
A Mini-Review on the Common Antiviral Drug Targets of Coronavirus


Abstract
:1. Introduction
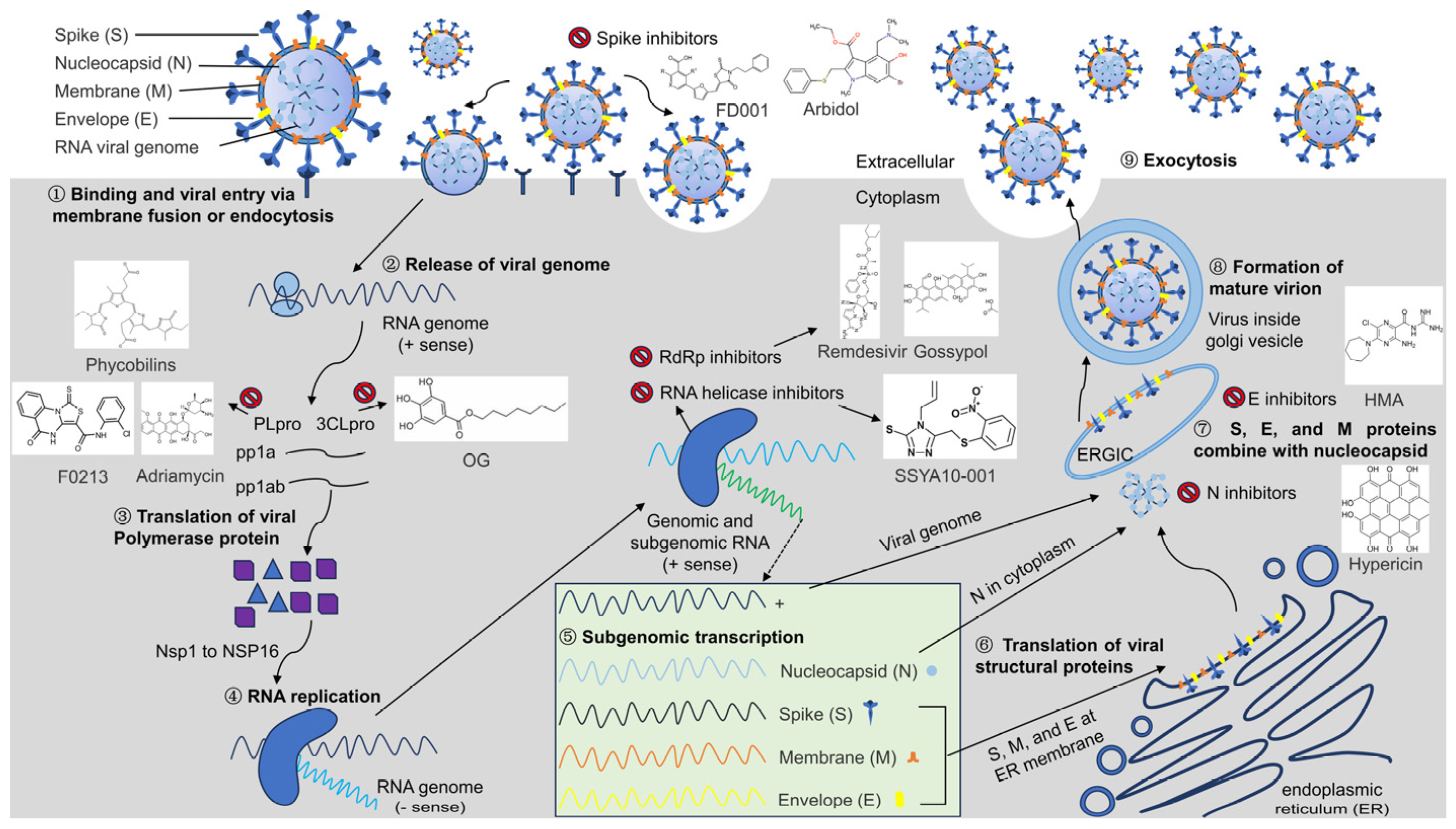
2. Pathogenic Mechanisms of Coronavirus
3. Common Drugs Targeting Coronavirus Entry into Host Cells
4. Common Drug Targets against Coronaviruses Based on Biosynthesis
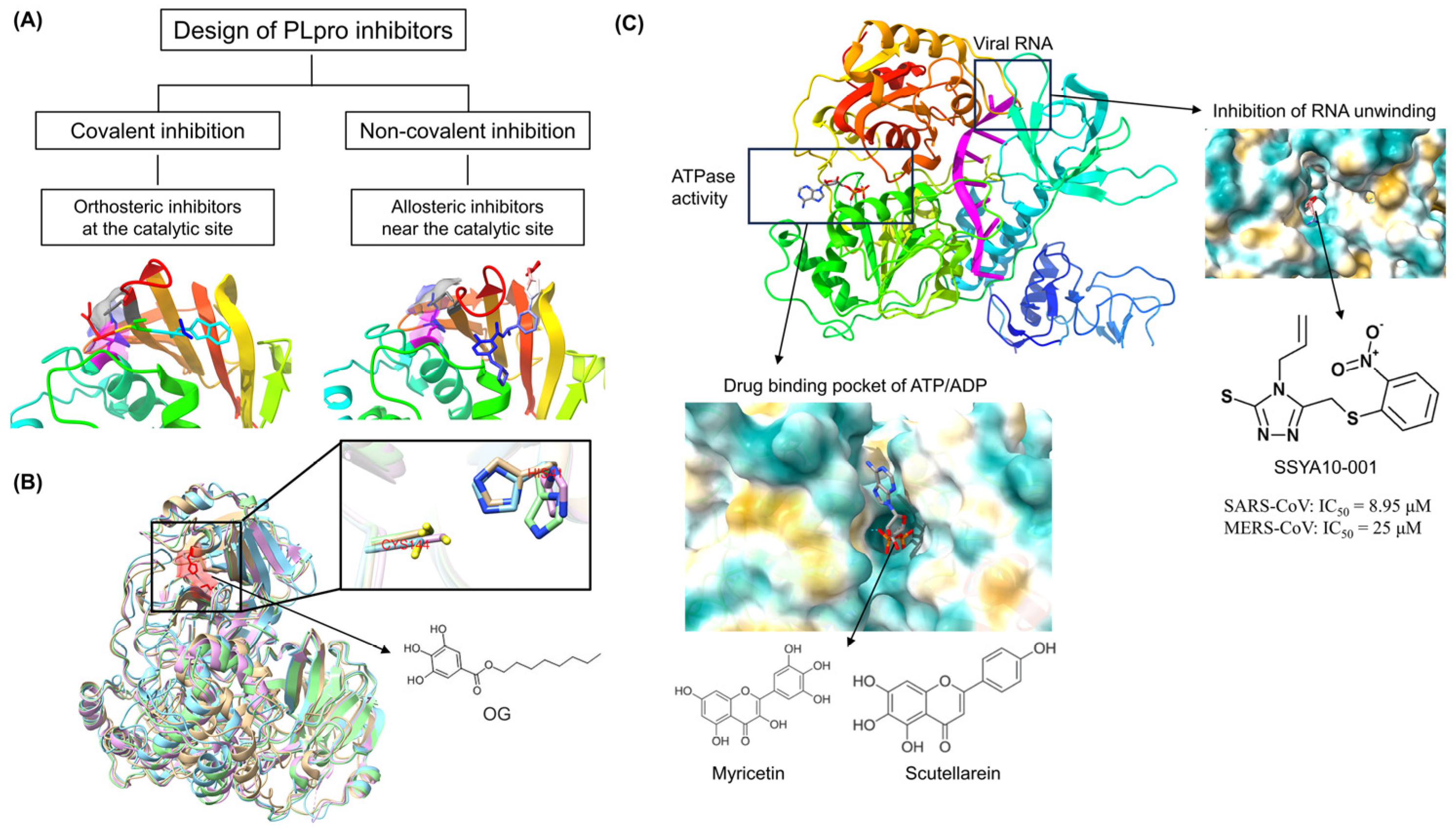
4.1. Coronavirus PLpro as a Common Target for Antivirals
4.2. Coronavirus 3CLpro as a Common Target for Antivirals
4.3. Coronavirus RdRp Protein as a Common Target for Antivirals
4.4. Coronavirus RNA Helicase as a Common Target for Antivirals
4.5. Coronavirus Exoribonuclease as a Common Target for Antivirals
5. Common Drug Targets against Coronaviruses Based on Viral Assembly and Release
6. Conclusions and Prospects
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Tompa, D.R.; Immanuel, A.; Srikanth, S.; Kadhirvel, S. Trends and strategies to combat viral infections: A review on FDA approved antiviral drugs. Int. J. Biol. Macromol. 2021, 172, 524–541. [Google Scholar] [CrossRef]
- Ton, A.T.; Gentile, F.; Hsing, M.; Ban, F.; Cherkasov, A. Rapid Identification of Potential Inhibitors of SARS-CoV-2 Main Protease by Deep Docking of 1.3 Billion Compounds. Mol. Inform. 2020, 39, e2000028. [Google Scholar] [CrossRef]
- Thomasy, S.M.; Maggs, D.J. A review of antiviral drugs and other compounds with activity against feline herpesvirus type 1. Vet. Ophthalmol. 2016, 19 (Suppl. 1), 119–130. [Google Scholar] [CrossRef]
- Zhang, Y.; Tang, L.V. Overview of Targets and Potential Drugs of SARS-CoV-2 According to the Viral Replication. J. Proteome Res. 2021, 20, 49–59. [Google Scholar] [CrossRef]
- Caly, L.; Druce, J.D.; Catton, M.G.; Jans, D.A.; Wagstaff, K.M. The FDA-approved drug ivermectin inhibits the replication of SARS-CoV-2 in vitro. Antivir. Res. 2020, 178, 104787. [Google Scholar] [CrossRef]
- Vincent, M.J.; Bergeron, E.; Benjannet, S.; Erickson, B.R.; Rollin, P.E.; Ksiazek, T.G.; Seidah, N.G.; Nichol, S.T. Chloroquine is a potent inhibitor of SARS coronavirus infection and spread. Virol. J. 2005, 2, 69. [Google Scholar] [CrossRef]
- Wang, X.; Cao, R.; Zhang, H.; Liu, J.; Xu, M.; Hu, H.; Li, Y.; Zhao, L.; Li, W.; Sun, X.; et al. The anti-influenza virus drug, arbidol is an efficient inhibitor of SARS-CoV-2 in vitro. Cell Discov. 2020, 6, 28. [Google Scholar] [CrossRef]
- Wang, C.; Xia, S.; Zhang, P.; Zhang, T.; Wang, W.; Tian, Y.; Meng, G.; Jiang, S.; Liu, K. Discovery of Hydrocarbon-Stapled Short alpha-Helical Peptides as Promising Middle East Respiratory Syndrome Coronavirus (MERS-CoV) Fusion Inhibitors. J. Med. Chem. 2018, 61, 2018–2026. [Google Scholar] [CrossRef]
- V’Kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170. [Google Scholar] [CrossRef]
- Hartenian, E.; Nandakumar, D.; Lari, A.; Ly, M.; Tucker, J.M.; Glaunsinger, B.A. The molecular virology of coronaviruses. J. Biol. Chem. 2020, 295, 12910–12934. [Google Scholar] [CrossRef]
- Masters, P.S. The molecular biology of coronaviruses. Adv. Virus Res. 2006, 66, 193–292. [Google Scholar]
- Fehr, A.R.; Perlman, S. Coronaviruses: An overview of their replication and pathogenesis. Methods Mol. Biol. 2015, 1282, 1–23. [Google Scholar]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, Q.; Guo, D. Emerging coronaviruses: Genome structure, replication, and pathogenesis. J. Med. Virol. 2020, 92, 2249. [Google Scholar] [CrossRef]
- Sharma, A.; Tiwari, S.; Deb, M.K.; Marty, J.L. Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2): A global pandemic and treatment strategies. Int. J. Antimicrob. Agents 2020, 56, 106054. [Google Scholar] [CrossRef]
- de Groot, R.J.; Baker, S.C.; Baric, R.S.; Brown, C.S.; Drosten, C.; Enjuanes, L.; Fouchier, R.A.; Galiano, M.; Gorbalenya, A.E.; Memish, Z.A.; et al. Middle East respiratory syndrome coronavirus (MERS-CoV): Announcement of the Coronavirus Study Group. J. Virol. 2013, 87, 7790–7792. [Google Scholar] [CrossRef]
- Cui, X.; Wang, Y.; Zhai, J.; Xue, M.; Zheng, C.; Yu, L. Future trajectory of SARS-CoV-2: Constant spillover back and forth between humans and animals. Virus Res. 2023, 328, 199075. [Google Scholar] [CrossRef]
- Silva, L.R.; da Silva Santos-Júnior, P.F.; de Andrade Brandão, J.; Anderson, L.; Bassi, Ê.J.; Xavier de Araújo-Júnior, J.; Cardoso, S.H.; da Silva-Júnior, E.F. Druggable targets from coronaviruses for designing new antiviral drugs. Bioorg. Med. Chem. 2020, 28, 115745. [Google Scholar] [CrossRef] [PubMed]
- Uma Reddy, B.; Routhu, N.K.; Kumar, A. Multifaceted roles of plant derived small molecule inhibitors on replication cycle of SARS-CoV-2. Microb. Pathog. 2022, 168, 105512. [Google Scholar] [CrossRef] [PubMed]
- Drożdżal, S.; Rosik, J.; Lechowicz, K.; Machaj, F.; Szostak, B.; Przybyciński, J.; Lorzadeh, S.; Kotfis, K.; Ghavami, S.; Łos, M.J. An update on drugs with therapeutic potential for SARS-CoV-2 (COVID-19) treatment. Drug Resist. Updates Rev. Comment. Antimicrob. Anticancer Chemother. 2021, 59, 100794. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Zheng, Y.; Zeng, X.; He, B.; Cheng, W. Structural biology of SARS-CoV-2: Open the door for novel therapies. Signal Transduct. Target. Ther. 2022, 7, 26. [Google Scholar] [CrossRef] [PubMed]
- Castillo, G.; Mora-Díaz, J.C.; Breuer, M.; Singh, P.; Nelli, R.K.; Giménez-Lirola, L.G. Molecular mechanisms of human coronavirus NL63 infection and replication. Virus Res. 2023, 327, 199078. [Google Scholar] [CrossRef]
- Upadhyay, M.; Gupta, S. Endoplasmic reticulum secretory pathway: Potential target against SARS-CoV-2. Virus Res. 2022, 320, 198897. [Google Scholar] [CrossRef]
- Pu, J.; He, X.; Xu, W.; Wang, C.; Lan, Q.; Hua, C.; Wang, K.; Lu, L.; Jiang, S. The Analogs of Furanyl Methylidene Rhodanine Exhibit Broad-Spectrum Inhibitory and Inactivating Activities against Enveloped Viruses, including SARS-CoV-2 and Its Variants. Viruses 2022, 14, 489. [Google Scholar] [CrossRef]
- Vankadari, N. Arbidol: A potential antiviral drug for the treatment of SARS-CoV-2 by blocking trimerization of the spike glycoprotein. Int. J. Antimicrob. Agents 2020, 56, 105998. [Google Scholar] [CrossRef]
- Pendyala, B.; Patras, A.; Dash, C. Phycobilins as Potent Food Bioactive Broad-Spectrum Inhibitors Against Proteases of SARS-CoV-2 and Other Coronaviruses: A Preliminary Study. Front. Microbiol. 2021, 12, 645713. [Google Scholar] [CrossRef]
- Alaofi, A.L.; Shahid, M.; Raish, M.; Ansari, M.A.; Syed, R.; Kalam, M.A. Identification of Doxorubicin as Repurposing Inhibitory Drug for MERS-CoV PLpro. Molecules 2022, 27, 7553. [Google Scholar] [CrossRef]
- Yuan, S.; Gao, X.; Tang, K.; Cai, J.P.; Hu, M.; Luo, P.; Wen, L.; Ye, Z.W.; Luo, C.; Tsang, J.O.; et al. Targeting papain-like protease for broad-spectrum coronavirus inhibition. Protein Cell 2022, 13, 940–953. [Google Scholar] [CrossRef]
- Lin, Y.; Zang, R.; Ma, Y.; Wang, Z.; Li, L.; Ding, S.; Zhang, R.; Wei, Z.; Yang, J.; Wang, X. Xanthohumol Is a Potent Pan-Inhibitor of Coronaviruses Targeting Main Protease. Int. J. Mol. Sci. 2021, 22, 12134. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, H.; Zou, M.; Oerlemans, R.; Shao, C.; Ren, Y.; Zhang, R.; Huang, X.; Li, G.; Cong, Y. Hypericin Inhibit Alpha-Coronavirus Replication by Targeting 3CL Protease. Viruses 2021, 13, 1825. [Google Scholar] [CrossRef]
- Wang, P.; Bai, J.; Liu, X.; Wang, M.; Wang, X.; Jiang, P. Tomatidine inhibits porcine epidemic diarrhea virus replication by targeting 3CL protease. Vet. Res. 2020, 51, 136. [Google Scholar] [CrossRef]
- Su, M.; Yin, B.; Xing, X.; Li, Z.; Zhang, J.; Feng, S.; Li, L.; Zhao, F.; Yang, X.; Yu, S.; et al. Octyl gallate targeting the 3C-like protease exhibits highly efficient antiviral activity against swine enteric coronavirus PEDV. Vet. Microbiol. 2023, 281, 109743. [Google Scholar] [CrossRef] [PubMed]
- Mahgoub, R.E.; Mohamed, F.E.; Ali, B.R.; Ferreira, J.; Rabeh, W.M.; Atatreh, N.; Ghattas, M.A. Discovery of pyrimidoindol and benzylpyrrolyl inhibitors targeting SARS-CoV-2 main protease (M(pro)) through pharmacophore modelling, covalent docking, and biological evaluation. J. Mol. Graph. Model. 2024, 127, 108672. [Google Scholar] [CrossRef]
- Runfeng, L.; Yunlong, H.; Jicheng, H.; Weiqi, P.; Qinhai, M.; Yongxia, S.; Chufang, L.; Jin, Z.; Zhenhua, J.; Haiming, J.; et al. Lianhuaqingwen exerts anti-viral and anti-inflammatory activity against novel coronavirus (SARS-CoV-2). Pharmacol. Res. 2020, 156, 104761. [Google Scholar] [CrossRef]
- Wang, W.; Li, W.; Wen, Z.; Wang, C.; Liu, W.; Zhang, Y.; Liu, J.; Ding, T.; Shuai, L.; Zhong, G.; et al. Gossypol Broadly Inhibits Coronaviruses by Targeting RNA-Dependent RNA Polymerases. Adv. Sci. 2022, 9, e2203499. [Google Scholar] [CrossRef]
- Adedeji, A.O.; Singh, K.; Kassim, A.; Coleman, C.M.; Elliott, R.; Weiss, S.R.; Frieman, M.B.; Sarafianos, S.G. Evaluation of SSYA10-001 as a replication inhibitor of severe acute respiratory syndrome, mouse hepatitis, and Middle East respiratory syndrome coronaviruses. Antimicrob. Agents Chemother. 2014, 58, 4894–4898. [Google Scholar] [CrossRef]
- Mandala, V.S.; McKay, M.J.; Shcherbakov, A.A.; Dregni, A.J.; Kolocouris, A.; Hong, M. Structure and drug binding of the SARS-CoV-2 envelope protein transmembrane domain in lipid bilayers. Nat. Struct. Mol. Biol. 2020, 27, 1202–1208. [Google Scholar] [CrossRef]
- Su, M.; Shi, D.; Xing, X.; Qi, S.; Yang, D.; Zhang, J.; Han, Y.; Zhu, Q.; Sun, H.; Wang, X.; et al. Coronavirus Porcine Epidemic Diarrhea Virus Nucleocapsid Protein Interacts with p53 To Induce Cell Cycle Arrest in S-Phase and Promotes Viral Replication. J. Virol. 2021, 95, e0018721. [Google Scholar] [CrossRef]
- Guo, L.; Lin, S.; Chen, Z.; Cao, Y.; He, B.; Lu, G. Targetable elements in SARS-CoV-2 S2 subunit for the design of pan-coronavirus fusion inhibitors and vaccines. Signal Transduct. Target. Ther. 2023, 8, 197. [Google Scholar] [CrossRef]
- Cai, Y.; Zhang, J.; Xiao, T.; Peng, H.; Sterling, S.M.; Walsh, R.M., Jr.; Rawson, S.; Rits-Volloch, S.; Chen, B. Distinct conformational states of SARS-CoV-2 spike protein. Science 2020, 369, 1586–1592. [Google Scholar] [CrossRef]
- Stincarelli, M.A.; Quagliata, M.; Di Santo, A.; Pacini, L.; Fernandez, F.R.; Arvia, R.; Rinaldi, S.; Papini, A.M.; Rovero, P.; Giannecchini, S. SARS-CoV-2 inhibitory activity of a short peptide derived from internal fusion peptide of S2 subunit of spike glycoprotein. Virus Res. 2023, 334, 199170. [Google Scholar] [CrossRef]
- Shin, D.; Mukherjee, R.; Grewe, D.; Bojkova, D.; Baek, K.; Bhattacharya, A.; Schulz, L.; Widera, M.; Mehdipour, A.R.; Tascher, G.; et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 2020, 587, 657–662. [Google Scholar] [CrossRef]
- Osipiuk, J.; Azizi, S.A.; Dvorkin, S.; Endres, M.; Jedrzejczak, R.; Jones, K.A.; Kang, S.; Kathayat, R.S.; Kim, Y.; Lisnyak, V.G.; et al. Structure of papain-like protease from SARS-CoV-2 and its complexes with non-covalent inhibitors. Nat. Commun. 2021, 12, 743. [Google Scholar] [CrossRef] [PubMed]
- Melo-Filho, C.C.; Bobrowski, T.; Martin, H.J.; Sessions, Z.; Popov, K.I.; Moorman, N.J.; Baric, R.S.; Muratov, E.N.; Tropsha, A. Conserved coronavirus proteins as targets of broad-spectrum antivirals. Antivir. Res. 2022, 204, 105360. [Google Scholar] [CrossRef]
- Shen, Z.; Ratia, K.; Cooper, L.; Kong, D.; Lee, H.; Kwon, Y.; Li, Y.; Alqarni, S.; Huang, F.; Dubrovskyi, O.; et al. Design of SARS-CoV-2 PLpro Inhibitors for COVID-19 Antiviral Therapy Leveraging Binding Cooperativity. J. Med. Chem. 2022, 65, 2940–2955. [Google Scholar] [CrossRef]
- Xiong, M.; Su, H.; Zhao, W.; Xie, H.; Shao, Q.; Xu, Y. What coronavirus 3C-like protease tells us: From structure, substrate selectivity, to inhibitor design. Med. Res. Rev. 2021, 41, 1965–1998. [Google Scholar] [CrossRef]
- Hillen, H.S.; Kokic, G.; Farnung, L.; Dienemann, C.; Tegunov, D.; Cramer, P. Structure of replicating SARS-CoV-2 polymerase. Nature 2020, 584, 154–156. [Google Scholar] [CrossRef]
- Subissi, L.; Posthuma, C.C.; Collet, A.; Zevenhoven-Dobbe, J.C.; Gorbalenya, A.E.; Decroly, E.; Snijder, E.J.; Canard, B.; Imbert, I. One severe acute respiratory syndrome coronavirus protein complex integrates processive RNA polymerase and exonuclease activities. Proc. Natl. Acad. Sci. USA 2014, 111, E3900-3909. [Google Scholar] [CrossRef]
- Zhang, W.F.; Stephen, P.; Thériault, J.F.; Wang, R.; Lin, S.X. Novel Coronavirus Polymerase and Nucleotidyl-Transferase Structures: Potential to Target New Outbreaks. J. Phys. Chem. Lett. 2020, 11, 4430–4435. [Google Scholar] [CrossRef]
- Tian, L.; Qiang, T.; Liang, C.; Ren, X.; Jia, M.; Zhang, J.; Li, J.; Wan, M.; YuWen, X.; Li, H.; et al. RNA-dependent RNA polymerase (RdRp) inhibitors: The current landscape and repurposing for the COVID-19 pandemic. Eur. J. Med. Chem. 2021, 213, 113201. [Google Scholar]
- Abou Baker, D.H.; Hassan, E.M.; El Gengaihi, S. An overview on medicinal plants used for combating coronavirus: Current potentials and challenges. J. Agric. Food Res. 2023, 13, 100632. [Google Scholar] [CrossRef]
- Reina, J. Remdesivir, the antiviral hope against SARS-CoV-2. Rev. Española Quimioter. 2020, 33, 176–179. [Google Scholar] [CrossRef] [PubMed]
- Şimşek Yavuz, S.; Ünal, S. Antiviral treatment of COVID-19. Turk. J. Med. Sci. 2020, 50, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Guo, X.; Hu, T.; Wei, D.; Ma, X.; Wu, J.; Huang, B.; Shen, J. Significant Inhibition of Porcine Epidemic Diarrhea Virus In Vitro by Remdesivir, Its Parent Nucleoside and β-D-N(4)-hydroxycytidine. Virol. Sin. 2021, 36, 997–1005. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Barauskas, O.; Kim, C.; Babusis, D.; Murakami, E.; Kornyeyev, D.; Lee, G.; Stepan, G.; Perron, M.; Bannister, R.; et al. Off-Target In Vitro Profiling Demonstrates that Remdesivir Is a Highly Selective Antiviral Agent. Antimicrob. Agents Chemother. 2021, 65, e02237-02220. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Yin, W.; Xu, H.E. RNA-dependent RNA polymerase: Structure, mechanism, and drug discovery for COVID-19. Biochem. Biophys. Res. Commun. 2021, 538, 47–53. [Google Scholar] [CrossRef]
- Tsai, S.C.; Lu, C.C.; Bau, D.T.; Chiu, Y.J.; Yen, Y.T.; Hsu, Y.M.; Fu, C.W.; Kuo, S.C.; Lo, Y.S.; Chiu, H.Y.; et al. Approaches towards fighting the COVID-19 pandemic (Review). Int. J. Mol. Med. 2021, 47, 3–22. [Google Scholar] [CrossRef] [PubMed]
- Jean, S.S.; Lee, P.I.; Hsueh, P.R. Treatment options for COVID-19: The reality and challenges. J. Microbiol. Immunol. Infect. Wei Mian Yu Gan Ran Za Zhi 2020, 53, 436–443. [Google Scholar] [CrossRef]
- Lu, C.C.; Chen, M.Y.; Lee, W.S.; Chang, Y.L. Potential therapeutic agents against COVID-19: What we know so far. J. Chin. Med. Assoc. JCMA 2020, 83, 534–536. [Google Scholar] [CrossRef]
- Persaud, K.E.; Sahu, R.R.; Neary, M.C.; Kapdi, A.R.; Lakshman, M.K. Two short approaches to the COVID-19 drug β-D-N(4)-hydroxycytidine and its prodrug molnupiravir. Org. Biomol. Chem. 2024, 22, 735–740. [Google Scholar] [CrossRef]
- Chen, J.; Malone, B.; Llewellyn, E.; Grasso, M.; Shelton, P.M.M.; Olinares, P.D.B.; Maruthi, K.; Eng, E.T.; Vatandaslar, H.; Chait, B.T.; et al. Structural Basis for Helicase-Polymerase Coupling in the SARS-CoV-2 Replication-Transcription Complex. Cell 2020, 182, 1560–1573.e1513. [Google Scholar] [CrossRef]
- Jia, Z.; Yan, L.; Ren, Z.; Wu, L.; Wang, J.; Guo, J.; Zheng, L.; Ming, Z.; Zhang, L.; Lou, Z.; et al. Delicate structural coordination of the Severe Acute Respiratory Syndrome coronavirus Nsp13 upon ATP hydrolysis. Nucleic Acids Res. 2019, 47, 6538–6550. [Google Scholar] [CrossRef]
- Yazdi, A.K.; Pakarian, P.; Perveen, S.; Hajian, T.; Santhakumar, V.; Bolotokova, A.; Li, F.; Vedadi, M. Kinetic Characterization of SARS-CoV-2 nsp13 ATPase Activity and Discovery of Small-Molecule Inhibitors. ACS Infect. Dis. 2022, 8, 1533–1542. [Google Scholar] [CrossRef]
- Zeng, J.; Weissmann, F.; Bertolin, A.P.; Posse, V.; Canal, B.; Ulferts, R.; Wu, M.; Harvey, R.; Hussain, S.; Milligan, J.C.; et al. Identifying SARS-CoV-2 antiviral compounds by screening for small molecule inhibitors of nsp13 helicase. Biochem. J. 2021, 478, 2405–2423. [Google Scholar] [CrossRef]
- Squeglia, F.; Romano, M.; Ruggiero, A.; Maga, G.; Berisio, R. Host DDX Helicases as Possible SARS-CoV-2 Proviral Factors: A Structural Overview of Their Hijacking Through Multiple Viral Proteins. Front. Chem. 2020, 8, 602162. [Google Scholar] [CrossRef]
- Shannon, A.; Chazot, A.; Feracci, M.; Falcou, C.; Fattorini, V.; Selisko, B.; Good, S.; Moussa, A.; Sommadossi, J.P.; Ferron, F.; et al. An exonuclease-resistant chain-terminating nucleotide analogue targeting the SARS-CoV-2 replicase complex. Nucleic Acids Res. 2024, 52, 1325–1340. [Google Scholar] [CrossRef]
- Singh, I.; Li, F.; Fink, E.A.; Chau, I.; Li, A.; Rodriguez-Hernández, A.; Glenn, I.; Zapatero-Belinchón, F.J.; Rodriguez, M.L.; Devkota, K.; et al. Structure-Based Discovery of Inhibitors of the SARS-CoV-2 Nsp14 N7-Methyltransferase. J. Med. Chem. 2023, 66, 7785–7803. [Google Scholar] [CrossRef]
- Asthana, A.; Corona, A.; Shin, W.J.; Kwak, M.J.; Gaughan, C.; Tramontano, E.; Jung, J.U.; Schobert, R.; Jha, B.K.; Silverman, R.H.; et al. Analogs of the Catechol Derivative Dynasore Inhibit HIV-1 Ribonuclease H, SARS-CoV-2 nsp14 Exoribonuclease, and Virus Replication. Viruses 2023, 15, 1539. [Google Scholar] [CrossRef]
- Kottur, J.; White, K.M.; Rodriguez, M.L.; Rechkoblit, O.; Quintana-Feliciano, R.; Nayar, A.; García-Sastre, A.; Aggarwal, A.K. Structures of SARS-CoV-2 N7-methyltransferase with DOT1L and PRMT7 inhibitors provide a platform for new antivirals. PLoS Pathog. 2023, 19, e1011546. [Google Scholar] [CrossRef]
- El Omari, K.; Li, S.; Kotecha, A.; Walter, T.S.; Bignon, E.A.; Harlos, K.; Somerharju, P.; De Haas, F.; Clare, D.K.; Molin, M.; et al. The structure of a prokaryotic viral envelope protein expands the landscape of membrane fusion proteins. Nat. Commun. 2019, 10, 846. [Google Scholar] [CrossRef]
- Cao, Y.; Yang, R.; Lee, I.; Zhang, W.; Sun, J.; Wang, W.; Meng, X. Characterization of the SARS-CoV-2 E Protein: Sequence, Structure, Viroporin, and Inhibitors. Protein Sci. A Publ. Protein Soc. 2021, 30, 1114–1130. [Google Scholar] [CrossRef]
- Borkotoky, S.; Banerjee, M. A computational prediction of SARS-CoV-2 structural protein inhibitors from Azadirachta indica (Neem). J. Biomol. Struct. Dyn. 2021, 39, 4111–4121. [Google Scholar] [CrossRef]
- McBride, R.; van Zyl, M.; Fielding, B.C. The coronavirus nucleocapsid is a multifunctional protein. Viruses 2014, 6, 2991–3018. [Google Scholar] [CrossRef]
- Peng, Y.; Du, N.; Lei, Y.; Dorje, S.; Qi, J.; Luo, T.; Gao, G.F.; Song, H. Structures of the SARS-CoV-2 nucleocapsid and their perspectives for drug design. EMBO J. 2020, 39, e105938. [Google Scholar] [CrossRef]
- Cubuk, J.; Alston, J.J.; Incicco, J.J.; Singh, S.; Stuchell-Brereton, M.D.; Ward, M.D.; Zimmerman, M.I.; Vithani, N.; Griffith, D.; Wagoner, J.A.; et al. The SARS-CoV-2 nucleocapsid protein is dynamic, disordered, and phase separates with RNA. Nat. Commun. 2021, 12, 1936. [Google Scholar] [CrossRef]
- Totura, A.L.; Bavari, S. Broad-spectrum coronavirus antiviral drug discovery. Expert Opin. Drug Discov. 2019, 14, 397–412. [Google Scholar] [CrossRef]
- Geraghty, R.J.; Aliota, M.T.; Bonnac, L.F. Broad-Spectrum Antiviral Strategies and Nucleoside Analogues. Viruses 2021, 13, 667. [Google Scholar] [CrossRef]
- Iwata-Yoshikawa, N.; Okamura, T.; Shimizu, Y.; Hasegawa, H.; Takeda, M.; Nagata, N. TMPRSS2 Contributes to Virus Spread and Immunopathology in the Airways of Murine Models after Coronavirus Infection. J. Virol. 2019, 93, 1128. [Google Scholar] [CrossRef]
- Mazzon, M.; Ortega-Prieto, A.M.; Imrie, D.; Luft, C.; Hess, L.; Czieso, S.; Grove, J.; Skelton, J.K.; Farleigh, L.; Bugert, J.J.; et al. Identification of Broad-Spectrum Antiviral Compounds by Targeting Viral Entry. Viruses 2019, 11, 176. [Google Scholar] [CrossRef]
- Sabbah, D.A.; Hajjo, R.; Bardaweel, S.K.; Zhong, H.A. An Updated Review on SARS-CoV-2 Main Proteinase (M(Pro)): Protein Structure and Small-Molecule Inhibitors. Curr. Top. Med. Chem. 2021, 21, 442–460. [Google Scholar] [CrossRef]
- Lu, L.; Su, S.; Yang, H.; Jiang, S. Antivirals with common targets against highly pathogenic viruses. Cell 2021, 184, 1604–1620. [Google Scholar] [CrossRef]
- Jones, J.C.; Yen, H.L.; Adams, P.; Armstrong, K.; Govorkova, E.A. Influenza antivirals and their role in pandemic preparedness. Antivir. Res. 2023, 210, 105499. [Google Scholar] [CrossRef]
- Muralidar, S.; Ambi, S.V.; Sekaran, S.; Krishnan, U.M. The emergence of COVID-19 as a global pandemic: Understanding the epidemiology, immune response and potential therapeutic targets of SARS-CoV-2. Biochimie 2020, 179, 85–100. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhang, Y.Q.; Xu, L.D.; Xiao, L.; Feng, Y.; Wang, B.; Huang, Y.W. Role of heat shock protein 90 as an antiviral target for swine enteric coronaviruses. Virus Res. 2023, 329, 199103. [Google Scholar] [CrossRef]
- Hu, X.; Cui, J.; Chen, J.; Du, S.; Wang, X.; Zhang, Y.; Qian, J.; Chen, H.; Wei, F.; Cai, Q.; et al. Identification of hACE2-interacting sites in SARS-CoV-2 spike receptor binding domain for antiviral drugs screening. Virus Res. 2022, 321, 198915. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drugs | Class | Virus | Targets | EC50 (µM) | IC50 (µM) | Experimental Setting | Reference |
|---|---|---|---|---|---|---|---|
| FD001 | Small-molecule compounds | SARS-CoV-2 | S | 1.58 | 0.18 | In vitro | [24] |
| Arbidol | Indolyl carboxylic acid | SARS-CoV-2 | S | 4.11 | / | In vitro | [25] |
| Phycobilin | Natural products | SARS-CoV-2 | PLpro | / | 62 | In vitro | [26] |
| Adriamycin | Anthracycline | MERS-CoV | PLpro | / | 1.67 | In vitro | [27] |
| F0213 | Lead inhibitor | SARS-CoV-2 | PLpro | 4.5 | 7.4 | In vitro | [28] |
| Xanthohumol | Chalcones | SARS-CoV-2 | 3CLpro | 5.93 | 1.53 | In vitro | [29] |
| Hypericin | Anthraquinones | PEDV | 3CLpro | 3.53 | 5.09 | In vitro | [30] |
| Tomatidine | Alkaloids | PEDV | 3CLpro | / | 3.45 | In vitro | [31] |
| Octyl gallate | Aliphatics | PEDV | 3CLpro | / | 22.15 | In vitro | [32] |
| Paxlovid | Compound antiviral drug | SARS-CoV-2 | 3CLpro | / | / | / | [33] |
| Remdesivir | Carboxylic ester | SARS-CoV-2 | RdRp | 0.62 | 0.65 | In vitro | [34] |
| Gossypol | Gossypol | SARS-CoV-2 | RdRp | 0.31 | 0.76 | In vitro | [35] |
| SSYA10-001 | Heterocyclic compounds | SARS-CoV | RNA helicase | 7 | 8.95 | In vitro | [36] |
| Hexamethylene amiloride | Pyrazines | HCoV-229E | E | 1.34 | / | In vitro | [37] |
| Hypericin | Anthraquinones | PEDV | N | / | / | / | [38] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Zhu, Q.; Xing, X.; Sun, D. A Mini-Review on the Common Antiviral Drug Targets of Coronavirus. Microorganisms 2024, 12, 600. https://doi.org/10.3390/microorganisms12030600
Wang J, Zhu Q, Xing X, Sun D. A Mini-Review on the Common Antiviral Drug Targets of Coronavirus. Microorganisms. 2024; 12(3):600. https://doi.org/10.3390/microorganisms12030600
Chicago/Turabian StyleWang, Jun, Qinghe Zhu, Xiaoxu Xing, and Dongbo Sun. 2024. "A Mini-Review on the Common Antiviral Drug Targets of Coronavirus" Microorganisms 12, no. 3: 600. https://doi.org/10.3390/microorganisms12030600
APA StyleWang, J., Zhu, Q., Xing, X., & Sun, D. (2024). A Mini-Review on the Common Antiviral Drug Targets of Coronavirus. Microorganisms, 12(3), 600. https://doi.org/10.3390/microorganisms12030600