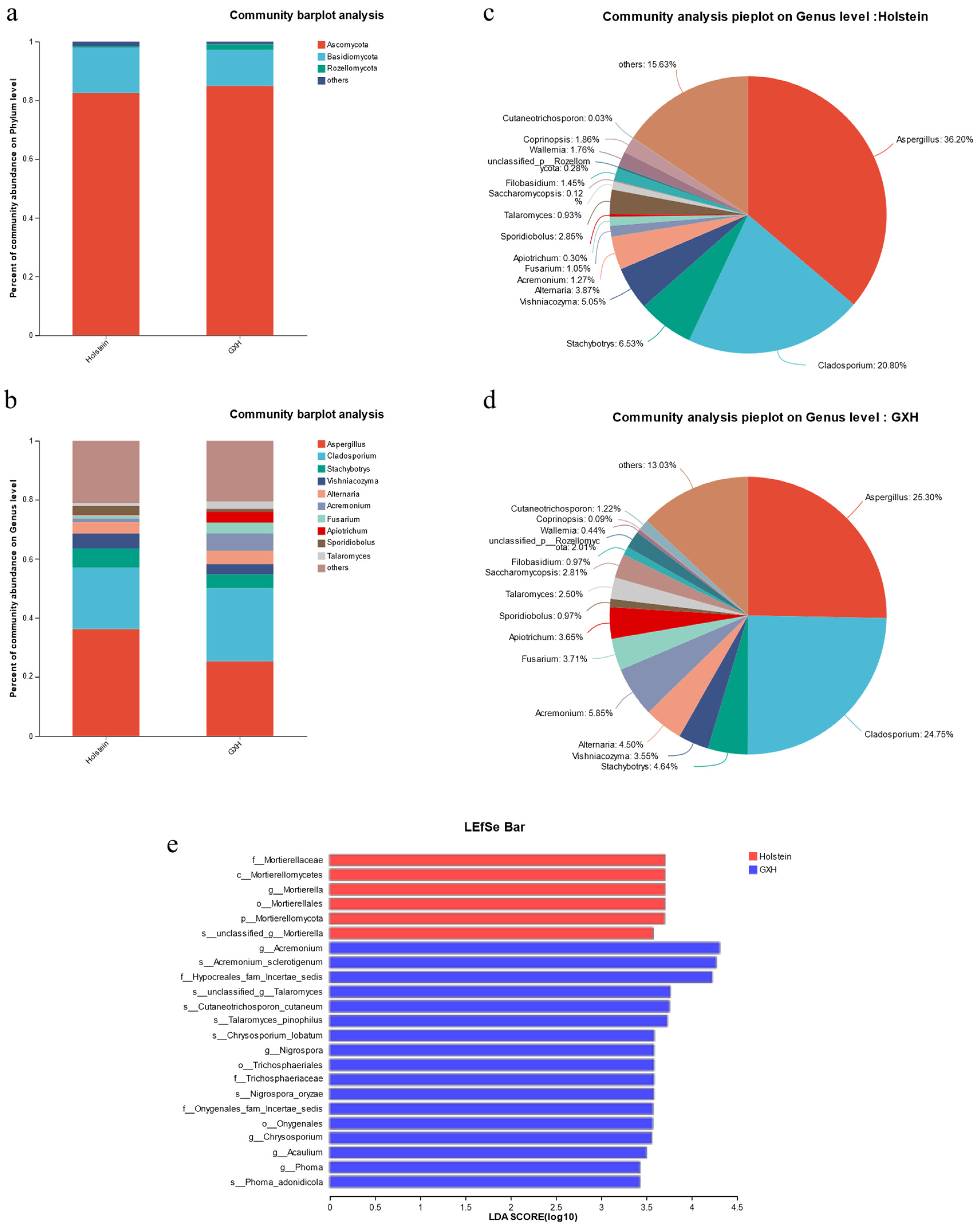
3.3.2. Taxonomic Composition and Differences of Fecal Fungal Community
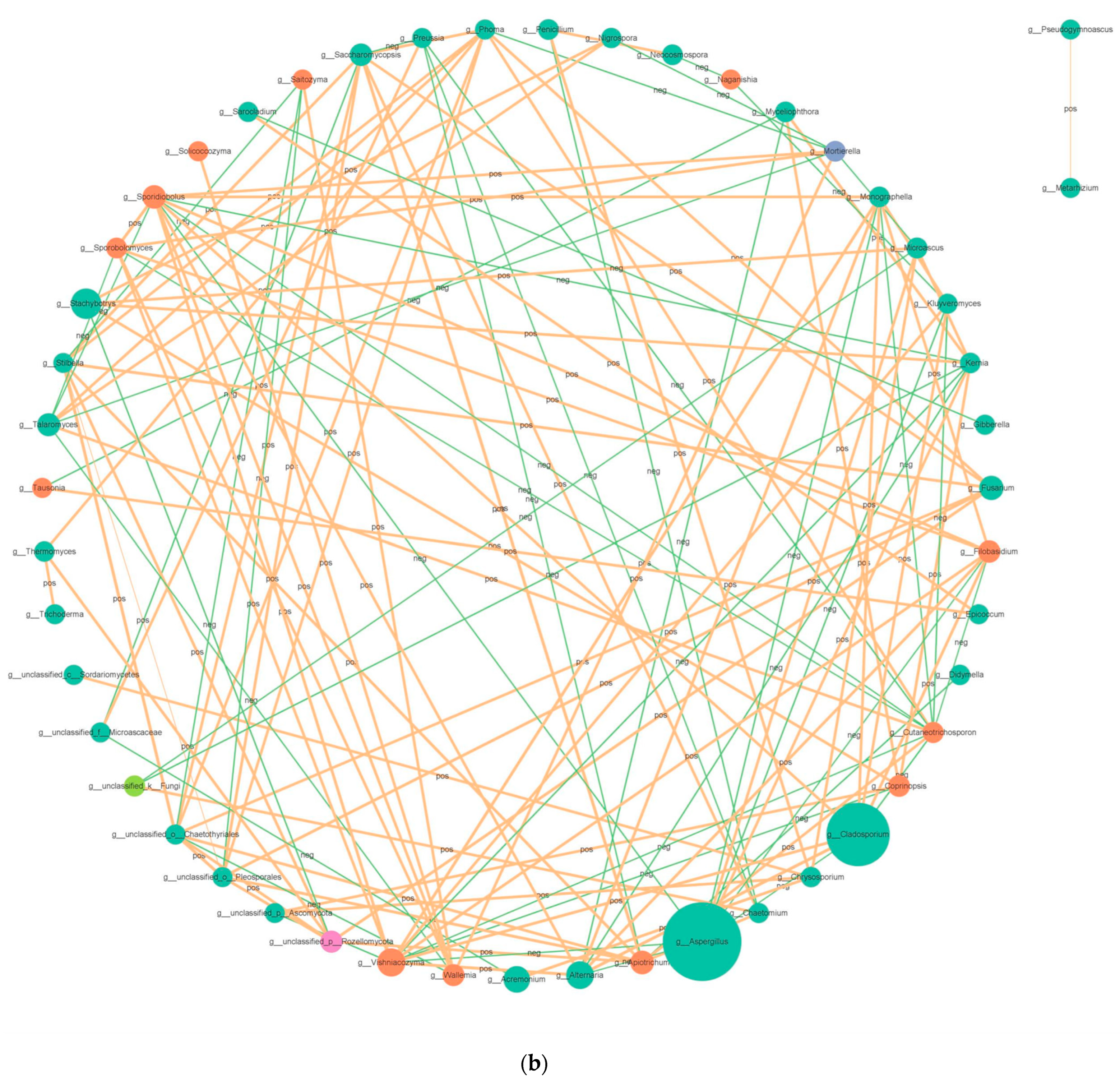
3.3.3. Correlation Network Analysis
3.4. Different Breeds Exhibited Variation in Bacterial–Fungal Associations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Malmuthuge, N.; Griebel, P.J.; Guan, L.L. The Gut Microbiome and Its Potential Role in the Development and Function of Newborn Calf Gastrointestinal Tract. Front. Vet. Sci. 2015, 2, 36. [Google Scholar] [CrossRef]
- Richard, M.L.; Sokol, H. The Gut Mycobiota: Insights into Analysis, Environmental Interactions and Role in Gastrointestinal Diseases. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 331–345. [Google Scholar] [CrossRef]
- Zmora, N.; Suez, J.; Elinav, E. You Are What You Eat: Diet, Health and the Gut Microbiota. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 35–56. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Nie, C.; Luo, R.; Qi, F.; Bai, X.; Chen, H.; Niu, J.; Chen, C.; Zhang, W. Effects of Multispecies Probiotic on Intestinal Microbiota and Mucosal Barrier Function of Neonatal Calves Infected with E. coli K99. Front. Microbiol. 2022, 12, 813245. [Google Scholar] [CrossRef]
- Chénard, T.; Prévost, K.; Dubé, J.; Massé, E. Immune System Modulations by Products of the Gut Microbiota. Vaccines 2020, 8, 461. [Google Scholar] [CrossRef]
- Liu, B.; Wang, C.; Huasai, S.; Han, A.; Zhang, J.; He, L.; Aorigele, C. Compound Probiotics Improve the Diarrhea Rate and Intestinal Microbiota of Newborn Calves. Animals 2022, 12, 322. [Google Scholar] [CrossRef]
- Ye, X.; Zhou, L.; Zhang, Y.; Xue, S.; Gan, Q.F.; Fang, S. Effect of Host Breeds on Gut Microbiome and Serum Metabolome in Meat Rabbits. BMC Vet. Res. 2021, 17, 24. [Google Scholar] [CrossRef]
- Sanna, S.; Kurilshikov, A.; van der Graaf, A.; Fu, J.; Zhernakova, A. Challenges and Future Directions for Studying Effects of Host Genetics on the Gut Microbiome. Nat. Genet. 2022, 54, 100–106. [Google Scholar] [CrossRef]
- Nash, A.K.; Auchtung, T.A.; Wong, M.C.; Smith, D.P.; Gesell, J.R.; Ross, M.C.; Stewart, C.J.; Metcalf, G.A.; Muzny, D.M.; Gibbs, R.A.; et al. The Gut Mycobiome of the Human Microbiome Project Healthy Cohort. Microbiome 2017, 5, 153. [Google Scholar] [CrossRef] [PubMed]
- Wensel, C.R.; Pluznick, J.L.; Salzberg, S.L.; Sears, C.L. Next-Generation Sequencing: Insights to Advance Clinical Investigations of the Microbiome. J. Clin. Investig. 2022, 132, e154944. [Google Scholar] [CrossRef] [PubMed]
- Fan, P.; Bian, B.; Teng, L.; Nelson, C.D.; Driver, J.; Elzo, M.A.; Jeong, K.C. Host Genetic Effects upon the Early Gut Microbiota in a Bovine Model with Graduated Spectrum of Genetic Variation. ISME J. 2020, 14, 302–317. [Google Scholar] [CrossRef]
- Imhann, F.; Vila, A.V.; Bonder, M.J.; Fu, J.; Gevers, D.; Visschedijk, M.C.; Spekhorst, L.M.; Alberts, R.; Franke, L.; van Dullemen, H.M.; et al. Interplay of Host Genetics and Gut Microbiota Underlying the Onset and Clinical Presentation of Inflammatory Bowel Disease. Gut 2018, 67, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Alipour, M.J.; Jalanka, J.; Pessa-Morikawa, T.; Kokkonen, T.; Satokari, R.; Hynönen, U.; Iivanainen, A.; Niku, M. The Composition of the Perinatal Intestinal Microbiota in Cattle. Sci. Rep. 2018, 8, 10437. [Google Scholar] [CrossRef] [PubMed]
- Tilocca, B.; Soggiu, A.; Greco, V.; Sacchini, F.; Garofolo, G.; Paci, V.; Bonizzi, L.; Urbani, A.; Tittarelli, M.; Roncada, P. Comparative Proteomics of Brucella melitensis Is a Useful Toolbox for Developing Prophylactic Interventions in a One-Health Context. One Health 2021, 13, 100253. [Google Scholar] [CrossRef] [PubMed]
- Larson, L.L.; Owen, F.G.; Albright, J.L.; Appleman, R.D.; Lamb, R.C.; Muller, L.D. Guidelines Toward More Uniformity in Measuring and Reporting Calf Experimental Data. J. Dairy Sci. 1977, 60, 989–991. [Google Scholar] [CrossRef]
- Rosa, F.; Michelotti, T.C.; St-Pierre, B.; Trevisi, E.; Osorio, J.S. Early Life Fecal Microbiota Transplantation in Neonatal Dairy Calves Promotes Growth Performance and Alleviates Inflammation and Oxidative Stress during Weaning. Animals 2021, 11, 2704. [Google Scholar] [CrossRef] [PubMed]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive Functional Profiling of Microbial Communities Using 16S rRNA Marker Gene Sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Flint, H.J.; Scott, K.P.; Louis, P.; Duncan, S.H. The Role of the Gut Microbiota in Nutrition and Health. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 577–589. [Google Scholar] [CrossRef]
- Wahlström, A.; Sayin, S.I.; Marschall, H.-U.; Bäckhed, F. Intestinal Crosstalk between Bile Acids and Microbiota and Its Impact on Host Metabolism. Cell Metab. 2016, 24, 41–50. [Google Scholar] [CrossRef]
- Li, A.; Wang, Y.; He, Y.; Liu, B.; Iqbal, M.; Mehmood, K.; Jamil, T.; Chang, Y.-F.; Hu, L.; Li, Y.; et al. Environmental Fluoride Exposure Disrupts the Intestinal Structure and Gut Microbial Composition in Ducks. Chemosphere 2021, 277, 130222. [Google Scholar] [CrossRef]
- Goodrich, J.K.; Waters, J.L.; Poole, A.C.; Sutter, J.L.; Koren, O.; Blekhman, R.; Beaumont, M.; Van Treuren, W.; Knight, R.; Bell, J.T.; et al. Human Genetics Shape the Gut Microbiome. Cell 2014, 159, 789–799. [Google Scholar] [CrossRef]
- Aagaard, K.; Ma, J.; Antony, K.M.; Ganu, R.; Petrosino, J.; Versalovic, J. The Placenta Harbors a Unique Microbiome. Sci. Transl. Med. 2014, 6, 237ra65. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Bello, M.G.; Costello, E.K.; Contreras, M.; Magris, M.; Hidalgo, G.; Fierer, N.; Knight, R. Delivery Mode Shapes the Acquisition and Structure of the Initial Microbiota across Multiple Body Habitats in Newborns. Proc. Natl. Acad. Sci. USA 2010, 107, 11971–11975. [Google Scholar] [CrossRef] [PubMed]
- Durso, L.M.; Harhay, G.P.; Smith, T.P.L.; Bono, J.L.; Desantis, T.Z.; Harhay, D.M.; Andersen, G.L.; Keen, J.E.; Laegreid, W.W.; Clawson, M.L. Animal-to-Animal Variation in Fecal Microbial Diversity among Beef Cattle. Appl. Environ. Microbiol. 2010, 76, 4858–4862. [Google Scholar] [CrossRef] [PubMed]
- Human Microbiome Project Consortium. Structure, Function and Diversity of the Healthy Human Microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Brulc, J.M.; Antonopoulos, D.A.; Miller, M.E.B.; Wilson, M.K.; Yannarell, A.C.; Dinsdale, E.A.; Edwards, R.E.; Frank, E.D.; Emerson, J.B.; Wacklin, P.; et al. Gene-Centric Metagenomics of the Fiber-Adherent Bovine Rumen Microbiome Reveals Forage Specific Glycoside Hydrolases. Proc. Natl. Acad. Sci. USA 2009, 106, 1948–1953. [Google Scholar] [CrossRef] [PubMed]
- Stojanov, S.; Berlec, A.; Štrukelj, B. The Influence of Probiotics on the Firmicutes/Bacteroidetes Ratio in the Treatment of Obesity and Inflammatory Bowel Disease. Microorganisms 2020, 8, 1715. [Google Scholar] [CrossRef]
- Liu, J.; Wang, X.; Zhang, W.; Kulyar, M.F.-E.-A.; Ullah, K.; Han, Z.; Qin, J.; Bi, C.; Wang, Y.; Li, K. Comparative Analysis of Gut Microbiota in Healthy and Diarrheic Yaks. Microb. Cell Factories 2022, 21, 111. [Google Scholar] [CrossRef]
- Rizzatti, G.; Lopetuso, L.R.; Gibiino, G.; Binda, C.; Gasbarrini, A. Proteobacteria: A Common Factor in Human Diseases. BioMed Res. Int. 2017, 2017, 9351507. [Google Scholar] [CrossRef]
- Savin, K.W.; Zawadzki, J.; Auldist, M.J.; Wang, J.; Ram, D.; Rochfort, S.; Cocks, B.G. Faecalibacterium Diversity in Dairy Cow Milk. PLoS ONE 2019, 14, e0221055. [Google Scholar] [CrossRef]
- Sokol, H.; Pigneur, B.; Watterlot, L.; Lakhdari, O.; Bermúdez-Humarán, L.G.; Gratadoux, J.-J.; Blugeon, S.; Bridonneau, C.; Furet, J.-P.; Corthier, G.; et al. Faecalibacterium prausnitzii Is an Anti-Inflammatory Commensal Bacterium Identified by Gut Microbiota Analysis of Crohn Disease Patients. Proc. Natl. Acad. Sci. USA 2008, 105, 16731–16736. [Google Scholar] [CrossRef]
- Litvak, Y.; Byndloss, M.X.; Tsolis, R.M.; Bäumler, A.J. Dysbiotic Proteobacteria Expansion: A Microbial Signature of Epithelial Dysfunction. Curr. Opin. Microbiol. 2017, 39, 1–6. [Google Scholar] [CrossRef]
- Shin, N.-R.; Whon, T.W.; Bae, J.-W. Proteobacteria: Microbial Signature of Dysbiosis in Gut Microbiota. Trends Biotechnol. 2015, 33, 496–503. [Google Scholar] [CrossRef]
- Lopes, D.R.G.; La Reau, A.J.; Duarte, M.d.S.; Detmann, E.; Bento, C.B.P.; Mercadante, M.E.Z.; Bonilha, S.F.M.; Suen, G.; Mantovani, H.C. The Bacterial and Fungal Microbiota of Nelore Steers Is Dynamic across the Gastrointestinal Tract and Its Fecal-Associated Microbiota Is Correlated to Feed Efficiency. Front. Microbiol. 2019, 10, 1263. [Google Scholar] [CrossRef] [PubMed]
- Iljazovic, A.; Roy, U.; Gálvez, E.J.C.; Lesker, T.R.; Zhao, B.; Gronow, A.; Amend, L.; Will, S.E.; Hofmann, J.D.; Pils, M.C.; et al. Perturbation of the Gut Microbiome by Prevotella spp. Enhances Host Susceptibility to Mucosal Inflammation. Mucosal Immunol. 2021, 14, 113–124. [Google Scholar] [CrossRef]
- Brunner, K.; Samassa, F.; Sansonetti, P.J.; Phalipon, A. Shigella-Mediated Immunosuppression in the Human Gut: Subversion Extends from Innate to Adaptive Immune Responses. Hum. Vaccines Immunother. 2019, 15, 1317–1325. [Google Scholar] [CrossRef]
- Benevides, L.; Burman, S.; Martin, R.; Robert, V.; Thomas, M.; Miquel, S.; Chain, F.; Sokol, H.; Bermudez-Humaran, L.G.; Morrison, M.; et al. New Insights into the Diversity of the Genus Faecalibacterium. Front. Microbiol. 2017, 8, 1790. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Wang, Y.; Li, H.; Dai, Y.; Chen, D.; Wang, M.; Jiang, X.; Huang, Z.; Yu, H.; Huang, J.; et al. Altered Fecal Microbiota Composition in Older Adults with Frailty. Front. Cell. Infect. Microbiol. 2021, 11, 696186. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Wang, J.; Fang, X.; Dou, T.; Han, L.; Yang, C. Combined Analysis of Metagenomic Data Revealed Consistent Changes of Gut Microbiome Structure and Function in Inflammatory Bowel Disease. J. Appl. Microbiol. 2021, 131, 3018–3031. [Google Scholar] [CrossRef]
- Upadhyay, R.; Littman, D.R. Provocateurs of Autoimmunity within the Gut Microbiota. Sci. Transl. Med. 2022, 14, eadd3901. [Google Scholar] [CrossRef]
- Hirano, A.; Umeno, J.; Okamoto, Y.; Shibata, H.; Ogura, Y.; Moriyama, T.; Torisu, T.; Fujioka, S.; Fuyuno, Y.; Kawarabayasi, Y.; et al. Comparison of the Microbial Community Structure between Inflamed and Non-Inflamed Sites in Patients with Ulcerative Colitis. J. Gastroenterol. Hepatol. 2018, 33, 1590–1597. [Google Scholar] [CrossRef]
- Brown, C.T.; Davis-Richardson, A.G.; Giongo, A.; Gano, K.A.; Crabb, D.B.; Mukherjee, N.; Casella, G.; Drew, J.C.; Ilonen, J.; Knip, M.; et al. Gut Microbiome Metagenomics Analysis Suggests a Functional Model for the Development of Autoimmunity for Type 1 Diabetes. PLoS ONE 2011, 6, e25792. [Google Scholar] [CrossRef]
- Wang, R.X.; Lee, J.S.; Campbell, E.L.; Colgan, S.P. Microbiota-Derived Butyrate Dynamically Regulates Intestinal Homeostasis through Regulation of Actin-Associated Protein Synaptopodin. Proc. Natl. Acad. Sci. USA 2020, 117, 11648–11657. [Google Scholar] [CrossRef]
- Verstreken, I.; Laleman, W.; Wauters, G.; Verhaegen, J. Desulfovibrio desulfuricans Bacteremia in an Immunocompromised Host with a Liver Graft and Ulcerative Colitis. J. Clin. Microbiol. 2012, 50, 199–201. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-R.; Jing, Q.-L.; Chen, F.-L.; Zheng, H.; Chen, L.-D.; Yang, Z.-C. Desulfovibrio Is Not Always Associated with Adverse Health Effects in the Guangdong Gut Microbiome Project. PeerJ 2021, 9, e12033. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, B.; Delgado, S.; Blanco-Míguez, A.; Lourenço, A.; Gueimonde, M.; Margolles, A. Probiotics, Gut Microbiota, and Their Influence on Host Health and Disease. Mol. Nutr. Food Res. 2017, 61, 1600240. [Google Scholar] [CrossRef] [PubMed]
- Moya, A.; Ferrer, M. Functional Redundancy-Induced Stability of Gut Microbiota Subjected to Disturbance. Trends Microbiol. 2016, 24, 402–413. [Google Scholar] [CrossRef] [PubMed]
- Vences, M.; Lyra, M.L.; Kueneman, J.G.; Bletz, M.C.; Archer, H.M.; Canitz, J.; Handreck, S.; Randrianiaina, R.-D.; Struck, U.; Bhuju, S.; et al. Gut Bacterial Communities across Tadpole Ecomorphs in Two Diverse Tropical Anuran Faunas. Naturwissenschaften 2016, 103, 25. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A Human Gut Microbial Gene Catalogue Established by Metagenomic Sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Suhr, M.J.; Hallen-Adams, H.E. The Human Gut Mycobiome: Pitfalls and Potentials—A Mycologist’s Perspective. Mycologia 2015, 107, 1057–1073. [Google Scholar] [CrossRef] [PubMed]
- Gruninger, R.J.; Puniya, A.K.; Callaghan, T.M.; Edwards, J.E.; Youssef, N.; Dagar, S.S.; Fliegerova, K.; Griffith, G.W.; Forster, R.; Tsang, A.; et al. Anaerobic Fungi (Phylum Neocallimastigomycota): Advances in Understanding Their Taxonomy, Life Cycle, Ecology, Role and Biotechnological Potential. FEMS Microbiol. Ecol. 2014, 90, 1–17. [Google Scholar] [CrossRef]
- Paterson, M.J.; Oh, S.; Underhill, D.M. Host-Microbe Interactions: Commensal Fungi in the Gut. Curr. Opin. Microbiol. 2017, 40, 131–137. [Google Scholar] [CrossRef]
- Gosiewski, T.; Salamon, D.; Szopa, M.; Sroka, A.; Malecki, M.T.; Bulanda, M. Quantitative Evaluation of Fungi of the Genus Candida in the Feces of Adult Patients with Type 1 and 2 Diabetes—A Pilot Study. Gut Pathog. 2014, 6, 43. [Google Scholar] [CrossRef] [PubMed]
- Eggimann, P.; Chevrolet, J.-C.; Starobinski, M.; Majno, P.; Totsch, M.; Chapuis, B.; Pittet, D. Primary Invasive Aspergillosis of the Digestive Tract: Report of Two Cases and Review of the Literature. Infection 2006, 34, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Limon, J.J.; Tang, J.; Li, D.; Wolf, A.J.; Michelsen, K.S.; Funari, V.; Gargus, M.; Nguyen, C.; Sharma, P.; Maymi, V.I.; et al. Malassezia Is Associated with Crohn’s Disease and Exacerbates Colitis in Mouse Models. Cell Host Microbe 2019, 25, 377–388.e6. [Google Scholar] [CrossRef]
- Li, K.; Mehmood, K.; Zhang, H.; Jiang, X.; Shahzad, M.; Dong, X.; Li, J. Characterization of Fungus Microbial Diversity in Healthy and Diarrheal Yaks in Gannan Region of Tibet Autonomous Prefecture. Acta Trop. 2018, 182, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Kong, Q.; Liu, S.; Li, A.; Wang, Y.; Zhang, L.; Iqbal, M.; Jamil, T.; Shang, Z.; Suo, L.-S.; Li, J. Characterization of Fungal Microbial Diversity in Healthy and Diarrheal Tibetan Piglets. BMC Microbiol. 2021, 21, 204. [Google Scholar] [CrossRef]
- Li, A.; Liu, B.; Li, F.; He, Y.; Wang, L.; Fakhar-e-Alam Kulyar, M.; Li, H.; Fu, Y.; Zhu, H.; Wang, Y.; et al. Integrated Bacterial and Fungal Diversity Analysis Reveals the Gut Microbial Alterations in Diarrheic Giraffes. Front. Microbiol. 2021, 12, 712092. [Google Scholar] [CrossRef]
- Iliev, I.D.; Funari, V.A.; Taylor, K.D.; Nguyen, Q.; Reyes, C.N.; Strom, S.P.; Brown, J.; Becker, C.A.; Fleshner, P.R.; Dubinsky, M.; et al. Interactions between Commensal Fungi and the C-Type Lectin Receptor Dectin-1 Influence Colitis. Science 2012, 336, 1314–1317. [Google Scholar] [CrossRef]
- Sokol, H.; Leducq, V.; Aschard, H.; Pham, H.-P.; Jegou, S.; Landman, C.; Cohen, D.; Liguori, G.; Bourrier, A.; Nion-Larmurier, I.; et al. Fungal Microbiota Dysbiosis in IBD. Gut 2017, 66, 1039–1048. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ingredient | Content |
|---|---|
| Corn | 55.200 |
| Soybean meal | 18.500 |
| Corn gluten meal | 10.000 |
| DGGS | 13.000 |
| Limestone | 1.800 |
| NaCl | 0.500 |
| Premix 1 | 1.000 |
| Total | 100.000 |
| Body Height (cm) | Body Oblique Length (cm) | Tube Circumference (cm) | Body Weight (kg) | Diarrhea Rate (%) | |
|---|---|---|---|---|---|
| GXH | 78.70 ± 2.36 a | 77.70 ± 2.54 a | 9.10 ± 1.79 | 43.30 ± 5.48 a | 11.42% (4/35) |
| Holstein | 75.70 ± 1.95 b | 74.00 ± 2.31 b | 8.00 ± 1.33 | 37.20 ± 5.12 b | 22.85% (8/35) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nan, S.; Li, J.; Kuang, Y.; Feng, J.; Wang, H.; Niu, J.; Wu, Y.; Zhang, W.; Nie, C. Differential Microbial Composition and Interkingdom Interactions in the Intestinal Microbiota of Holstein and German Simmental × Holstein Cross F1 Calves: A Comprehensive Analysis of Bacterial and Fungal Diversity. Microorganisms 2024, 12, 486. https://doi.org/10.3390/microorganisms12030486
Nan S, Li J, Kuang Y, Feng J, Wang H, Niu J, Wu Y, Zhang W, Nie C. Differential Microbial Composition and Interkingdom Interactions in the Intestinal Microbiota of Holstein and German Simmental × Holstein Cross F1 Calves: A Comprehensive Analysis of Bacterial and Fungal Diversity. Microorganisms. 2024; 12(3):486. https://doi.org/10.3390/microorganisms12030486
Chicago/Turabian StyleNan, Shanshan, Jiacheng Li, Yu Kuang, Jiaqi Feng, Hailiang Wang, Junli Niu, Yanyan Wu, Wenju Zhang, and Cunxi Nie. 2024. "Differential Microbial Composition and Interkingdom Interactions in the Intestinal Microbiota of Holstein and German Simmental × Holstein Cross F1 Calves: A Comprehensive Analysis of Bacterial and Fungal Diversity" Microorganisms 12, no. 3: 486. https://doi.org/10.3390/microorganisms12030486
APA StyleNan, S., Li, J., Kuang, Y., Feng, J., Wang, H., Niu, J., Wu, Y., Zhang, W., & Nie, C. (2024). Differential Microbial Composition and Interkingdom Interactions in the Intestinal Microbiota of Holstein and German Simmental × Holstein Cross F1 Calves: A Comprehensive Analysis of Bacterial and Fungal Diversity. Microorganisms, 12(3), 486. https://doi.org/10.3390/microorganisms12030486