
Cefotaxime Exposure-Caused Oxidative Stress, Intestinal Damage and Gut Microbial Disruption in Artemia sinica

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Animal Culture, Solution Prepare and Antibiotic Exposure
2.2. Oxidative Stress Assessment
2.3. Morphology and Gut Histomorphology
2.4. DNA Extraction and PacBio Sequencing of 16S rRNA Gene
2.5. Bioinformatics and Statistics
3. Result
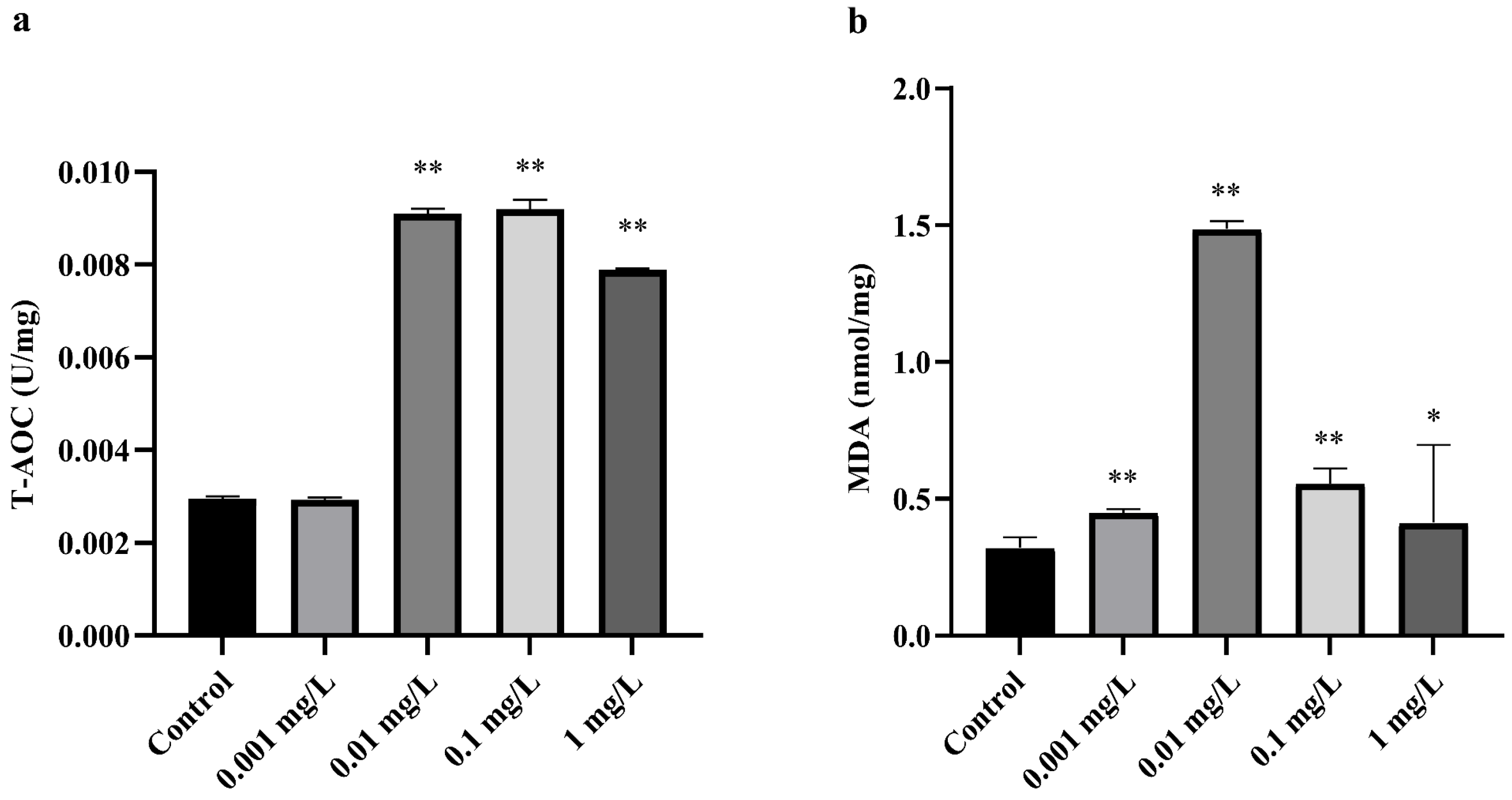
3.1. Effect of CTX on Oxidative Stress in A. sinica
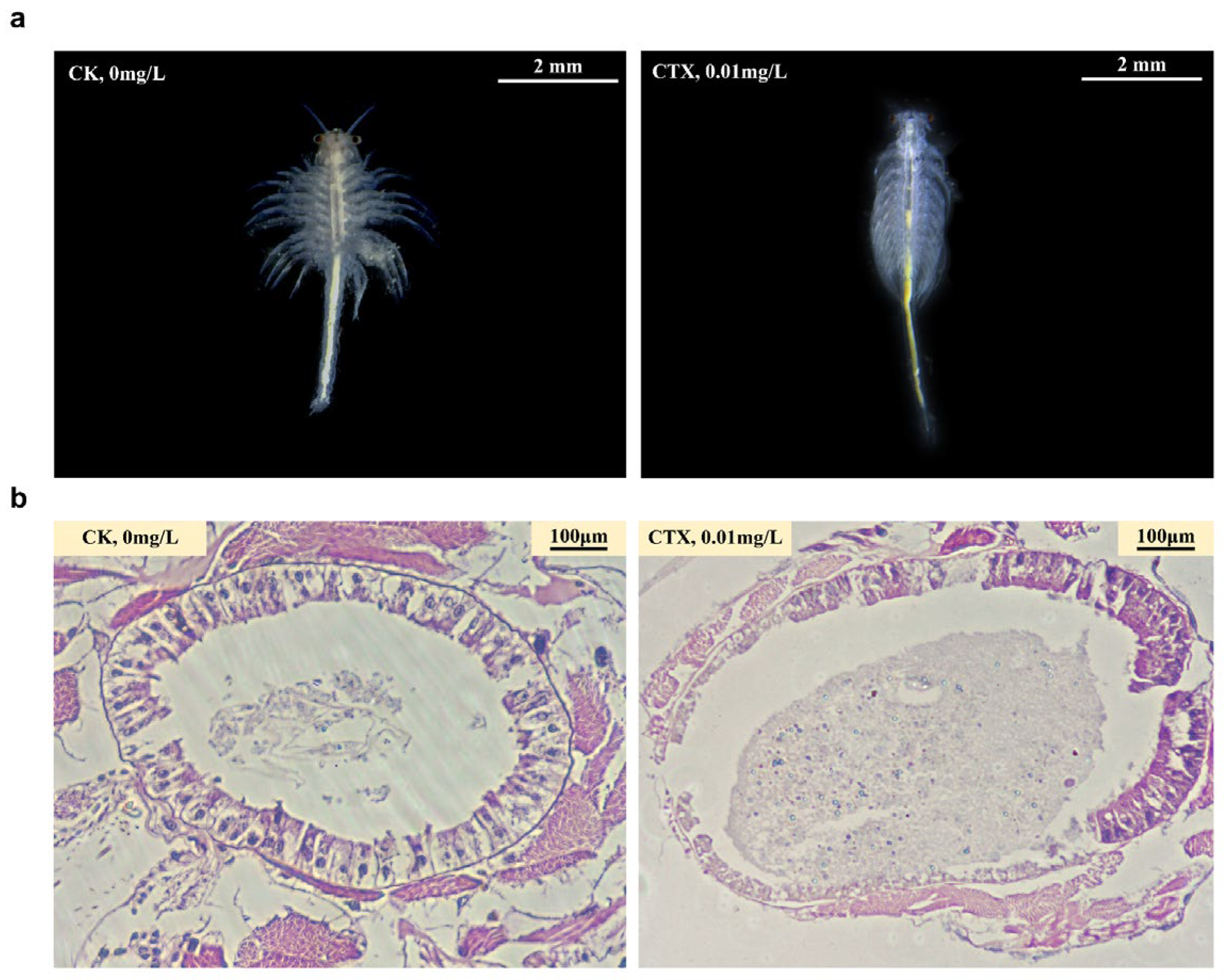
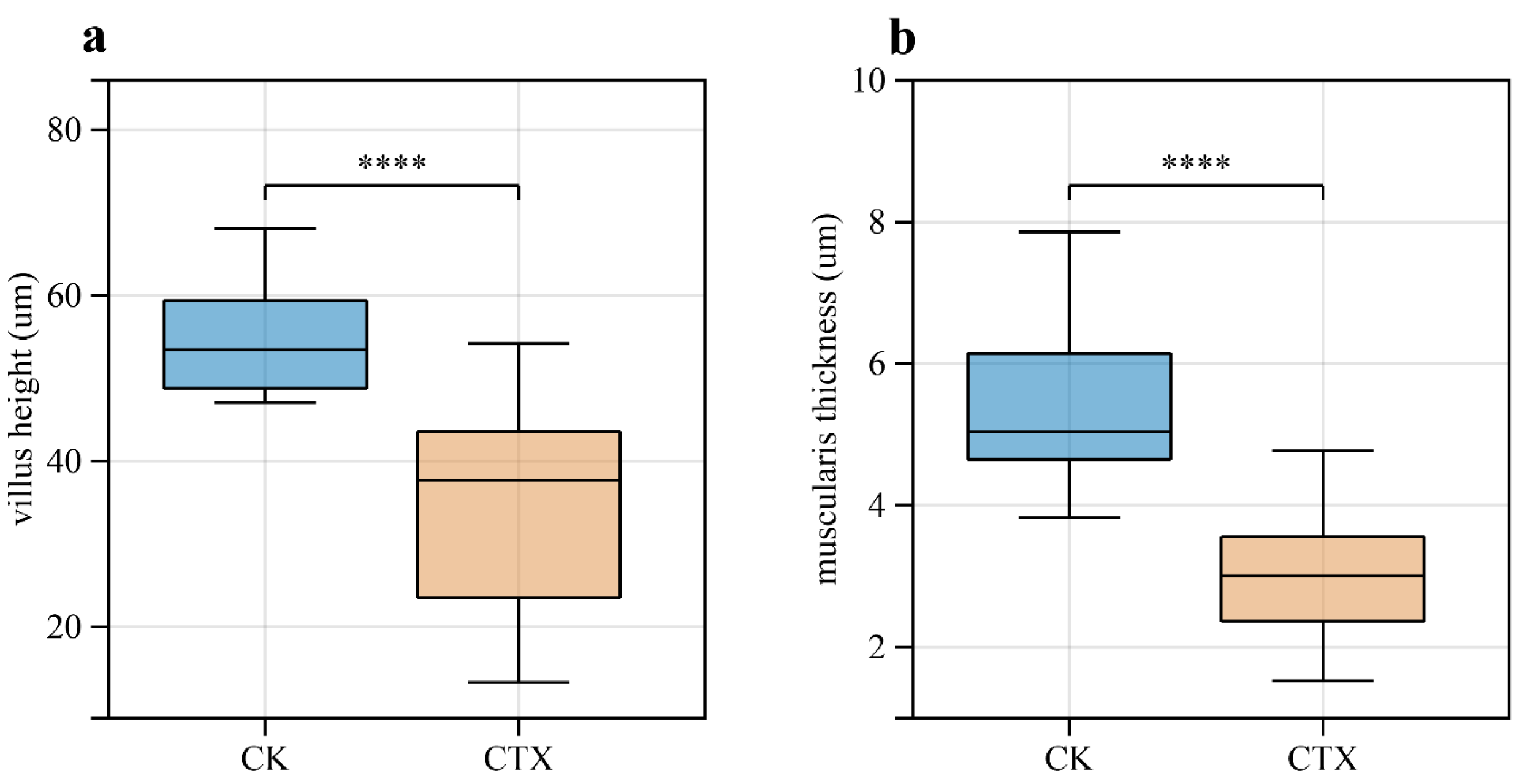
3.2. Morphology Changes and Destruction of Intestinal Structural Integrity
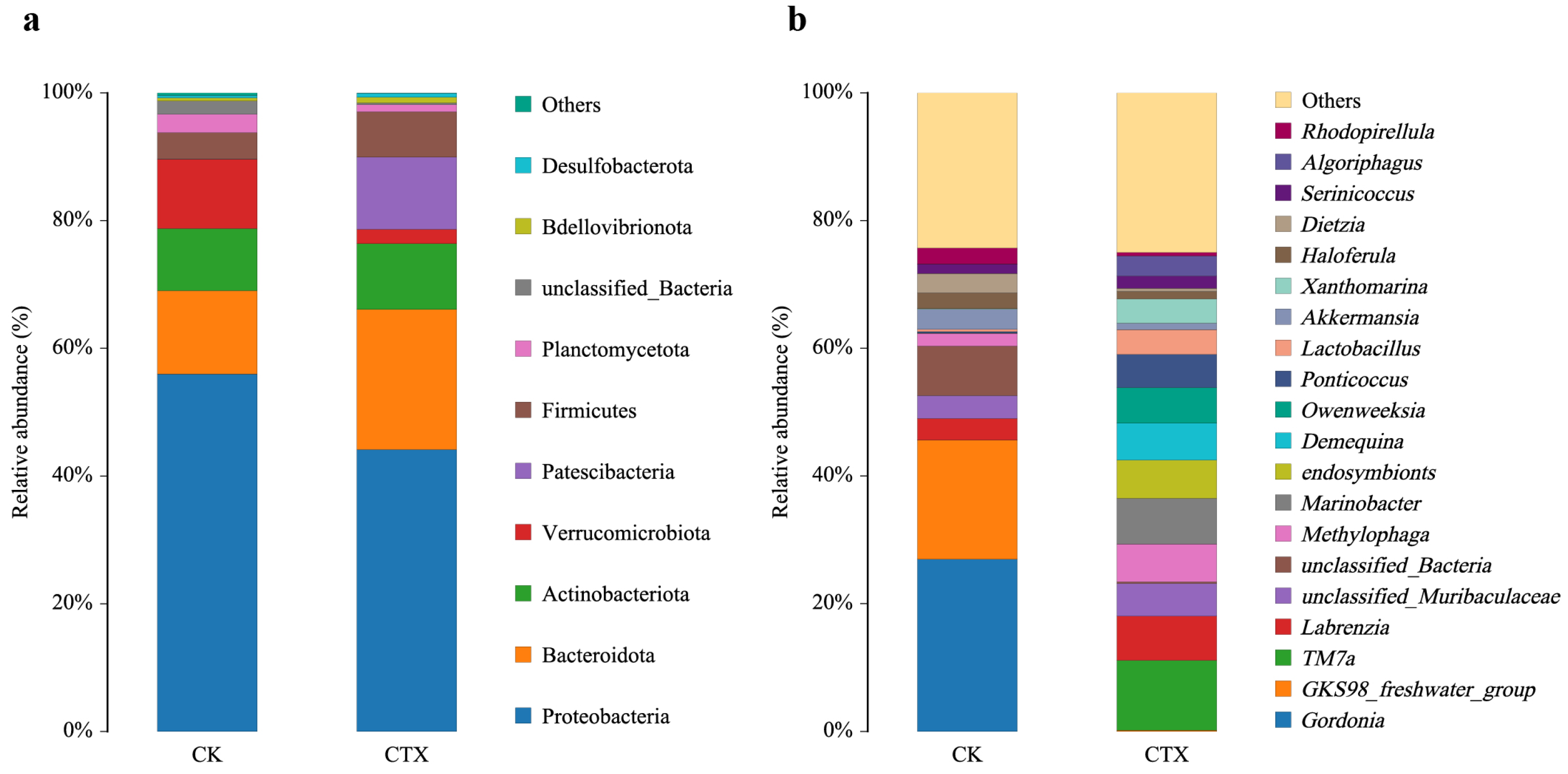
3.3. Gut Microbial Community Composition and Diversity
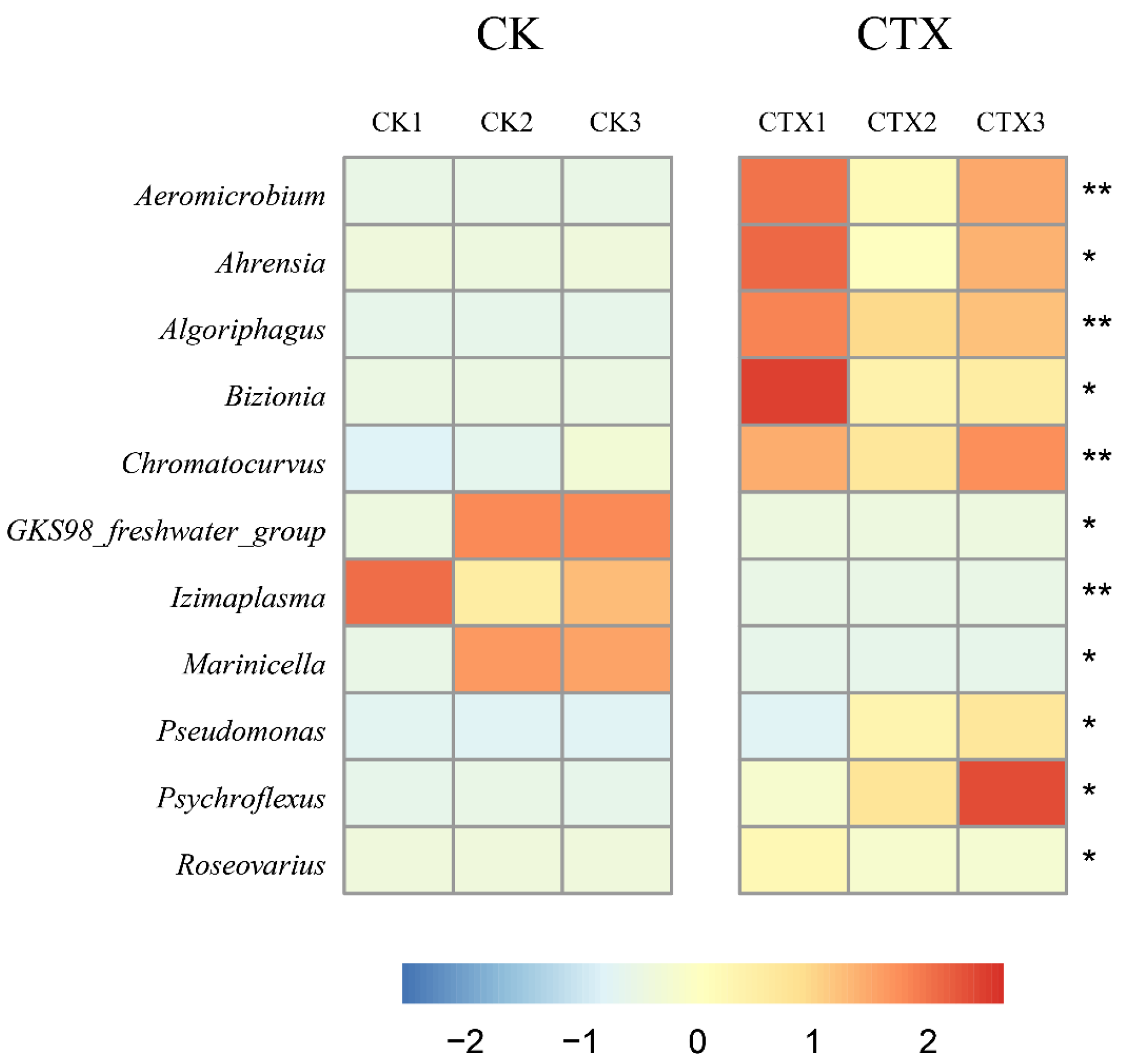
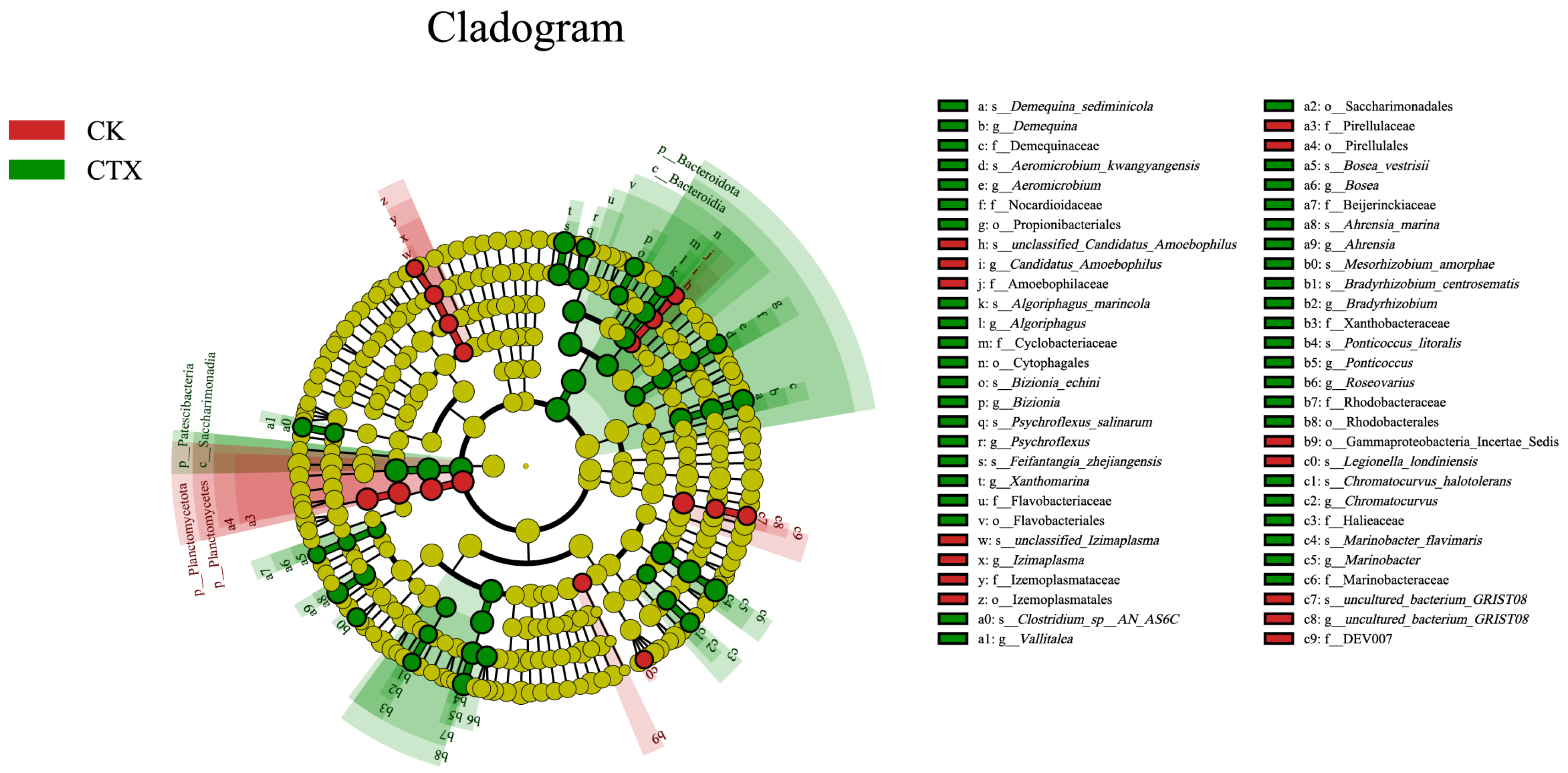
3.4. Differences in the Gut Microbiome between Groups
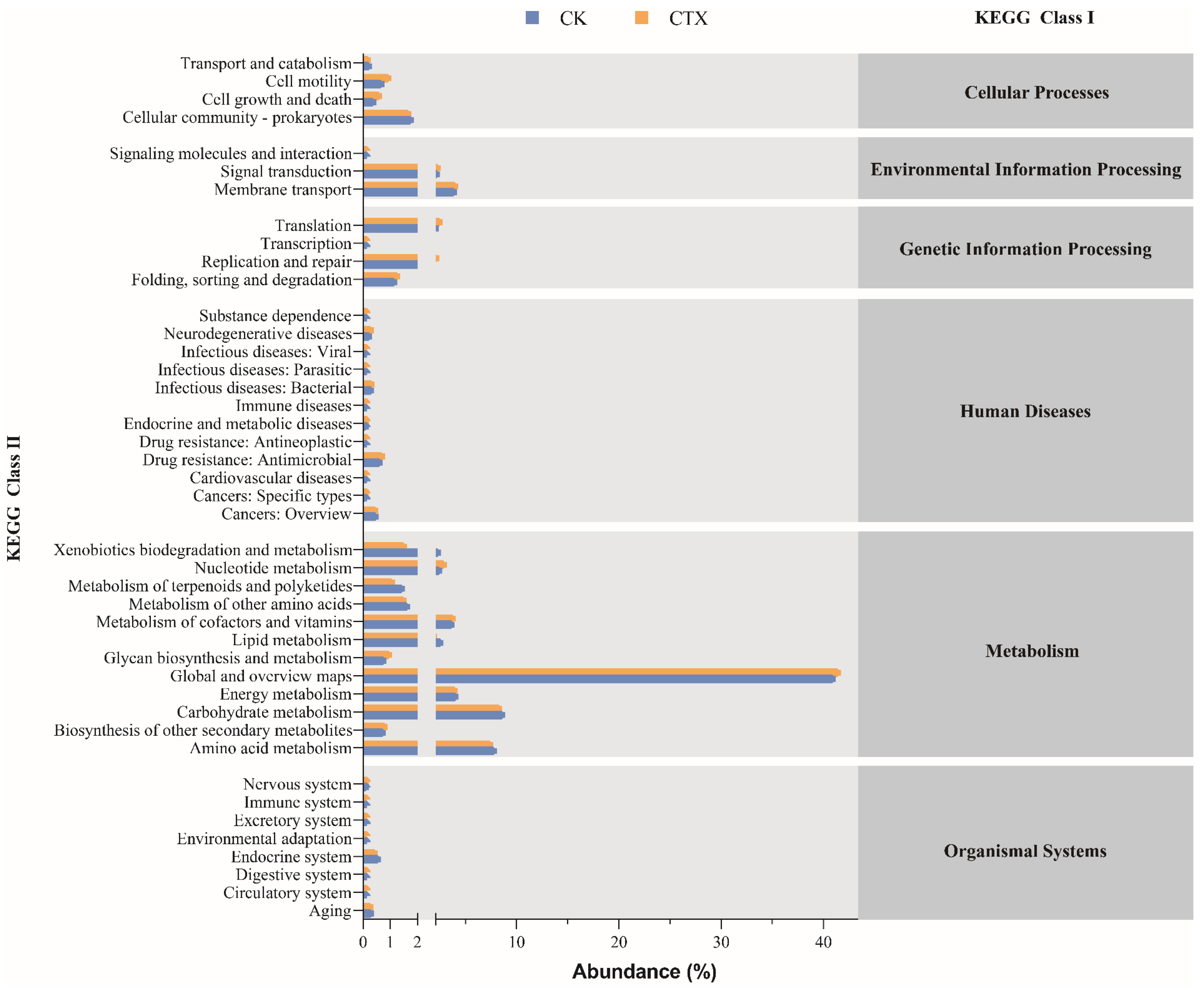
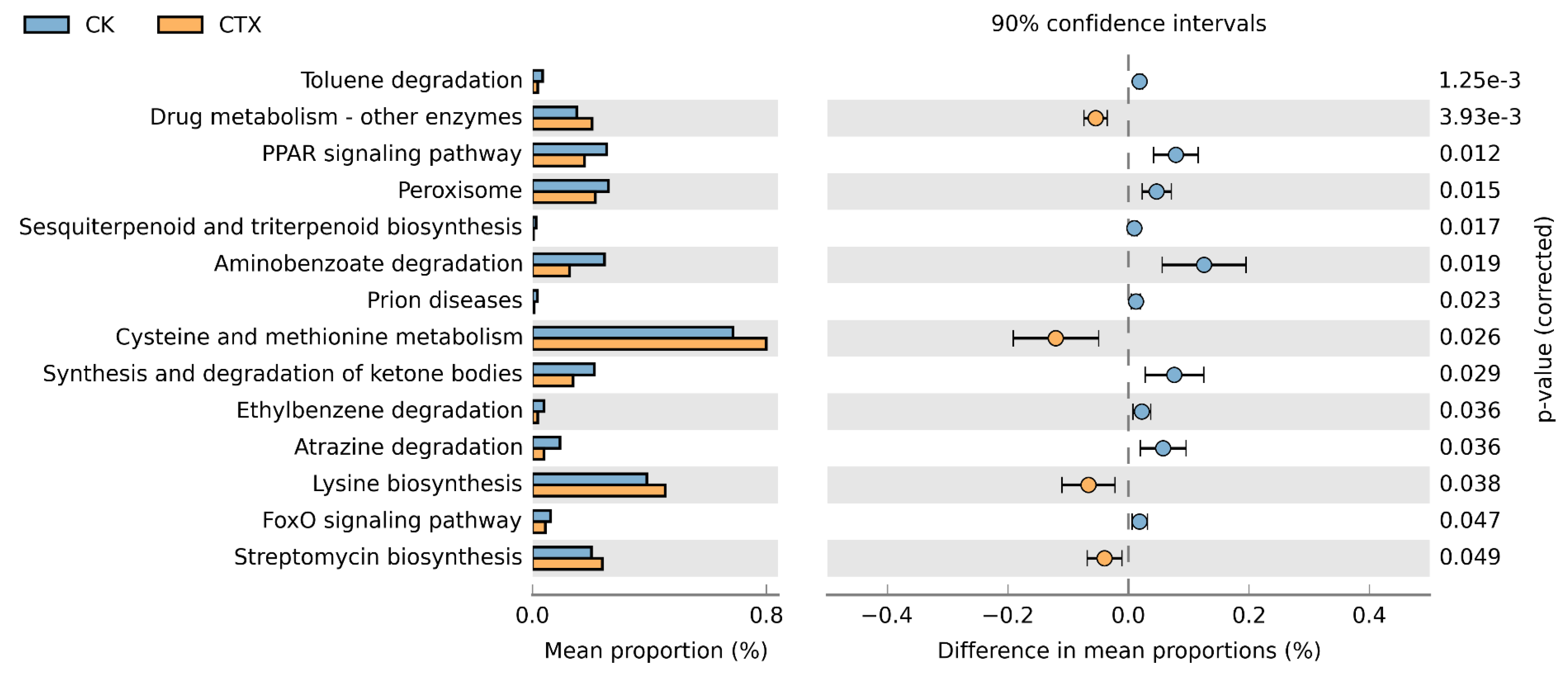
3.5. Function Prediction of Gut Microbiota
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kolkovski, S.; Curnow, J.; King, J. Intensive rearing system for fish larvae research II: Artemia hatching and enriching system. Aquac. Eng. 2004, 31, 309–317. [Google Scholar] [CrossRef]
- Batel, A.; Linti, F.; Scherer, M.; Erdinger, L.; Braunbeck, T. Transfer of benzo[a]pyrene from microplastics to Artemia nauplii and further to zebrafish via a trophic food web experiment: CYP1A induction and visual tracking of persistent organic pollutants. Environ. Toxicol. Chem. 2016, 35, 1656–1666. [Google Scholar] [CrossRef]
- Mirzaei VandKhanghah, M.; Hedayati, A.; Nazeri, S.; Mohammadi Azarm, H.; Ghorbani, R. Biomagnification of Copper Along the Aquatic Food Chain (Artemia franciscana, Danio rerio, and Astronotus ocellatus). Biol. Trace Elem. Res. 2022, 200, 1854–1860. [Google Scholar] [CrossRef]
- Ntungwe, N.E.; Dominguez-Martin, E.M.; Roberto, A.; Tavares, J.; Isca, V.M.S.; Pereira, P.; Cebola, M.J.; Rijo, P. Artemia species: An Important Tool to Screen General Toxicity Samples. Curr. Pharm Des. 2020, 26, 2892–2908. [Google Scholar] [CrossRef]
- Bergami, E.; Bocci, E.; Vannuccini, M.L.; Monopoli, M.; Salvati, A.; Dawson, K.A.; Corsi, I. Nano-sized polystyrene affects feeding, behavior and physiology of brine shrimp Artemia franciscana larvae. Ecotoxicol. Environ. Saf. 2016, 123, 18–25. [Google Scholar] [CrossRef]
- Zhu, B.; Zhu, S.; Li, J.; Hui, X.; Wang, G.-X. The developmental toxicity, bioaccumulation and distribution of oxidized single walled carbon nanotubes in Artemia salina. Toxicol. Res. 2018, 7, 897–906. [Google Scholar] [CrossRef]
- Banti, C.N.; Hadjikakou, S.K. Evaluation of Toxicity with Brine Shrimp Assay. Bio-Protocol 2021, 11, e3895. [Google Scholar] [CrossRef]
- Libralato, G.; Prato, E.; Migliore, L.; Cicero, A.M.; Manfra, L. A review of toxicity testing protocols and endpoints with Artemia spp. Ecol. Indic. 2016, 69, 35–49. [Google Scholar] [CrossRef]
- Albarano, L.; Ruocco, N.; Lofrano, G.; Guida, M.; Libralato, G. Genotoxicity in Artemia spp.: An old model with new sensitive endpoints. Aquat. Toxicol. 2022, 252, 106320. [Google Scholar] [CrossRef]
- Martinez, J.L. Environmental pollution by antibiotics and by antibiotic resistance determinants. Environ. Pollut. 2009, 157, 2893–2902. [Google Scholar] [CrossRef]
- Kraemer, S.A.; Ramachandran, A.; Perron, G.G. Antibiotic Pollution in the Environment: From Microbial Ecology to Public Policy. Microorganisms 2019, 7, 180. [Google Scholar] [CrossRef]
- Dwyer, D.J.; Belenky, P.A.; Yang, J.H.; MacDonald, I.C.; Martell, J.D.; Takahashi, N.; Chan, C.T.Y.; Lobritz, M.A.; Braff, D.; Schwarz, E.G.; et al. Antibiotics induce redox-related physiological alterations as part of their lethality. Proc. Natl. Acad. Sci. USA 2014, 111, E2100–E2109. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, L.; Zhuang, H.; Li, X.; Gao, X.; An, Z.; Liu, X.; Yang, H.; Wei, W.; Zhang, X. Excessive use of enrofloxacin leads to growth inhibition of juvenile giant freshwater prawn Macrobrachium rosenbergii. Ecotoxicol. Environ. Saf. 2019, 169, 344–352. [Google Scholar] [CrossRef]
- Liang, X.; Wang, F.; Li, K.; Nie, X.; Fang, H. Effects of norfloxacin nicotinate on the early life stage of zebrafish (Danio rerio): Developmental toxicity, oxidative stress and immunotoxicity. Fish Shellfish. Immunol. 2020, 96, 262–269. [Google Scholar] [CrossRef]
- Jin, M.K.; Yang, Y.T.; Zhao, C.X.; Huang, X.R.; Chen, H.M.; Zhao, W.L.; Yang, X.R.; Zhu, Y.G.; Liu, H.J. ROS as a key player in quinolone antibiotic stress on Arabidopsis thaliana: From the perspective of photosystem function, oxidative stress and phyllosphere microbiome. Sci. Total Environ. 2022, 848, 157821. [Google Scholar] [CrossRef]
- Huang, J.; Liao, L.; Wang, G.; Du, Z.; Wu, Z. Reproductive toxicity of enrofloxacin in Caenorhabditis elegans involves oxidative stress-induced cell apoptosis. J Environ. Sci. 2023, 127, 726–737. [Google Scholar] [CrossRef]
- Relman, D.A.; Lipsitch, M. Microbiome as a tool and a target in the effort to address antimicrobial resistance. Proc. Natl. Acad. Sci. USA 2018, 115, 12902–12910. [Google Scholar] [CrossRef]
- Quinlan, E.L.; Nietch, C.T.; Blocksom, K.; Lazorchak, J.M.; Batt, A.L.; Griffiths, R.; Klemm, D.J. Temporal Dynamics of Periphyton Exposed to Tetracycline in Stream Mesocosms. Environ. Sci. Technol. 2011, 45, 10684–10690. [Google Scholar] [CrossRef]
- Ribeiro, A.R.; Sures, B.; Schmidt, T.C. Cephalosporin antibiotics in the aquatic environment: A critical review of occurrence, fate, ecotoxicity and removal technologies. Environ. Pollut. 2018, 241, 1153–1166. [Google Scholar] [CrossRef]
- Meropoulis, S.; Giannoulia, S.; Skandalis, S.; Rassias, G.; Aggelopoulos, C.A. Key-study on plasma-induced degradation of cephalosporins in water: Process optimization, assessment of degradation mechanisms and residual toxicity. Sep. Purif. Technol. 2022, 298, 121639. [Google Scholar] [CrossRef]
- Bu, Q.; Wang, B.; Huang, J.; Deng, S.; Yu, G. Pharmaceuticals and personal care products in the aquatic environment in China: A review. J. Hazard Mater. 2013, 262, 189–211. [Google Scholar] [CrossRef]
- Song, C.; Zhang, C.; Fan, L.; Qiu, L.; Wu, W.; Meng, S.; Hu, G.; Kamira, B.; Chen, J. Occurrence of antibiotics and their impacts to primary productivity in fishponds around Tai Lake, China. Chemosphere 2016, 161, 127–135. [Google Scholar] [CrossRef]
- Lin, H.; Chen, L.; Li, H.; Luo, Z.; Lu, J.; Yang, Z. Pharmaceutically active compounds in the Xiangjiang River, China: Distribution pattern, source apportionment, and risk assessment. Sci. Total Environ. 2018, 636, 975–984. [Google Scholar] [CrossRef]
- Tang, X. Study on Residues and Removal Technologies of Cephalosporin Antibiotics in Pharmaceutical Wastewater Treatment Plant. Master’s Thesis, Tsinghua University, Beijing, China, 2015. [Google Scholar]
- Baquero, F.; Martinez, J.L.; Canton, R. Antibiotics and antibiotic resistance in water environments. Curr. Opin. Biotechnol. 2008, 19, 260–265. [Google Scholar] [CrossRef]
- Yang, J.; Ying, G.; Zhao, J.; Tao, R.; Haochang, S. Pollution characteristics of antibiotics in complete sets of farming system. Environ. Chem. 2015, 34, 54–59. [Google Scholar]
- He, X.; Xu, Y.; Chen, J.; Ling, J.; Li, Y.; Huang, L.; Zhou, X.; Zheng, L.; Xie, G. Evolution of corresponding resistance genes in the water of fish tanks with multiple stresses of antibiotics and heavy metals. Water Res. 2017, 124, 39–48. [Google Scholar] [CrossRef]
- Zhou, M.; Xu, Y.; Ouyang, P.; Ling, J.; Cai, Q.; Huang, L.; Zhou, X.; Zheng, L. Evolution and distribution of resistance genes and bacterial community in water and biofilm of a simulated fish-duck integrated pond with stress. Chemosphere 2020, 245, 125549. [Google Scholar] [CrossRef]
- Jijie, R.; Mihalache, G.; Balmus, I.-M.; Strungaru, S.-A.; Baltag, E.S.; Ciobica, A.; Nicoara, M.; Faggio, C. Zebrafish as a Screening Model to Study the Single and Joint Effects of Antibiotics. Pharmaceuticals 2021, 14, 578. [Google Scholar] [CrossRef]
- Han, Y.; Zheng, Y.; Zhang, J.; Hu, C. Neurobehavioral Effects of Cephalosporins: Assessment of Locomotors Activity, Motor and Sensory Development in Zebrafish. Front. Pharmacol. 2018, 9, 160. [Google Scholar] [CrossRef]
- Qiu, W.; Liu, X.; Yang, F.; Li, R.; Xiong, Y.; Fu, C.; Li, G.; Liu, S.; Zheng, C. Single and joint toxic effects of four antibiotics on some metabolic pathways of zebrafish (Danio rerio) larvae. Sci. Total Environ. 2020, 716, 137062. [Google Scholar] [CrossRef]
- Wang, Y.; Shen, H.-Y. Effect of Cefotaxime on the CAT Activities and GSH Contents of Zebrafish. IOP Conf. Ser. Earth Environ. Sci. 2018, 153, 062071. [Google Scholar] [CrossRef]
- Xue, X.; Li, X.; Liu, J.; Zhu, L.; Zhou, L.; Jia, J.; Wang, Z. Field-realistic dose of cefotaxime enhances potential mobility of β-lactam resistance genes in the gut microbiota of zebrafish (Danio rerio). Aquat. Toxicol. 2023, 257, 106459. [Google Scholar] [CrossRef]
- Ahmad, A.; Dutta, J.J.T.I. Ecotoxicological Studies of Cephalosporin Antibiotics on Daphnia Magna. Toxicol. Int. 2019, 25, 169–178. [Google Scholar] [CrossRef]
- Kunst, C.; Schmid, S.; Michalski, M.; Tumen, D.; Buttenschon, J.; Muller, M.; Gulow, K. The Influence of Gut Microbiota on Oxidative Stress and the Immune System. Biomedicines 2023, 11, 1388. [Google Scholar] [CrossRef]
- Palleja, A.; Mikkelsen, K.H.; Forslund, S.K.; Kashani, A.; Allin, K.H.; Nielsen, T.; Hansen, T.H.; Liang, S.; Feng, Q.; Zhang, C.; et al. Recovery of gut microbiota of healthy adults following antibiotic exposure. Nat. Microbiol. 2018, 3, 1255–1265. [Google Scholar] [CrossRef]
- Lekang, K.; Shekhar, S.; Berild, D.; Petersen, F.C.; Winther-Larsen, H.C. Effects of different amoxicillin treatment durations on microbiome diversity and composition in the gut. PLoS ONE 2022, 17, e0275737. [Google Scholar] [CrossRef]
- Limbu, S.M.; Zhou, L.; Sun, S.X.; Zhang, M.L.; Du, Z.Y. Chronic exposure to low environmental concentrations and legal aquaculture doses of antibiotics cause systemic adverse effects in Nile tilapia and provoke differential human health risk. Environ. Int. 2018, 115, 205–219. [Google Scholar] [CrossRef]
- Wang, Z.; Sun, X.; Wang, W.; Zheng, M.; Zhang, D.; Yin, H. NF-κB-coupled IL17 mediates inflammatory signaling and intestinal inflammation in Artemia sinica. Fish Shellfish Immunol. 2022, 128, 38–49. [Google Scholar] [CrossRef]
- Kalghatgi, S.; Spina, C.S.; Costello, J.C.; Liesa, M.; Morones-Ramirez, J.R.; Slomovic, S.; Molina, A.; Shirihai, O.S.; Collins, J.J. Bactericidal antibiotics induce mitochondrial dysfunction and oxidative damage in Mammalian cells. Sci. Transl. Med. 2013, 5, 192ra185. [Google Scholar] [CrossRef]
- Deavall, D.G.; Martin, E.A.; Horner, J.M.; Roberts, R. Drug-induced oxidative stress and toxicity. J. Toxicol. 2012, 2012, 645460. [Google Scholar] [CrossRef]
- Dzoyem, J.P.; Kuete, V.; Eloff, J.N. 23—Biochemical Parameters in Toxicological Studies in Africa: Significance, Principle of Methods, Data Interpretation, and Use in Plant Screenings. In Toxicological Survey of African Medicinal Plants; Kuete, V., Ed.; Elsevier: Amsterdam, The Netherlands, 2014; pp. 659–715. [Google Scholar]
- Cordiano, R.; Di Gioacchino, M.; Mangifesta, R.; Panzera, C.; Gangemi, S.; Minciullo, P.L. Malondialdehyde as a Potential Oxidative Stress Marker for Allergy-Oriented Diseases: An Update. Molecules 2023, 28, 5979. [Google Scholar] [CrossRef]
- Shi, D.; Hao, H.; Wei, Z.; Yang, D.; Yin, J.; Li, H.; Chen, Z.; Yang, Z.; Chen, T.; Zhou, S.; et al. Combined exposure to non-antibiotic pharmaceutics and antibiotics in the gut synergistically promote the development of multi-drug-resistance in Escherichia coli. Gut Microbes 2022, 14, 2018901. [Google Scholar] [CrossRef]
- Siu, G.M.; Draper, H.H. Metabolism of malonaldehyde in vivo and in vitro. Lipids 1982, 17, 349–355. [Google Scholar] [CrossRef]
- Esterbauer, H.; Schaur, R.J.; Zollner, H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic. Biol. Med. 1991, 11, 81–128. [Google Scholar] [CrossRef]
- Piazza, R.S.; Trevisan, R.; Flores-Nunes, F.; Toledo-Silva, G.; Wendt, N.; Mattos, J.J.; Lima, D.; Taniguchi, S.; Sasaki, S.T.; Mello, A.C.; et al. Exposure to phenanthrene and depuration: Changes on gene transcription, enzymatic activity and lipid peroxidation in gill of scallops Nodipecten nodosus. Aquat. Toxicol. 2016, 177, 146–155. [Google Scholar] [CrossRef]
- Lima, D.; Zacchi, F.L.; Mattos, J.J.; Flores-Nunes, F.; Gomes, C.; de Mello, A.C.P.; Siebert, M.N.; Piazza, C.E.; Taniguchi, S.; Sasaki, S.T.; et al. Molecular and cellular effects of temperature in oysters Crassostrea brasiliana exposed to phenanthrene. Chemosphere 2018, 209, 307–318. [Google Scholar] [CrossRef]
- Garcia, D.; Lima, D.; da Silva, D.G.H.; de Almeida, E.A. Decreased malondialdehyde levels in fish (Astyanax altiparanae) exposed to diesel: Evidence of metabolism by aldehyde dehydrogenase in the liver and excretion in water. Ecotoxicol. Environ. Saf. 2020, 190, 110107. [Google Scholar] [CrossRef]
- Diaz de Barboza, G.; Guizzardi, S.; Moine, L.; Tolosa de Talamoni, N. Oxidative stress, antioxidants and intestinal calcium absorption. World J. Gastroenterol. 2017, 23, 2841–2853. [Google Scholar] [CrossRef]
- Li, L.; Peng, P.; Ding, N.; Jia, W.; Huang, C.; Tang, Y. Oxidative Stress, Inflammation, Gut Dysbiosis: What Can Polyphenols Do in Inflammatory Bowel Disease? Antioxidants 2023, 12, 967. [Google Scholar] [CrossRef]
- Cheng, R.; Guo, J.; Pu, F.; Wan, C.; Shi, L.; Li, H.; Yang, Y.; Huang, C.; Li, M.; He, F. Loading ceftriaxone, vancomycin, and Bifidobacteria bifidum TMC3115 to neonatal mice could differently and consequently affect intestinal microbiota and immunity in adulthood. Sci. Rep. 2019, 9, 3254. [Google Scholar] [CrossRef]
- Wan, Q.; Cheng, R.; Guo, J.; Wang, K.; Shen, X.; Pu, F.; Li, M.; Fang, H. Effect of ceftriaxone on the intestinal epithelium and microbiota in neonatal mice. Chin. J. Contemp. Pediatr. 2018, 20, 318–325. [Google Scholar]
- Kim, J.W.; Choi, C.S.; Kim, K.C.; Park, J.H.; Seung, H.; Joo, S.H.; Yang, S.M.; Shin, C.Y.; Park, S.H. Gastrointestinal tract abnormalities induced by prenatal valproic Acid exposure in rat offspring. Toxicol. Res. 2013, 29, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Chelakkot, C.; Ghim, J.; Ryu, S.H. Mechanisms regulating intestinal barrier integrity and its pathological implications. Exp. Mol. Med. 2018, 50, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhu, X.; Yu, X.; Novak, P.; Gui, Q.; Yin, K. Enhancing intestinal barrier efficiency: A novel metabolic diseases therapy. Front. Nutr. 2023, 10, 1120168. [Google Scholar] [CrossRef]
- Charo-Karisa, H.; Ali, S.E.; Marijani, E.; Ibrahim, N.A.; Trinh, T.Q.; Chadag, M.V.; Benzie, J.A.H. Genetic parameters for black spot disease (diplopstomiasis) caused by Uvulifer sp. infection in Nile tilapia (Oreochromis niloticus L.). Aquaculture 2021, 532, 736039. [Google Scholar] [CrossRef]
- Bhalodi, A.A.; van Engelen, T.S.R.; Virk, H.S.; Wiersinga, W.J. Impact of antimicrobial therapy on the gut microbiome. J. Antimicrob. Chemother. 2019, 74, i6–i15. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Chen, L.; Mai, G.; Zhang, H.; Yang, J.; Deng, D.; Ma, Y. Dynamics of the gut microbiota in developmental stages of Litopenaeus vannamei reveal its association with body weight. Sci. Rep. 2019, 9, 734. [Google Scholar] [CrossRef] [PubMed]
- Shui, Y.; Guan, Z.B.; Liu, G.F.; Fan, L.M. Gut microbiota of red swamp crayfish Procambarus clarkii in integrated crayfish-rice cultivation model. AMB Express 2020, 10, 5. [Google Scholar] [CrossRef] [PubMed]
- Holt, C.C.; Bass, D.; Stentiford, G.D.; van der Giezen, M. Understanding the role of the shrimp gut microbiome in health and disease. J. Invertebr. Pathol. 2021, 186, 107387. [Google Scholar] [CrossRef]
- Quiroz, M.; Triado-Margarit, X.; Casamayor, E.O.; Gajardo, G. Comparison of Artemia-bacteria associations in brines, laboratory cultures and the gut environment: A study based on Chilean hypersaline environments. Extremophiles 2015, 19, 135–147. [Google Scholar] [CrossRef]
- Nixon, S.L.; Daly, R.A.; Borton, M.A.; Solden, L.M.; Welch, S.A.; Cole, D.R.; Mouser, P.J.; Wilkins, M.J.; Wrighton, K.C. Genome-Resolved Metagenomics Extends the Environmental Distribution of the Verrucomicrobia Phylum to the Deep Terrestrial Subsurface. mSphere 2019, 4, 613–619. [Google Scholar] [CrossRef]
- King, G.M.; Judd, C.; Kuske, C.R.; Smith, C. Analysis of Stomach and Gut Microbiomes of the Eastern Oyster (Crassostrea virginica) from Coastal Louisiana, USA. PLoS ONE 2012, 7, e51475. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Wang, Z.; Chen, M.; Qu, Y.; Li, J.; Zhou, A.; Xie, S.; Zeng, F.; Zou, J. Microbiota comparison of Pacific white shrimp intestine and sediment at freshwater and marine cultured environment. Sci. Total Environ. 2019, 657, 1194–1204. [Google Scholar] [CrossRef]
- Sun, Y.; Han, W.; Liu, J.; Huang, X.; Zhou, W.; Zhang, J.; Cheng, Y. Bacterial community compositions of crab intestine, surrounding water, and sediment in two different feeding modes of Eriocheir sinensis. Aquac. Rep. 2020, 16, 100236. [Google Scholar] [CrossRef]
- Lein, E.Y.; Mohamad Lal, M.T.; Venmathi Maran, B.A.; Ch’ng, C.L.; Hamasaki, K.; Sano, M.; Tuzan, A.D. Gastrointestinal Microbiota of Spiny Lobster: A Review. Fishes 2022, 7, 108. [Google Scholar] [CrossRef]
- Glover, J.S.; Ticer, T.D.; Engevik, M.A. Characterizing the mucin-degrading capacity of the human gut microbiota. Sci. Rep. 2022, 12, 8456. [Google Scholar] [CrossRef]
- Yu, W.; Dai, W.; Tao, Z.; Jinbo, X. Characterizing the compositional and functional structures of intestinal microflora between healthy and diseased Litopenaeus vannamei. J. Fish. China 2018, 42, 399–409. [Google Scholar]
- Sun, Y.; Wang, X.; Li, L.; Zhong, C.; Zhang, Y.; Yang, X.; Li, M.; Yang, C. The role of gut microbiota in intestinal disease: From an oxidative stress perspective. Front. Microbiol. 2024, 15, 1328324. [Google Scholar] [CrossRef]
- Cabral, D.J.; Penumutchu, S.; Reinhart, E.M.; Zhang, C.; Korry, B.J.; Wurster, J.I.; Nilson, R.; Guang, A.; Sano, W.H.; Rowan-Nash, A.D.; et al. Microbial Metabolism Modulates Antibiotic Susceptibility within the Murine Gut Microbiome. Cell Metab. 2019, 30, 800–823.e807. [Google Scholar] [CrossRef]
- Hildebrand, F.; Moitinho-Silva, L.; Blasche, S.; Jahn, M.T.; Gossmann, T.I.; Huerta-Cepas, J.; Hercog, R.; Luetge, M.; Bahram, M.; Pryszlak, A.; et al. Antibiotics-induced monodominance of a novel gut bacterial order. Gut 2019, 68, 1781–1790. [Google Scholar] [CrossRef]
- Wu, S.; Bhat, Z.F.; Gounder, R.S.; Mohamed Ahmed, I.A.; Al-Juhaimi, F.Y.; Ding, Y.; Bekhit, A.E.A. Effect of Dietary Protein and Processing on Gut Microbiota-A Systematic Review. Nutrients 2022, 14, 453. [Google Scholar] [CrossRef] [PubMed]
- Mokhtari, P.; Metos, J.; Anandh Babu, P.V. Impact of type 1 diabetes on the composition and functional potential of gut microbiome in children and adolescents: Possible mechanisms, current knowledge, and challenges. Gut Microbes 2021, 13, 1926841. [Google Scholar] [CrossRef] [PubMed]
- Tlaskalova-Hogenova, H.; Stepankova, R.; Kozakova, H.; Hudcovic, T.; Vannucci, L.; Tuckova, L.; Rossmann, P.; Hrncir, T.; Kverka, M.; Zakostelska, Z.; et al. The role of gut microbiota (commensal bacteria) and the mucosal barrier in the pathogenesis of inflammatory and autoimmune diseases and cancer: Contribution of germ-free and gnotobiotic animal models of human diseases. Cell Mol Immunol. 2011, 8, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Boursier, J.; Mueller, O.; Barret, M.; Machado, M.; Fizanne, L.; Araujo-Perez, F.; Guy, C.D.; Seed, P.C.; Rawls, J.F.; David, L.A.; et al. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology 2016, 63, 764–775. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-L.; Zhang, M.-M.; Zhou, H.; Su, G.-Q.; Ding, Y.; Xu, G.-H.; Wang, X.; Li, C.-F.; Huang, W.-F.; Yi, L.-T. Inhibition of NLRP3 attenuates sodium dextran sulfate-induced inflammatory bowel disease through gut microbiota regulation. Biomed. J. 2023, 46, 100580. [Google Scholar] [CrossRef] [PubMed]
- Hudson, J.; Egan, S. Opportunistic diseases in marine eukaryotes: Could Bacteroidota be the next threat to ocean life? Environ. Microbiol. 2022, 24, 4505–4518. [Google Scholar] [CrossRef] [PubMed]
- Waśkiewicz, A.; Irzykowska, L. Flavobacterium spp.—Characteristics, Occurrence, and Toxicity. In Encyclopedia of Food Microbiology, 2nd ed.; Batt, C.A., Tortorello, M.L., Eds.; Academic Press: Oxford, UK, 2014; pp. 938–942. [Google Scholar]
- Loch, T.P.; Faisal, M. Emerging flavobacterial infections in fish: A review. J. Adv. Res. 2015, 6, 283–300. [Google Scholar] [CrossRef] [PubMed]
- Jeyachandran, S.; Kiyun, P.; Ihn–Sil, K.; Vaseeharan, B. Bacterial Disease Control Methods in Shrimp (Penaeus 1798) Farming Sector in Asian Countries. In Arthropods; Ramón Eduardo Rebolledo, R., Ed.; IntechOpen: Rijeka, Croatia, 2021; Chapter 4. [Google Scholar]
- Kayani, M.U.R.; Yu, K.; Qiu, Y.; Shen, Y.; Gao, C.; Feng, R.; Zeng, X.; Wang, W.; Chen, L.; Su, H.L. Environmental concentrations of antibiotics alter the zebrafish gut microbiome structure and potential functions. Environ. Pollut. 2021, 278, 116760. [Google Scholar] [CrossRef] [PubMed]
- Weitekamp, C.A.; Kvasnicka, A.; Keely, S.P.; Brinkman, N.E.; Howey, X.M.; Gaballah, S.; Phelps, D.; Catron, T.; Zurlinden, T.; Wheaton, E.; et al. Monoassociation with bacterial isolates reveals the role of colonization, community complexity and abundance on locomotor behavior in larval zebrafish. Anim. Microbiome 2021, 3, 12. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, Y.; Quan, M.; Zhao, H.; Jia, J. Gut Microbiota Changes and Their Correlation with Cognitive and Neuropsychiatric Symptoms in Alzheimer’s Disease. J. Alzheimer’s Dis. 2021, 81, 583–595. [Google Scholar] [CrossRef]
- Shin, N.R.; Whon, T.W.; Bae, J.W. Proteobacteria: Microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 2015, 33, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.-Y.; Yang, H.-X.; He, W.; Tian, D.-Y.; Kou, H.-Y.; Wu, K.; Yang, C.-G.; Cheng, Z.-Q.; Song, X.-H. A grass carp model with an antibiotic-disrupted intestinal microbiota. Aquaculture 2021, 541, 736790. [Google Scholar] [CrossRef]
- Litvak, Y.; Byndloss, M.X.; Tsolis, R.M.; Bäumler, A.J. Dysbiotic Proteobacteria expansion: A microbial signature of epithelial dysfunction. Curr. Opin. Microbiol. 2017, 39, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Hu, Y.; Li, X.; Lu, W.; Li, J. Function prediction and network analysis to investigate the response of microbial communities to a single environmental factor. J. Freshw. Ecol. 2020, 35, 271–289. [Google Scholar] [CrossRef]
- Banda, J.F.; Zhang, Q.; Ma, L.; Pei, L.; Du, Z.; Hao, C.; Dong, H. Both pH and salinity shape the microbial communities of the lakes in Badain Jaran Desert, NW China. Sci. Total Environ. 2021, 791, 148108. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Z.; He, X.; Zhang, H.X.; Liao, Y.; Wang, Q.; Li, L.W.; Yu, J.W. Factors Driving Microbial Community Dynamics and Potential Health Effects of Bacterial Pathogen on Landscape Lakes with Reclaimed Water Replenishment in Beijing, PR China. Int. J. Environ. Res. Public Health 2022, 19, 5127. [Google Scholar] [CrossRef] [PubMed]
- Ali, N.G.; Ali, T.E.-S.; Aboyadak, I.M.; Elbakry, M.A. Controlling Pseudomonas aeruginosa infection in Oreochromis niloticus spawners by cefotaxime sodium. Aquaculture 2021, 544, 737107. [Google Scholar] [CrossRef]
- Chen, J.; Su, Z.; Yao, P.; Huang, B.; Zhang, Y.; Wen, D. Effects of wastewater discharge on antibiotic resistance genes and microbial community in a coastal area. Environ. Sci. 2022, 43, 4616–4624. [Google Scholar]
- Kang, Y.; Tian, L.; Gu, X.; Chen, Y.; Ma, X.; Lin, S.; Li, Z.; Lou, Y.; Zheng, M. Characterization of the Ocular Surface Microbiome in Keratitis Patients after Repeated Ophthalmic Antibiotic Exposure. Microbiol. Spectr. 2022, 10, e0216221. [Google Scholar] [CrossRef]
- Akinduro, A.; Onyekwelu, C.I.; Oyelumade, T.; Ajibade, O.A.; Odetoyin, B.; Olaniyi, O.O. Impact of soil supplemented with pig manure on the abundance of antibiotic resistant bacteria and their associated genes. J. Antibiot. 2023, 76, 548–562. [Google Scholar] [CrossRef]
- Qiu, X.; Zhou, G.; Wang, H. Nanoscale zero-valent iron inhibits the horizontal gene transfer of antibiotic resistance genes in chicken manure compost. J. Hazard Mater. 2022, 422, 126883. [Google Scholar] [CrossRef] [PubMed]
- Bai, H.; He, L.Y.; Gao, F.Z.; Wu, D.L.; Yao, K.S.; Zhang, M.; Jia, W.L.; He, L.X.; Zou, H.Y.; Yao, M.S.; et al. Airborne antibiotic resistome and human health risk in railway stations during COVID-19 pandemic. Environ. Int. 2023, 172, 107784. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pang, H.; Zheng, K.; Wang, W.; Zheng, M.; Liu, Y.; Yin, H.; Zhang, D. Cefotaxime Exposure-Caused Oxidative Stress, Intestinal Damage and Gut Microbial Disruption in Artemia sinica. Microorganisms 2024, 12, 675. https://doi.org/10.3390/microorganisms12040675
Pang H, Zheng K, Wang W, Zheng M, Liu Y, Yin H, Zhang D. Cefotaxime Exposure-Caused Oxidative Stress, Intestinal Damage and Gut Microbial Disruption in Artemia sinica. Microorganisms. 2024; 12(4):675. https://doi.org/10.3390/microorganisms12040675
Chicago/Turabian StylePang, Huizhong, Kaixuan Zheng, Wenbo Wang, Mingjuan Zheng, Yudan Liu, Hong Yin, and Daochuan Zhang. 2024. "Cefotaxime Exposure-Caused Oxidative Stress, Intestinal Damage and Gut Microbial Disruption in Artemia sinica" Microorganisms 12, no. 4: 675. https://doi.org/10.3390/microorganisms12040675
APA StylePang, H., Zheng, K., Wang, W., Zheng, M., Liu, Y., Yin, H., & Zhang, D. (2024). Cefotaxime Exposure-Caused Oxidative Stress, Intestinal Damage and Gut Microbial Disruption in Artemia sinica. Microorganisms, 12(4), 675. https://doi.org/10.3390/microorganisms12040675