
Diversity Analysis of Intestinal Bifidobacteria in the Hohhot Population
Abstract
:1. Introduction
2. Materials and Methods
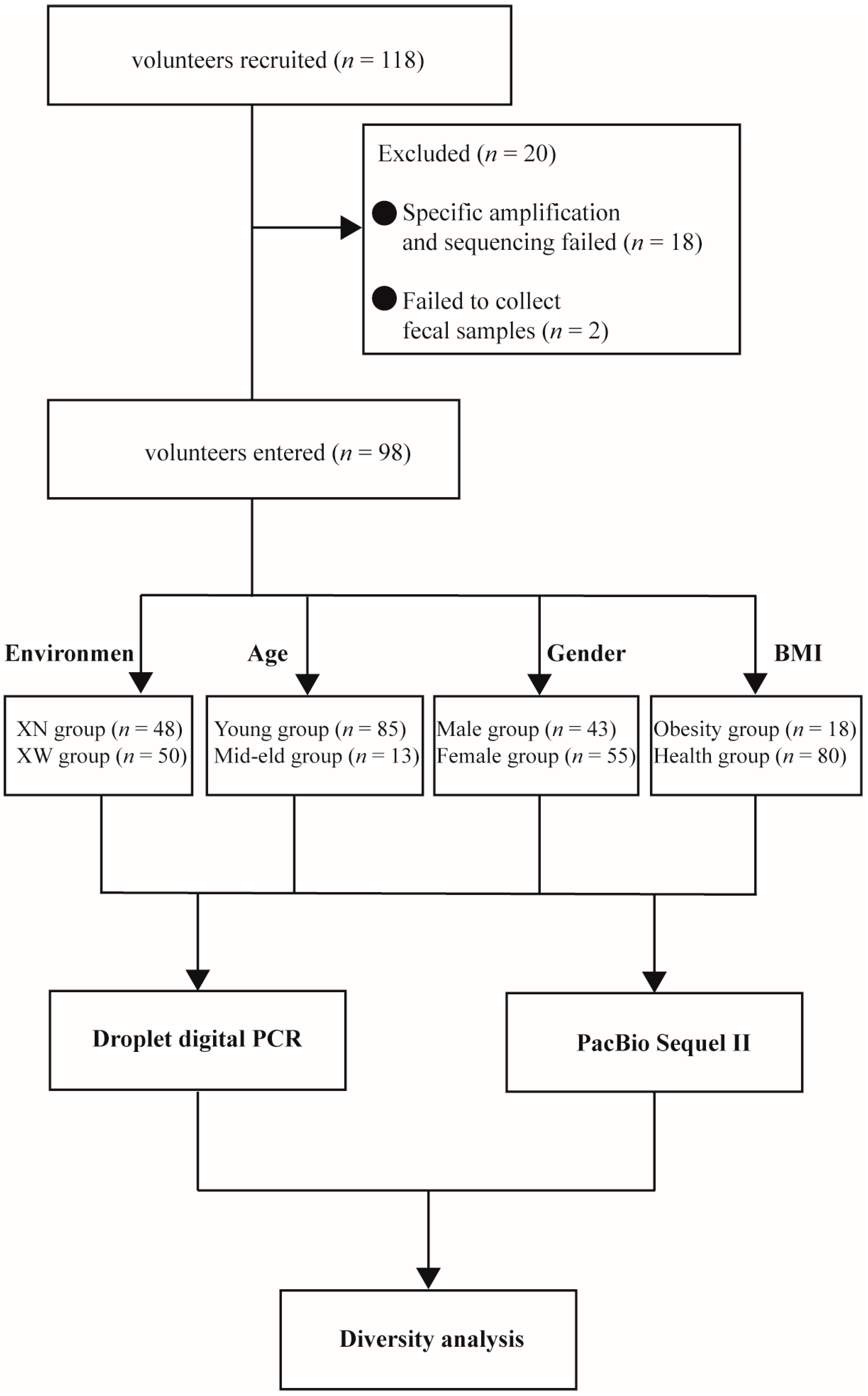
2.1. Subject Recruitment and Fecal Sample Collection
2.2. DNA Extraction and PacBio Sequel II Sequencing
2.3. dd-PCR
2.4. Bioinformatics Analyses and Statistical Analyses
3. Results
3.1. Volunteer Data and Grouping Information
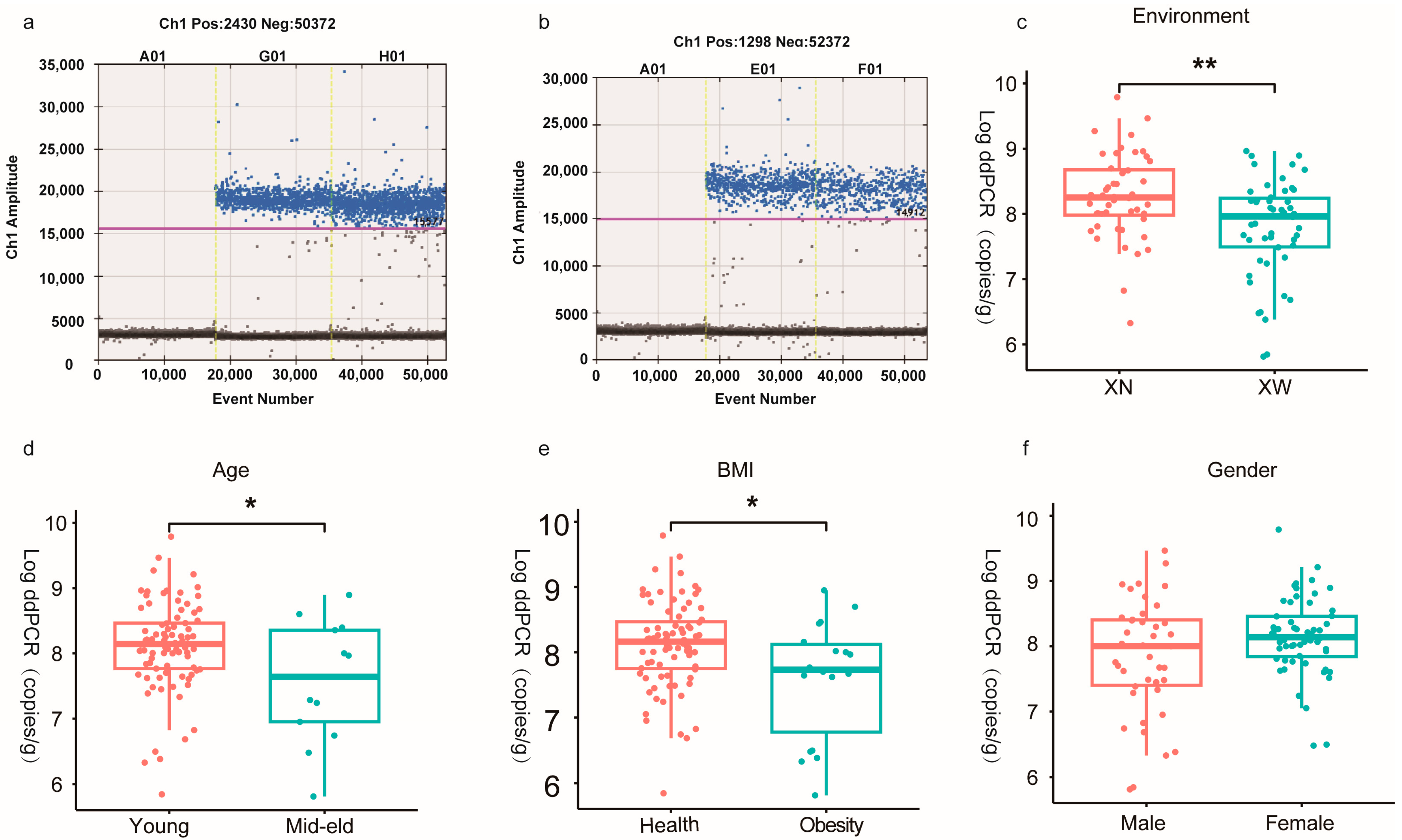
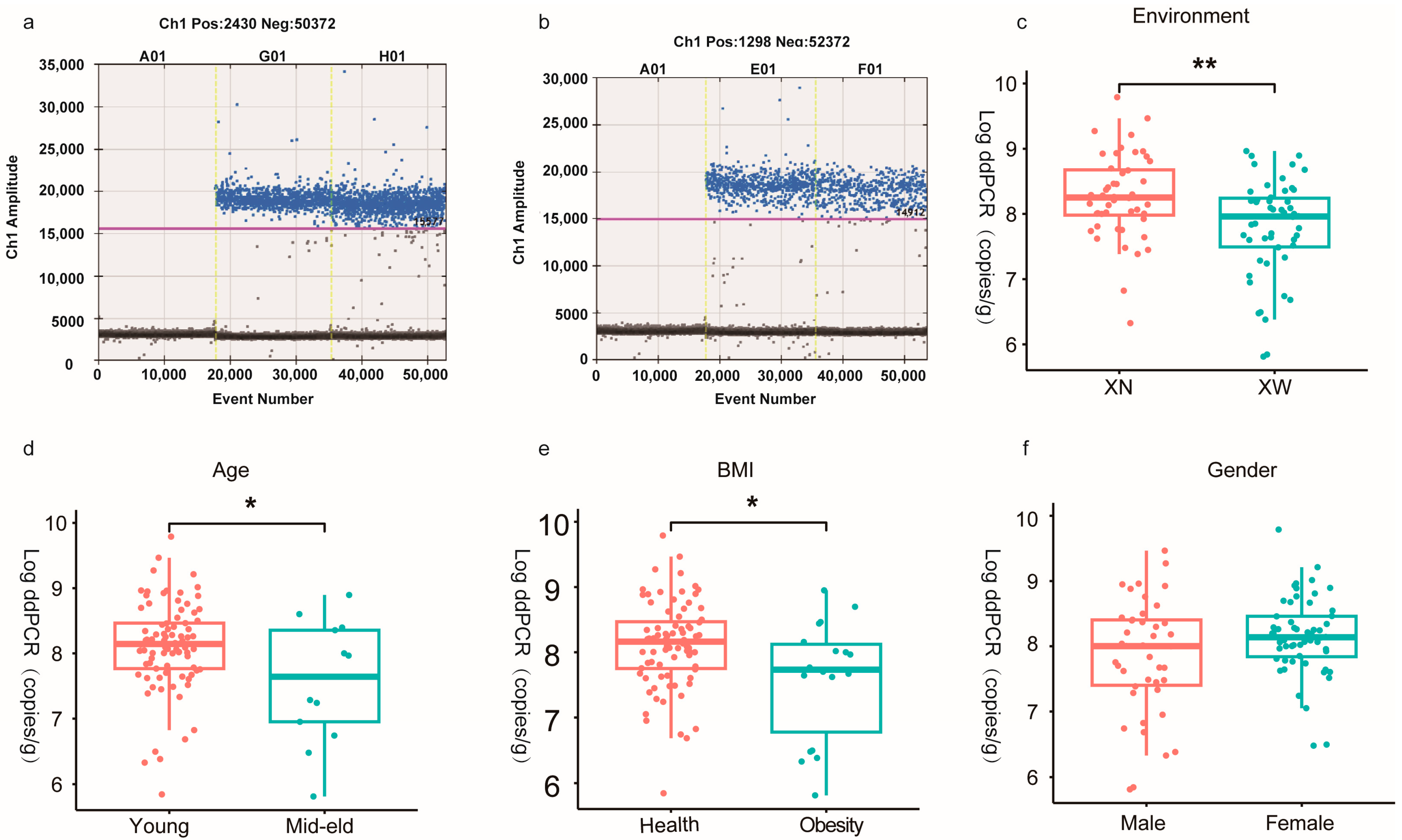
3.2. Absolute Quantitative Analysis of Bifidobacterium
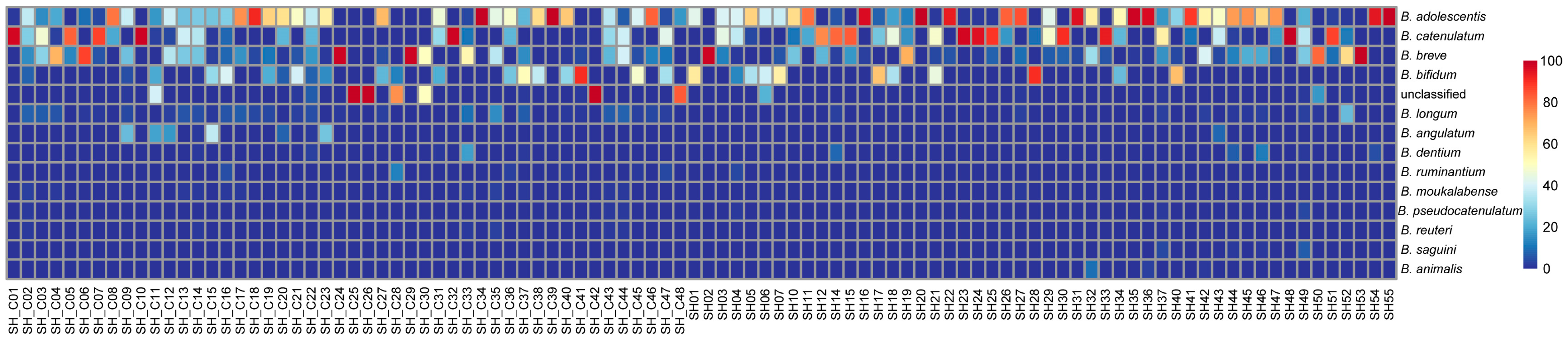
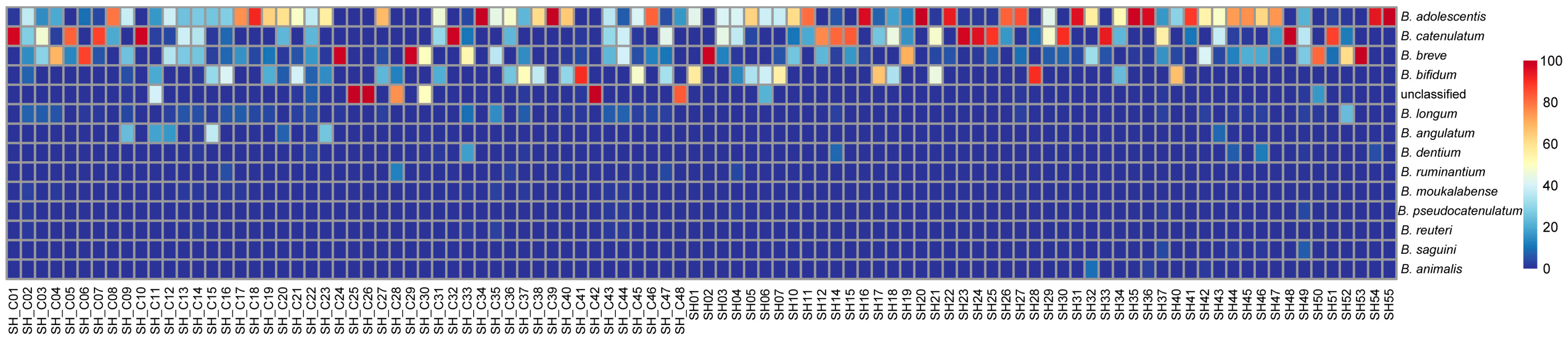
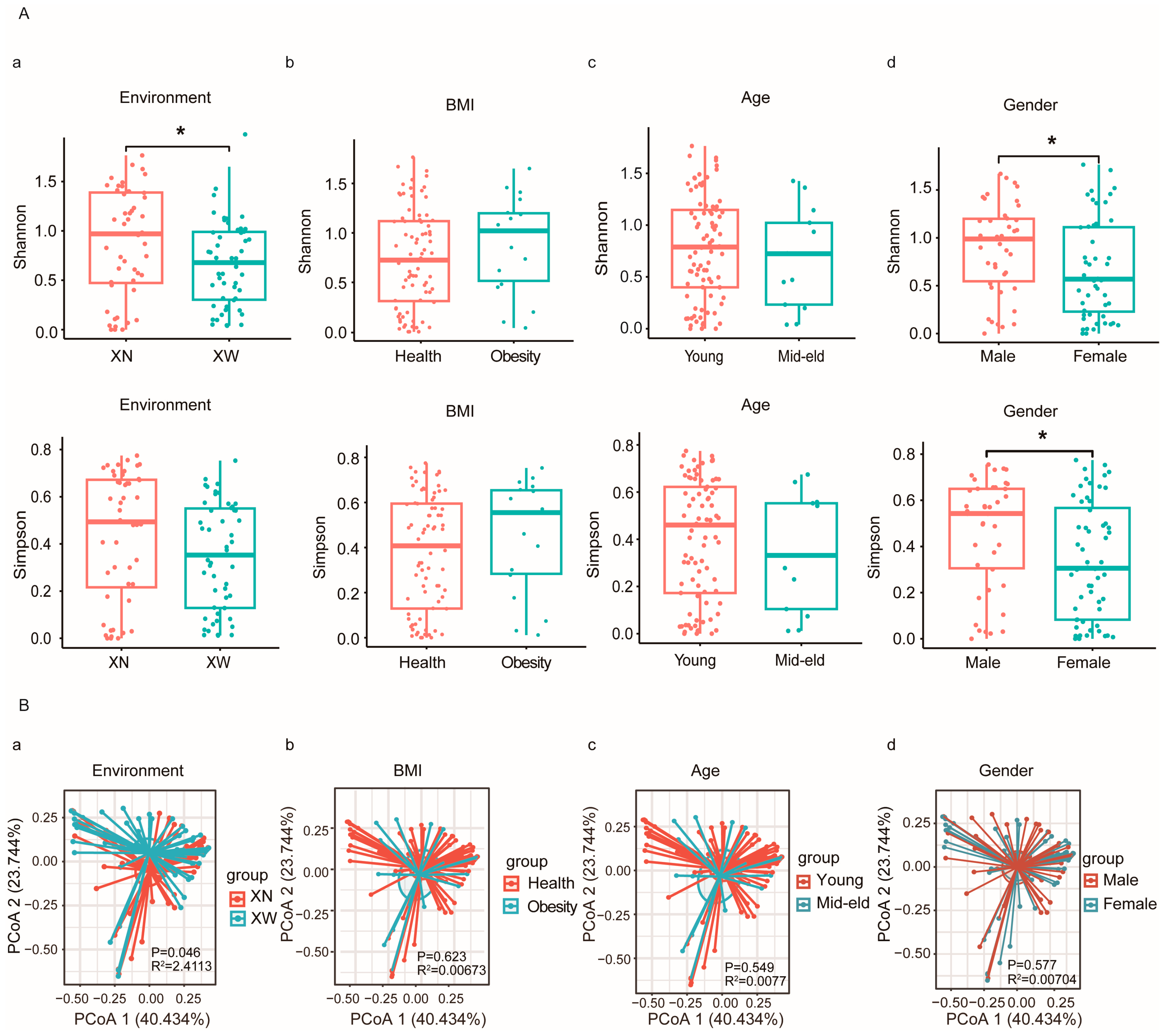
3.3. Diversity Analysis of Bifidobacterium
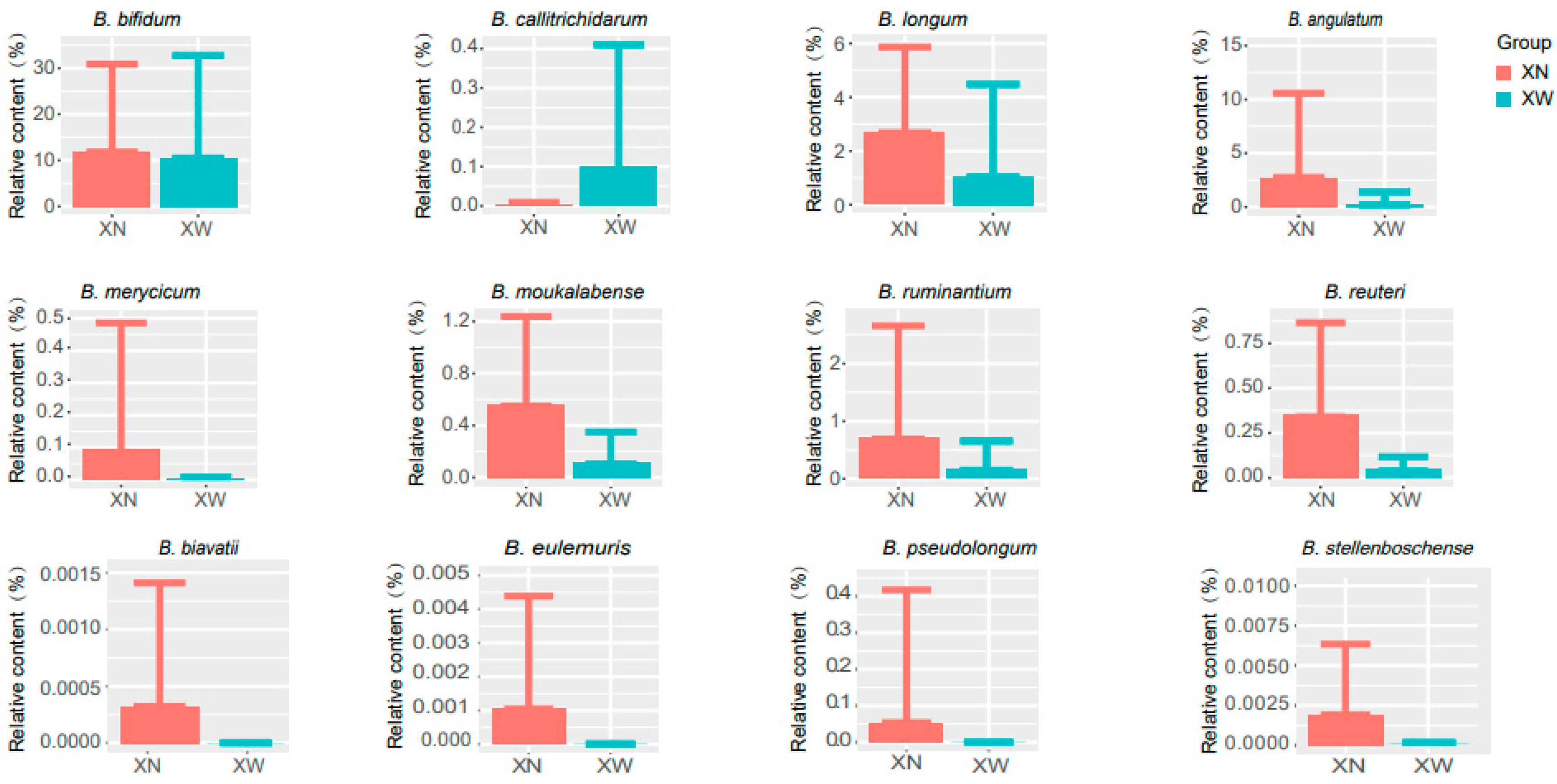
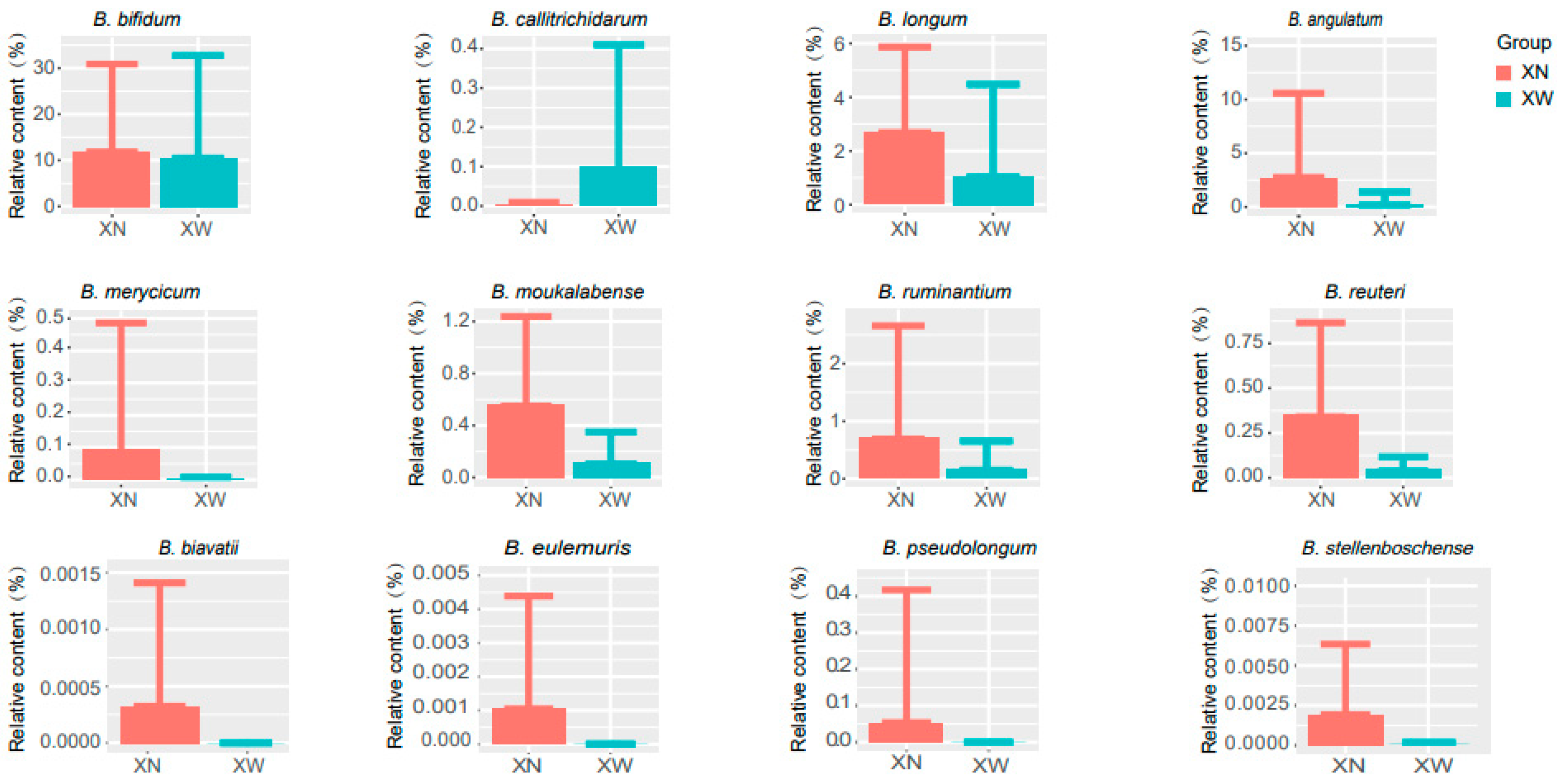
3.4. Analysis of Differential Bacteria
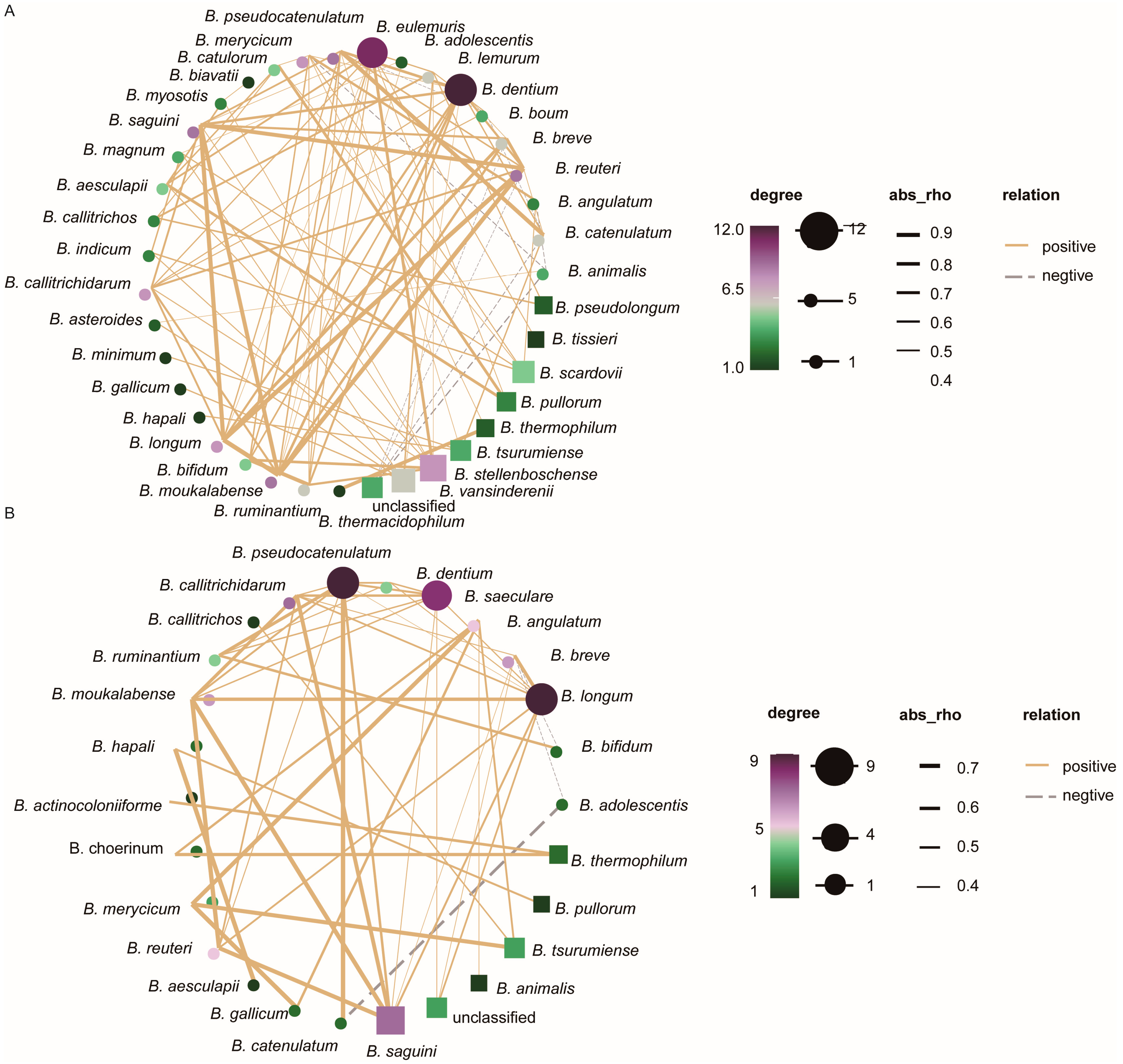
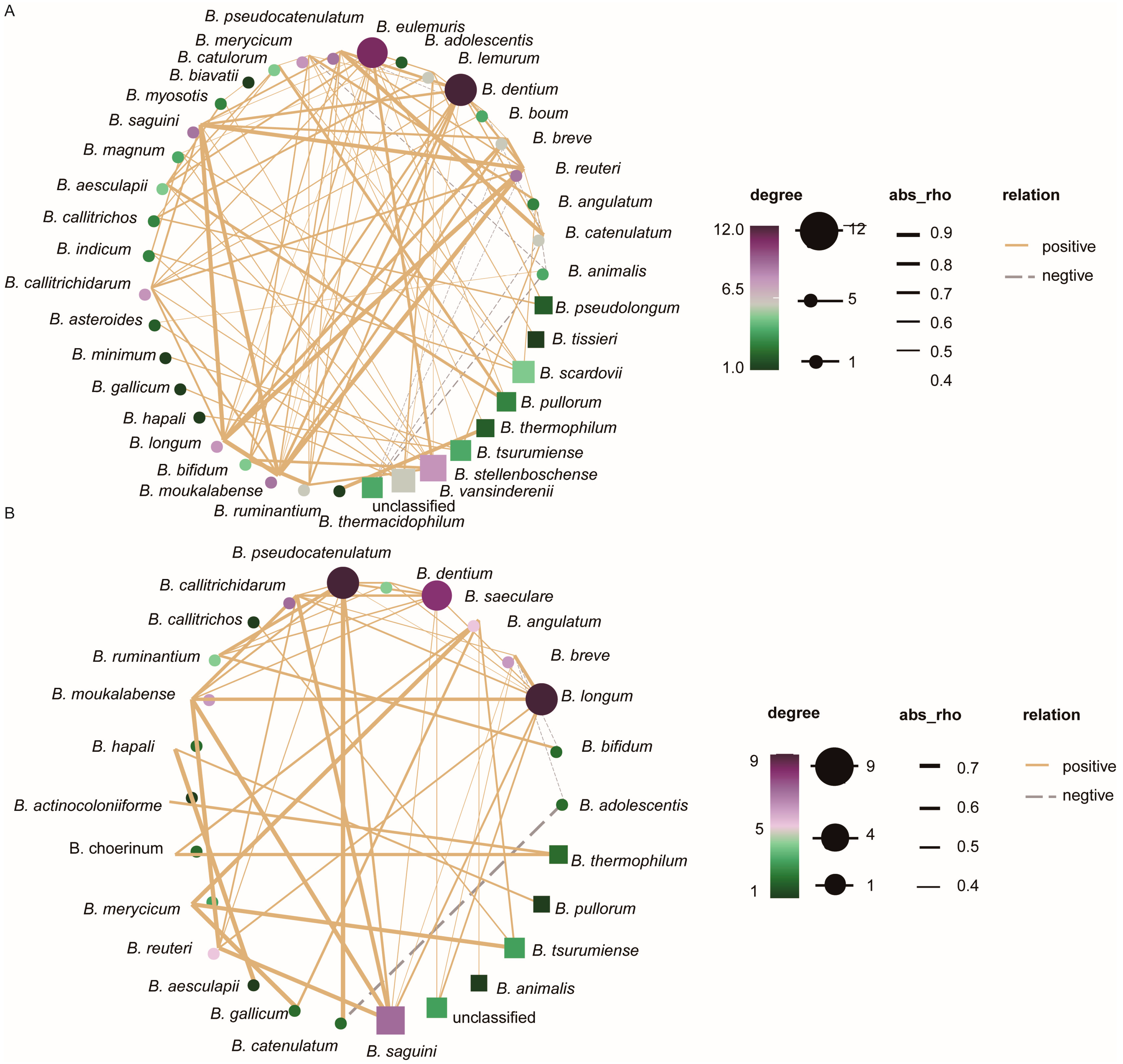
3.5. Analysis of Interaction Relationships of Flora Network
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Age | Gender | Height (m) | Weight (kg) | BMI |
|---|---|---|---|---|---|
| SH_C01 | 18 | Female | 1.7 | 78 | 26.98962 |
| SH_C02 | 18 | Male | 1.76 | 57 | 18.40134 |
| SH_C03 | 19 | Male | 1.8 | 95 | 29.32099 |
| SH_C04 | 19 | Female | 1.6 | 65 | 25.39063 |
| SH_C05 | 19 | Female | 1.58 | 51 | 20.42942 |
| SH_C06 | 19 | Female | 1.66 | 51 | 18.50777 |
| SH_C07 | 19 | Female | 1.75 | 60 | 19.59184 |
| SH_C08 | 20 | Female | 1.66 | 60 | 21.77384 |
| SH_C09 | 20 | Male | 1.83 | 65 | 19.40936 |
| SH_C10 | 20 | Male | 1.78 | 60 | 18.937 |
| SH_C11 | 20 | Female | 1.65 | 55 | 20.20202 |
| SH_C12 | 20 | Female | 1.75 | 80 | 26.12245 |
| SH_C13 | 20 | Female | 1.65 | 52 | 19.10009 |
| SH_C14 | 20 | Male | 1.71 | 55 | 18.80921 |
| SH_C15 | 20 | Male | 1.85 | 106 | 30.97151 |
| SH_C16 | 20 | Male | 1.75 | 63 | 20.57143 |
| SH_C17 | 20 | Male | 1.86 | 70 | 20.23355 |
| SH_C18 | 20 | Male | 1.78 | 70 | 22.09317 |
| SH_C19 | 20 | Male | 1.7 | 60 | 20.76125 |
| SH_C20 | 20 | Male | 1.8 | 75 | 23.14815 |
| SH_C21 | 20 | Female | 1.65 | 47 | 17.26354 |
| SH_C22 | 20 | Female | 1.7 | 60 | 20.76125 |
| SH_C23 | 20 | Female | 1.58 | 50 | 20.02884 |
| SH_C24 | 20 | Male | 1.84 | 80 | 23.62949 |
| SH_C25 | 20 | Female | 1.64 | 52 | 19.33373 |
| SH_C26 | 20 | Female | 1.65 | 62 | 22.77319 |
| SH_C27 | 20 | Female | 1.58 | 48 | 19.22769 |
| SH_C28 | 20 | Male | 1.84 | 107 | 31.60444 |
| SH_C29 | 20 | Female | 1.64 | 53 | 19.70553 |
| SH_C30 | 20 | Male | 1.8 | 60 | 18.51852 |
| SH_C31 | 21 | Male | 1.85 | 125 | 36.52301 |
| SH_C32 | 21 | Male | 1.78 | 65 | 20.51509 |
| SH_C33 | 21 | Female | 1.73 | 70 | 23.38869 |
| SH_C34 | 21 | Female | 1.58 | 54 | 21.63115 |
| SH_C35 | 21 | Male | 1.79 | 60 | 18.72601 |
| SH_C36 | 22 | Male | 1.78 | 60 | 18.937 |
| SH_C37 | 22 | Female | 1.58 | 45 | 18.02596 |
| SH_C38 | 22 | Female | 1.66 | 68 | 24.67702 |
| SH_C39 | 22 | Female | 1.61 | 55 | 21.21832 |
| SH_C40 | 22 | Male | 1.9 | 72 | 19.9446 |
| SH_C41 | 22 | Male | 1.78 | 115 | 36.29592 |
| SH_C42 | 22 | Male | 1.8 | 65 | 20.06173 |
| SH_C43 | 22 | Female | 1.72 | 57 | 19.26717 |
| SH_C44 | 22 | Male | 1.85 | 65 | 18.99196 |
| SH_C45 | 22 | Male | 1.75 | 66 | 21.55102 |
| SH_C46 | 23 | Male | 1.9 | 105 | 29.08587 |
| SH_C47 | 23 | Male | 1.81 | 100 | 30.5241 |
| SH_C48 | 23 | Female | 1.69 | 83 | 29.06061 |
| SH01 | 53 | Male | 1.75 | 72.5 | 23.67347 |
| SH02 | 25 | Male | 1.76 | 87.6 | 28.27996 |
| SH03 | 25 | Female | 1.62 | 48.2 | 18.3661 |
| SH04 | 41 | Female | 1.5 | 58 | 25.77778 |
| SH05 | 31 | Female | 1.7 | 70 | 24.22145 |
| SH06 | 37 | Female | 1.61 | 58.9 | 22.72289 |
| SH07 | 25 | Male | 1.79 | 81.2 | 25.34253 |
| SH10 | 27 | Male | 1.75 | 76.2 | 24.88163 |
| SH11 | 33 | Male | 1.77 | 83.6 | 26.68454 |
| SH12 | 33 | Male | 1.75 | 84.5 | 27.59184 |
| SH14 | 55 | Female | 1.57 | 59.5 | 24.13891 |
| SH15 | 53 | Female | 1.59 | 50 | 19.7777 |
| SH16 | 38 | Male | 1.71 | 71.7 | 24.52037 |
| SH17 | 38 | Female | 1.62 | 69.9 | 26.63466 |
| SH18 | 26 | Male | 1.85 | 124.2 | 36.28926 |
| SH19 | 31 | Female | 1.56 | 69 | 28.35306 |
| SH20 | 48 | Female | 1.54 | 71 | 29.93759 |
| SH21 | 33 | Male | 1.68 | 84.7 | 30.00992 |
| SH22 | 24 | Female | 1.68 | 60 | 21.2585 |
| SH23 | 23 | Female | 1.57 | 47 | 19.06771 |
| SH24 | 23 | Female | 1.68 | 53 | 18.77834 |
| SH25 | 28 | Female | 1.65 | 57.7 | 21.19376 |
| SH26 | 26 | Female | 1.71 | 81.3 | 27.80343 |
| SH27 | 27 | Female | 1.57 | 41.5 | 16.83638 |
| SH28 | 26 | Female | 1.58 | 50.6 | 20.26919 |
| SH29 | 24 | Male | 1.9 | 68.8 | 19.05817 |
| SH30 | 28 | Female | 1.57 | 57.6 | 23.36809 |
| SH31 | 27 | Female | 1.62 | 55.5 | 21.14769 |
| SH32 | 25 | Male | 1.79 | 65.8 | 20.53619 |
| SH33 | 29 | Female | 1.6 | 66.2 | 25.85938 |
| SH34 | 35 | Male | 1.78 | 94.9 | 29.95203 |
| SH35 | 36 | Female | 1.59 | 62 | 24.52435 |
| SH36 | 61 | Female | 1.59 | 79.8 | 31.56521 |
| SH37 | 53 | Male | 1.72 | 82 | 27.71769 |
| SH40 | 25 | Female | 1.64 | 47.6 | 17.6978 |
| SH41 | 24 | Male | 1.85 | 74.1 | 21.65084 |
| SH42 | 63 | Male | 1.61 | 63.6 | 24.53609 |
| SH43 | 23 | Female | 1.7 | 62.5 | 21.6263 |
| SH44 | 24 | Female | 1.65 | 72.1 | 26.48301 |
| SH45 | 37 | Female | 1.61 | 59.6 | 22.99294 |
| SH46 | 64 | Female | 1.57 | 69.6 | 28.23644 |
| SH47 | 37 | Female | 1.58 | 54.2 | 21.71126 |
| SH48 | 38 | Female | 1.66 | 61 | 22.13674 |
| SH49 | 33 | Female | 1.56 | 74.6 | 30.65417 |
| SH50 | 25 | Female | 1.64 | 51.1 | 18.99911 |
| SH51 | 59 | Male | 1.65 | 68.4 | 25.12397 |
| SH52 | 64 | Male | 1.72 | 90 | 30.42185 |
| SH53 | 24 | Female | 1.71 | 58.2 | 19.90356 |
| SH54 | 64 | Male | 1.62 | 64.4 | 24.53894 |
| SH55 | 62 | Female | 1.58 | 57.7 | 23.11328 |
| Sample | Fecal Quantity (g) | Return Solution Volume (μL) | Dilution Multiple | Copy Number (Copies/μL) | N (CFU/g) |
|---|---|---|---|---|---|
| SH_C01 | 0.12 | 100 | 200 | 117 | 1.95 × 108 |
| SH_C02 | 0.14 | 100 | 100 | 144 | 1.03 × 108 |
| SH_C03 | 0.13 | 100 | 100 | 1092.91 | 8.41 × 108 |
| SH_C04 | 0.1 | 100 | 1000 | 18.31 | 1.83 × 108 |
| SH_C05 | 0.1 | 100 | 100 | 54.7 | 5.47 × 107 |
| SH_C06 | 0.12 | 100 | 100 | 599 | 4.99 × 108 |
| SH_C07 | 0.12 | 100 | 1000 | 740 | 6.17 × 109 |
| SH_C08 | 0.15 | 100 | 1000 | 17.5 | 1.17 × 108 |
| SH_C09 | 0.13 | 100 | 1000 | 13.01 | 1.00 × 108 |
| SH_C10 | 0.1 | 100 | 1000 | 91.43 | 9.14 × 108 |
| SH_C11 | 0.11 | 100 | 100 | 1139 | 1.04 × 109 |
| SH_C12 | 0.26 | 100 | 200 | 374 | 2.88 × 108 |
| SH_C13 | 0.13 | 100 | 100 | 1110.91 | 8.55 × 108 |
| SH_C14 | 0.11 | 100 | 1000 | 28.11 | 2.56 × 108 |
| SH_C15 | 0.11 | 100 | 100 | 2.33 | 2.12 × 106 |
| SH_C16 | 0.18 | 100 | 200 | 6 | 6.67 × 106 |
| SH_C17 | 0.13 | 100 | 1000 | 242.93 | 1.87 × 109 |
| SH_C18 | 0.23 | 100 | 1000 | 31.43 | 1.37 × 108 |
| SH_C19 | 0.11 | 100 | 1000 | 321.93 | 2.93 × 109 |
| SH_C20 | 0.1 | 100 | 1000 | 31.63 | 3.16 × 108 |
| SH_C21 | 0.09 | 100 | 1000 | 147 | 1.63 × 109 |
| SH_C22 | 0.12 | 100 | 100 | 559 | 4.66 × 108 |
| SH_C23 | 0.12 | 100 | 100 | 774 | 6.45 × 108 |
| SH_C24 | 0.12 | 100 | 1000 | 6.83 | 5.69 × 107 |
| SH_C25 | 0.17 | 100 | 100 | 178.93 | 1.05 × 108 |
| SH_C26 | 0.19 | 100 | 100 | 83.7 | 4.41 × 107 |
| SH_C27 | 0.14 | 100 | 100 | 268 | 1.91 × 108 |
| SH_C28 | 0.11 | 100 | 1000 | 15.83 | 1.44 × 108 |
| SH_C29 | 0.19 | 100 | 1000 | 11.1 | 5.84 × 107 |
| SH_C30 | 0.13 | 100 | 1000 | 115.73 | 8.90 × 108 |
| SH_C31 | 0.11 | 100 | 100 | 837 | 7.61 × 108 |
| SH_C32 | 0.12 | 100 | 200 | 253 | 4.22 × 108 |
| SH_C33 | 0.12 | 100 | 100 | 219 | 1.83 × 108 |
| SH_C34 | 0.25 | 100 | 1000 | 43.93 | 1.76 × 108 |
| SH_C35 | 0.15 | 100 | 100 | 164 | 1.09 × 108 |
| SH_C36 | 0.19 | 100 | 1000 | 44.6 | 2.35 × 108 |
| SH_C37 | 0.12 | 100 | 1000 | 34.91 | 2.91 × 108 |
| SH_C38 | 0.15 | 100 | 100 | 126 | 8.40 × 107 |
| SH_C39 | 0.12 | 100 | 1000 | 16.71 | 1.39 × 108 |
| SH_C40 | 0.18 | 100 | 100 | 182 | 1.01 × 108 |
| SH_C41 | 0.1 | 100 | 100 | 41.8 | 4.18 × 107 |
| SH_C42 | 0.12 | 100 | 200 | 119.4 | 1.99 × 108 |
| SH_C43 | 0.19 | 100 | 100 | 192 | 1.01 × 108 |
| SH_C44 | 0.2 | 100 | 1000 | 5.63 | 2.82 × 107 |
| SH_C45 | 0.14 | 100 | 100 | 34.03 | 2.43 × 107 |
| SH_C46 | 0.12 | 100 | 100 | 195 | 1.63 × 108 |
| SH_C47 | 0.14 | 100 | 100 | 42.33 | 3.02 × 107 |
| SH_C48 | 0.2 | 100 | 100 | 128.93 | 6.45 × 107 |
| SH01 | 0.27 | 100 | 1000 | 5.2 | 1.93 × 107 |
| SH02 | 0.13 | 100 | 1000 | 6.1 | 4.70 × 107 |
| SH03 | 0.11 | 200 | 1000 | 50.7 | 9.22 × 108 |
| SH04 | 0.1 | 200 | 100 | 21.9 | 4.38 × 107 |
| SH05 | 0.1 | 200 | 100 | 16.4 | 3.28 × 107 |
| SH06 | 0.12 | 200 | 100 | 95.4 | 1.59 × 108 |
| SH07 | 0.13 | 200 | 100 | 20 | 3.08 × 107 |
| SH10 | 0.18 | 200 | 1000 | 13.7 | 1.52 × 108 |
| SH11 | 0.15 | 100 | 1000 | 10.3 | 6.87 × 107 |
| SH12 | 0.11 | 100 | 100 | 5.3 | 4.82 × 106 |
| SH14 | 0.13 | 100 | 100 | 1022 | 7.86 × 108 |
| SH15 | 0.17 | 100 | 100 | 29.6 | 1.74 × 107 |
| SH16 | 0.18 | 100 | 100 | 38.7 | 2.15 × 107 |
| SH17 | 0.12 | 100 | 100 | 13.5 | 1.13 × 107 |
| SH18 | 0.12 | 100 | 100 | 331 | 2.76 × 108 |
| SH19 | 0.17 | 100 | 1000 | 0.53 | 3.12 × 106 |
| SH20 | 0.11 | 100 | 100 | 110.2 | 1.00 × 108 |
| SH21 | 0.16 | 100 | 100 | 80.6 | 5.04 × 107 |
| SH22 | 0.1 | 100 | 500 | 47 | 2.35 × 108 |
| SH23 | 0.29 | 100 | 500 | 23.4 | 4.03 × 107 |
| SH24 | 0.11 | 100 | 500 | 19.9 | 9.05 × 107 |
| SH25 | 0.13 | 100 | 500 | 46.4 | 1.78 × 108 |
| SH26 | 0.17 | 100 | 100 | 209 | 1.23 × 108 |
| SH27 | 0.13 | 100 | 100 | 458 | 3.52 × 108 |
| SH28 | 0.13 | 100 | 500 | 40.1 | 1.54 × 108 |
| SH29 | 0.11 | 100 | 500 | 10.4 | 4.73 × 107 |
| SH30 | 0.12 | 100 | 500 | 28.1 | 1.17 × 108 |
| SH31 | 0.08 | 100 | 500 | 26.2 | 1.64 × 108 |
| SH32 | 0.1 | 100 | 500 | 0.14 | 7.00 × 105 |
| SH33 | 0.09 | 100 | 500 | 20.8 | 1.16 × 108 |
| SH34 | 0.15 | 100 | 200 | 1.8 | 2.40 × 106 |
| SH35 | 0.1 | 100 | 500 | 44 | 2.20 × 108 |
| SH36 | 0.1 | 100 | 500 | 0.6 | 3.00 × 106 |
| SH37 | 0.12 | 100 | 100 | 272 | 2.27 × 108 |
| SH40 | 0.11 | 100 | 100 | 173 | 1.57 × 108 |
| SH41 | 0.11 | 100 | 500 | 126.7 | 5.76 × 108 |
| SH42 | 0.14 | 100 | 500 | 2.5 | 8.93 × 106 |
| SH43 | 0.12 | 100 | 100 | 149 | 1.24 × 108 |
| SH44 | 0.08 | 100 | 100 | 381 | 4.76 × 108 |
| SH45 | 0.1 | 100 | 100 | 60.6 | 6.06 × 107 |
| SH46 | 0.18 | 100 | 500 | 33.3 | 9.25 × 107 |
| SH47 | 0.1 | 100 | 500 | 116.5 | 5.83 × 108 |
| SH48 | 0.06 | 100 | 500 | 93.2 | 7.77 × 108 |
| SH49 | 0.14 | 100 | 100 | 146 | 1.04 × 108 |
| SH50 | 0.11 | 100 | 500 | 9.3 | 4.23 × 107 |
| SH51 | 0.2 | 100 | 500 | 2.2 | 5.50 × 106 |
| SH52 | 0.1 | 100 | 500 | 0.13 | 6.50 × 105 |
| SH53 | 0.2 | 100 | 100 | 142 | 7.10 × 107 |
| SH54 | 0.11 | 100 | 500 | 54.8 | 2.49 × 108 |
| SH55 | 0.17 | 100 | 500 | 13.6 | 4.00 × 107 |
References
- Peterson, J.; Garges, S.; Giovanni, M.; McInnes, P.; Wang, L.; Schloss, J.A.; Bonazzi, V.; McEwen, J.E.; Wetterstrand, K.A.; Deal, C.; et al. The NIH Human Microbiome Project. Genome Res. 2009, 19, 2317–2323. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Tseng, C.H.; Pei, Z.; Blaser, M.J. Molecular analysis of human forearm superficial skin bacterial biota. Proc. Natl. Acad. Sci. USA 2007, 104, 2927–2932. [Google Scholar] [CrossRef] [PubMed]
- Faveri, M.; Mayer, M.P.; Feres, M.; de Figueiredo, L.C.; Dewhirst, F.E.; Paster, B.J. Microbiological diversity of generalized aggressive periodontitis by 16S rRNA clonal analysis. Oral. Microbiol. Immunol. 2008, 23, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Hyman, R.W.; Fukushima, M.; Diamond, L.; Kumm, J.; Giudice, L.C.; Davis, R.W. Microbes on the human vaginal epithelium. Proc. Natl. Acad. Sci. USA 2005, 102, 7952–7957. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.W.; Hoyles, L. Human microbiome myths and misconceptions. Nat. Microbiol. 2023, 8, 1392–1396. [Google Scholar] [CrossRef]
- Kuziel, G.A.; Rakoff-Nahoum, S. The gut microbiome. Curr. Biol. 2022, 32, R257–R264. [Google Scholar] [CrossRef]
- Heintz-Buschart, A.; Wilmes, P. Human Gut Microbiome: Function Matters. Trends Microbiol. 2018, 26, 563–574. [Google Scholar] [CrossRef]
- Fan, Y.; Pedersen, O. Gut microbiota in human metabolic health and disease. Nat. Rev. Microbiol. 2021, 19, 55–71. [Google Scholar] [CrossRef] [PubMed]
- Bäckhed, F.; Fraser, C.M.; Ringel, Y.; Sanders, M.E.; Sartor, R.B.; Sherman, P.M.; Versalovic, J.; Young, V.; Finlay, B.B. Defining a healthy human gut microbiome: Current concepts, future directions, and clinical applications. Cell Host Microbe 2012, 12, 611–622. [Google Scholar] [CrossRef]
- Lee, J.H.; O’Sullivan, D.J. Genomic insights into bifidobacteria. Microbiol. Mol. Biol. Rev. 2010, 74, 378–416. [Google Scholar] [CrossRef]
- Turroni, F.; Foroni, E.; Pizzetti, P.; Giubellini, V.; Ribbera, A.; Merusi, P.; Cagnasso, P.; Bizzarri, B.; de’Angelis, G.L.; Shanahan, F.; et al. Exploring the diversity of the bifidobacterial population in the human intestinal tract. Appl. Environ. Microbiol. 2009, 75, 1534–1545. [Google Scholar] [CrossRef] [PubMed]
- Ventura, M.; Turroni, F.; Zomer, A.; Foroni, E.; Giubellini, V.; Bottacini, F.; Canchaya, C.; Claesson, M.J.; He, F.; Mantzourani, M.; et al. The Bifidobacterium dentium Bd1 genome sequence reflects its genetic adaptation to the human oral cavity. PLoS Genet. 2009, 5, e1000785. [Google Scholar] [CrossRef] [PubMed]
- Tojo, R.; Suárez, A.; Clemente, M.G.; de los Reyes-Gavilán, C.G.; Margolles, A.; Gueimonde, M.; Ruas-Madiedo, P. Intestinal microbiota in health and disease: Role of bifidobacteria in gut homeostasis. World J. Gastroenterol. 2014, 20, 15163–15176. [Google Scholar] [CrossRef] [PubMed]
- Staudacher, H.M.; Lomer, M.C.E.; Farquharson, F.M.; Louis, P.; Fava, F.; Franciosi, E.; Scholz, M.; Tuohy, K.M.; Lindsay, J.O.; Irving, P.M.; et al. A Diet Low in FODMAPs Reduces Symptoms in Patients With Irritable Bowel Syndrome and A Probiotic Restores Bifidobacterium Species: A Randomized Controlled Trial. Gastroenterology 2017, 153, 936–947. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Chen, S.; Lv, H.; Peng, L.; Yang, W.; Chen, J.; Wu, Z.; Wan, C. Effect of Bifidobacterium animalis subsp. lactis SF on enhancing the tumor suppression of irinotecan by regulating the intestinal flora. Pharmacol. Res. 2022, 184, 106406. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Jin, H.; Kwok, L.Y.; Sun, Z.; Liong, M.T.; Zhang, H. Probiotic consumption relieved human stress and anxiety symptoms possibly via modulating the neuroactive potential of the gut microbiota. Neurobiol. Stress. 2021, 14, 100294. [Google Scholar] [CrossRef]
- Azuma, N.; Mawatari, T.; Saito, Y.; Tsukamoto, M.; Sampei, M.; Iwama, Y. Effect of Continuous Ingestion of Bifidobacteria and Dietary Fiber on Improvement in Cognitive Function: A Randomized, Double-Blind, Placebo-Controlled Trial. Nutrients 2023, 15, 4175. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, R.; Ding, J.; Huang, Y.; Zheng, F.; Zheng, S.; Ye, Y.; Li, Q.; Wang, X.; Ma, X.; Zou, Y.; et al. Maturation of the gut metabolome during the first year of life in humans. Gut Microbes 2023, 15, 2231596. [Google Scholar] [CrossRef] [PubMed]
- Ventura, M.; Canchaya, C.; Tauch, A.; Chandra, G.; Fitzgerald, G.F.; Chater, K.F.; van Sinderen, D. Genomics of Actinobacteria: Tracing the evolutionary history of an ancient phylum. Microbiol. Mol. Biol. Rev. 2007, 71, 495–548. [Google Scholar] [CrossRef]
- Olm, M.R.; Dahan, D.; Carter, M.M.; Merrill, B.D.; Yu, F.B.; Jain, S.; Meng, X.; Tripathi, S.; Wastyk, H.; Neff, N.; et al. Robust variation in infant gut microbiome assembly across a spectrum of lifestyles. Science 2022, 376, 1220–1223. [Google Scholar] [CrossRef]
- de la Cuesta-Zuluaga, J.; Kelley, S.T.; Chen, Y.; Escobar, J.S.; Mueller, N.T.; Ley, R.E.; McDonald, D.; Huang, S.; Swafford, A.D.; Knight, R.; et al. Age- and Sex-Dependent Patterns of Gut Microbial Diversity in Human Adults. mSystems 2019, 4, e00261-19. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.A.; Jeffery, I.B.; Beaumont, M.; Bell, J.T.; Clark, A.G.; Ley, R.E.; O’Toole, P.W.; Spector, T.D.; Steves, C.J. Signatures of early frailty in the gut microbiota. Genome Med. 2016, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Pang, S.; Chen, X.; Lu, Z.; Meng, L.; Huang, Y.; Yu, X.; Huang, L.; Ye, P.; Chen, X.; Liang, J.; et al. Longevity of centenarians is reflected by the gut microbiome with youth-associated signatures. Nat. Aging 2023, 3, 436–449. [Google Scholar] [CrossRef] [PubMed]
- Drago, L.; Toscano, M.; Rodighiero, V.; De Vecchi, E.; Mogna, G. Cultivable and pyrosequenced fecal microflora in centenarians and young subjects. J. Clin. Gastroenterol. 2012, 46, S81–S84. [Google Scholar] [CrossRef] [PubMed]
- Hansen, M.E.B.; Rubel, M.A.; Bailey, A.G.; Ranciaro, A.; Thompson, S.R.; Campbell, M.C.; Beggs, W.; Dave, J.R.; Mokone, G.G.; Mpoloka, S.W.; et al. Population structure of human gut bacteria in a diverse cohort from rural Tanzania and Botswana. Genome Biol. 2019, 20, 16. [Google Scholar] [CrossRef]
- Shin, J.H.; Sim, M.; Lee, J.Y.; Shin, D.M. Lifestyle and geographic insights into the distinct gut microbiota in elderly women from two different geographic locations. J. Physiol. Anthropol. 2016, 35, 31. [Google Scholar] [CrossRef]
- Zhang, J.; Guo, Z.; Xue, Z.; Sun, Z.; Zhang, M.; Wang, L.; Wang, G.; Wang, F.; Xu, J.; Cao, H.; et al. A phylo-functional core of gut microbiota in healthy young Chinese cohorts across lifestyles, geography and ethnicities. ISME J. 2015, 9, 1979–1990. [Google Scholar] [CrossRef]
- Parizadeh, M.; Arrieta, M.C. The global human gut microbiome: Genes, lifestyles, and diet. Trends Mol. Med. 2023, 29, 789–801. [Google Scholar] [CrossRef]
- Gupta, V.K.; Paul, S.; Dutta, C. Geography, Ethnicity or Subsistence-Specific Variations in Human Microbiome Composition and Diversity. Front. Microbiol. 2017, 8, 1162. [Google Scholar] [CrossRef]
- Michels, N.; Van de Wiele, T.; Fouhy, F.; O’Mahony, S.; Clarke, G.; Keane, J. Gut microbiome patterns depending on children’s psychosocial stress: Reports versus biomarkers. Brain Behav. Immun. 2019, 80, 751–762. [Google Scholar] [CrossRef]
- Sudo, N.; Chida, Y.; Aiba, Y.; Sonoda, J.; Oyama, N.; Yu, X.N.; Kubo, C.; Koga, Y. Postnatal microbial colonization programs the hypothalamic-pituitary-adrenal system for stress response in mice. J. Physiol. 2004, 558, 263–275. [Google Scholar] [CrossRef]
- Mackos, A.R.; Varaljay, V.A.; Maltz, R.; Gur, T.L.; Bailey, M.T. Role of the Intestinal Microbiota in Host Responses to Stressor Exposure. Int. Rev. Neurobiol. 2016, 131, 1–19. [Google Scholar] [CrossRef]
- Stecher, B. The Roles of Inflammation, Nutrient Availability and the Commensal Microbiota in Enteric Pathogen Infection. Microbiol. Spectr. 2015, 3, 297–320. [Google Scholar] [CrossRef]
- Rada, V.; Petr, J. A new selective medium for the isolation of glucose non-fermenting bifidobacteria from hen caeca. J. Microbiol. Methods 2000, 43, 127–132. [Google Scholar] [CrossRef]
- Ishikawa, E.; Matsuki, T.; Kubota, H.; Makino, H.; Sakai, T.; Oishi, K.; Kushiro, A.; Fujimoto, J.; Watanabe, K.; Watanuki, M.; et al. Ethnic diversity of gut microbiota: Species characterization of Bacteroides fragilis group and genus Bifidobacterium in healthy Belgian adults, and comparison with data from Japanese subjects. J. Biosci. Bioeng. 2013, 116, 265–270. [Google Scholar] [CrossRef]
- Austin, B. The value of cultures to modern microbiology. Antonie Leeuwenhoek 2017, 110, 1247–1256. [Google Scholar] [CrossRef]
- Arvizu, I.S.; Murray, S.R. A simple, quantitative assay for the detection of viable but non-culturable (VBNC) bacteria. STAR Protoc. 2021, 2, 100738. [Google Scholar] [CrossRef]
- Junick, J.; Blaut, M. Quantification of human fecal bifidobacterium species by use of quantitative real-time PCR analysis targeting the groEL gene. Appl. Environ. Microbiol. 2012, 78, 2613–2622. [Google Scholar] [CrossRef]
- Morisset, D.; Štebih, D.; Milavec, M.; Gruden, K.; Žel, J. Quantitative analysis of food and feed samples with droplet digital PCR. PLoS ONE 2013, 8, e62583. [Google Scholar] [CrossRef]
- Chen, S.; He, F.; Cheng, X. Analysis of subgingival bacterial and fungal diversity in patients with peri-implantitis based on 16sRNA and internal transcribed spacer sequencing. Future Microbiol. 2023, 19. [Google Scholar] [CrossRef]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef]
- Turroni, F.; Marchesi, J.R.; Foroni, E.; Gueimonde, M.; Shanahan, F.; Margolles, A.; van Sinderen, D.; Ventura, M. Microbiomic analysis of the bifidobacterial population in the human distal gut. ISME J. 2009, 3, 745–751. [Google Scholar] [CrossRef]
- Hu, L.; Lu, W.; Wang, L.; Pan, M.; Zhang, H.; Zhao, J.; Chen, W. Assessment of Bifidobacterium Species Using groEL Gene on the Basis of Illumina MiSeq High-Throughput Sequencing. Genes 2017, 8, 336. [Google Scholar] [CrossRef]
- Zheng, H.; Bai, X.; Gao, X.; Sun, Z.; Zhang, H. Screening and optimization of Bifidobacterium-specific sequencing primers. Microbiol. China 2019, 46, 1736–1747. [Google Scholar]
- Athanasopoulou, K.; Boti, M.A.; Adamopoulos, P.G.; Skourou, P.C.; Scorilas, A. Third-Generation Sequencing: The Spearhead towards the Radical Transformation of Modern Genomics. Life 2021, 12, 30. [Google Scholar] [CrossRef]
- Mosher, J.J.; Bowman, B.; Bernberg, E.L.; Shevchenko, O.; Kan, J.; Korlach, J.; Kaplan, L.A. Improved performance of the PacBio SMRT technology for 16S rDNA sequencing. J. Microbiol. Methods 2014, 104, 59–60. [Google Scholar] [CrossRef]
- Zhao, F.; Bai, X.; Zhang, J.; Kwok, L.Y.; Shen, L.; Jin, H.; Sun, T.; Sun, Z.; Zhang, H. Gut Bifidobacterium responses to probiotic Lactobacillus casei Zhang administration vary between subjects from different geographic regions. Appl. Microbiol. Biotechnol. 2022, 106, 2665–2675. [Google Scholar] [CrossRef]
- Gao, X.; Bai, X.; Zheng, H.; Sun, Z.; Zhang, H.; Zhong, Z. Design and evaluation of Bifidobacterium genus-specific primer for quantification. Acta Microbiol. Sin. 2020, 60, 545–555. [Google Scholar]
- Yang, C.; Zhao, F.; Hou, Q.; Wang, J.; Li, M.; Sun, Z. PacBio sequencing reveals bacterial community diversity in cheeses collected from different regions. J. Dairy Sci. 2020, 103, 1238–1249. [Google Scholar] [CrossRef]
- Zhao, F.; Guo, Z.; Kwok, L.Y.; Zhao, Z.; Wang, K.; Li, Y.; Sun, Z.; Zhao, J.; Zhang, H. Bifidobacterium lactis Probio-M8 improves bone metabolism in patients with postmenopausal osteoporosis, possibly by modulating the gut microbiota. Eur. J. Nutr. 2023, 62, 965–976. [Google Scholar] [CrossRef]
- Silva, F.M.; Petrica, J.; Serrano, J.; Paulo, R.; Ramalho, A.; Lucas, D.; Ferreira, J.P.; Duarte-Mendes, P. The Sedentary Time and Physical Activity Levels on Physical Fitness in the Elderly: A Comparative Cross Sectional Study. Int. J. Environ. Res. Public Health 2019, 16, 3697. [Google Scholar] [CrossRef]
- Mäkeläinen, H.; Tahvonen, R.; Salminen, S.; Ouwehand, A.C. In vivo safety assessment of two Bifidobacterium longum strains. Microbiol. Immunol. 2003, 47, 911–914. [Google Scholar] [CrossRef]
- Zhang, X.; Zhong, H.; Li, Y.; Shi, Z.; Ren, H.; Zhang, Z.; Zhou, X.; Tang, S.; Han, X.; Lin, Y.; et al. Sex- and age-related trajectories of the adult human gut microbiota shared across populations of different ethnicities. Nat. Aging 2021, 1, 87–100. [Google Scholar] [CrossRef]
- Arboleya, S.; Watkins, C.; Stanton, C.; Ross, R.P. Gut Bifidobacteria Populations in Human Health and Aging. Front. Microbiol. 2016, 7, 1204. [Google Scholar] [CrossRef]
- Kivimäki, M.; Strandberg, T.; Pentti, J.; Nyberg, S.T.; Frank, P.; Jokela, M.; Ervasti, J.; Suominen, S.B.; Vahtera, J.; Sipilä, P.N.; et al. Body-mass index and risk of obesity-related complex multimorbidity: An observational multicohort study. Lancet Diabetes Endocrinol. 2022, 10, 253–263. [Google Scholar] [CrossRef]
- Kappel, B.A.; De Angelis, L.; Puetz, A.; Ballanti, M.; Menghini, R.; Marx, N.; Federici, M. Antibiotic-induced gut microbiota depletion exacerbates host hypercholesterolemia. Pharmacol. Res. 2023, 187, 106570. [Google Scholar] [CrossRef]
- Su, Q.; Tun, H.M.; Liu, Q.; Yeoh, Y.K.; Mak, J.W.Y.; Chan, F.K.; Ng, S.C. Gut microbiome signatures reflect different subtypes of irritable bowel syndrome. Gut Microbes 2023, 15, 2157697. [Google Scholar] [CrossRef]
- Mahdavinia, M.; Fyolek, J.P.; Jiang, J.; Thivalapill, N.; Bilaver, L.A.; Warren, C.; Fox, S.; Nimmagadda, S.R.; Newmark, P.J.; Sharma, H.; et al. Gut microbiome is associated with asthma and race in children with food allergy. J. Allergy Clin. Immunol. 2023, 152, 1541–1549.e1541. [Google Scholar] [CrossRef]
- Roslund, M.I.; Puhakka, R.; Grönroos, M.; Nurminen, N.; Oikarinen, S.; Gazali, A.M.; Cinek, O.; Kramná, L.; Siter, N.; Vari, H.K.; et al. Biodiversity intervention enhances immune regulation and health-associated commensal microbiota among daycare children. Sci. Adv. 2020, 6, eaba2578. [Google Scholar] [CrossRef]
- Zhou, B.; Jin, G.; Pang, X.; Mo, Q.; Bao, J.; Liu, T.; Wu, J.; Xie, R.; Liu, X.; Liu, J.; et al. Lactobacillus rhamnosus GG colonization in early life regulates gut-brain axis and relieves anxiety-like behavior in adulthood. Pharmacol. Res. 2022, 177, 106090. [Google Scholar] [CrossRef]
- Song, Q.; Zhang, X.; Liu, W.; Wei, H.; Liang, W.; Zhou, Y.; Ding, Y.; Ji, F.; Ho-Kwan Cheung, A.; Wong, N.; et al. Bifidobacterium pseudolongum-generated acetate suppresses non-alcoholic fatty liver disease-associated hepatocellular carcinoma. J. Hepatol. 2023, 79, 1352–1365. [Google Scholar] [CrossRef]
- Ye, L.; Hou, Y.; Hu, W.; Wang, H.; Yang, R.; Zhang, Q.; Feng, Q.; Zheng, X.; Yao, G.; Hao, H. Repressed Blautia-acetate immunological axis underlies breast cancer progression promoted by chronic stress. Nat. Commun. 2023, 14, 6160. [Google Scholar] [CrossRef]
- Kim, C.S.; Cha, L.; Sim, M.; Jung, S.; Chun, W.Y.; Baik, H.W.; Shin, D.M. Probiotic Supplementation Improves Cognitive Function and Mood with Changes in Gut Microbiota in Community-Dwelling Older Adults: A Randomized, Double-Blind, Placebo-Controlled, Multicenter Trial. J. Gerontol. A Biol. Sci. Med. Sci. 2021, 76, 32–40. [Google Scholar] [CrossRef]
- Derrien, M.; Turroni, F.; Ventura, M.; van Sinderen, D. Insights into endogenous Bifidobacterium species in the human gut microbiota during adulthood. Trends Microbiol. 2022, 30, 940–947. [Google Scholar] [CrossRef]
- Engevik, M.A.; Danhof, H.A.; Hall, A.; Engevik, K.A.; Horvath, T.D.; Haidacher, S.J.; Hoch, K.M.; Endres, B.T.; Bajaj, M.; Garey, K.W.; et al. The metabolic profile of Bifidobacterium dentium reflects its status as a human gut commensal. BMC Microbiol. 2021, 21, 154. [Google Scholar] [CrossRef] [PubMed]
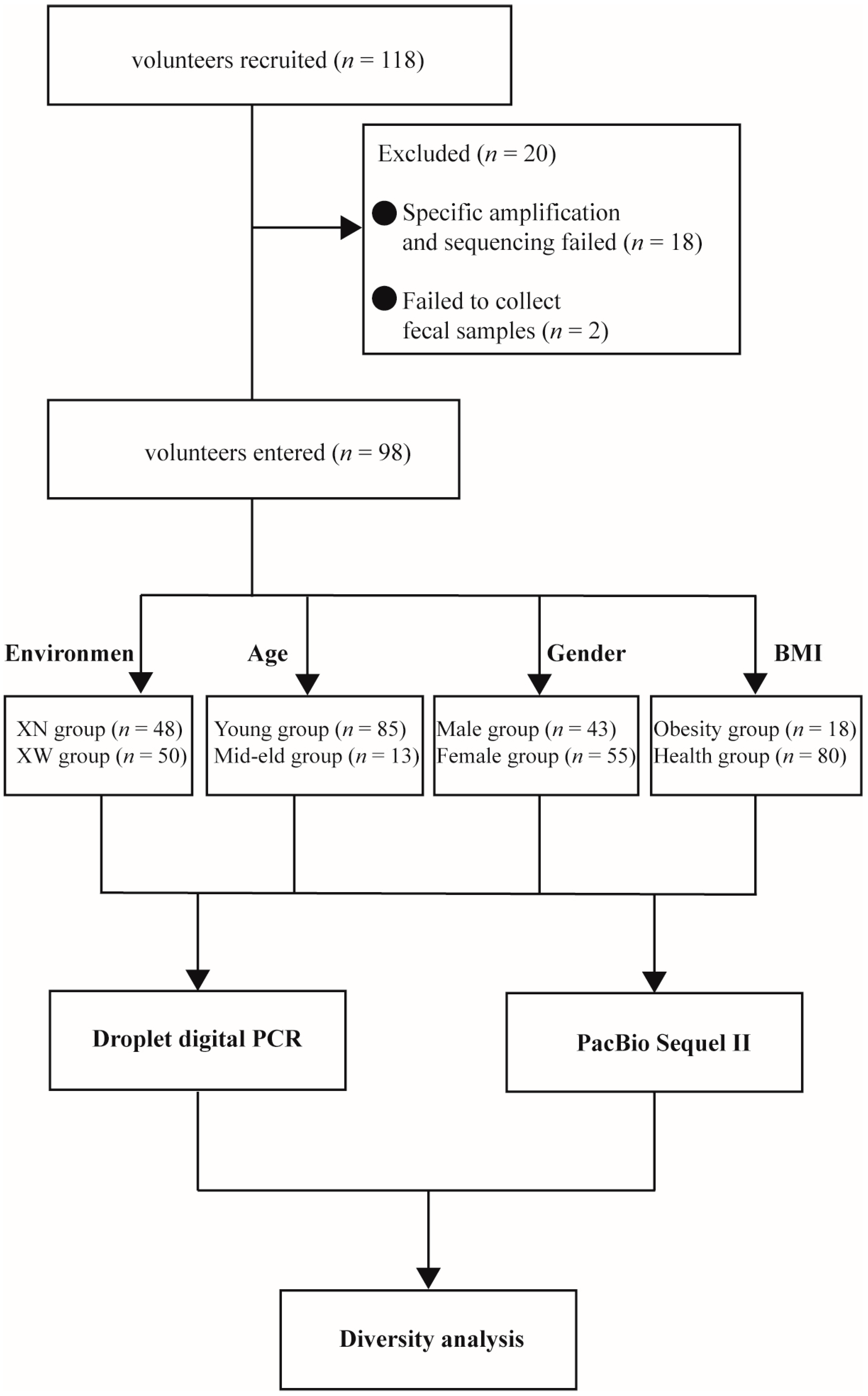
| Group | Living Environment | Age | Gender | BMI | ||||
|---|---|---|---|---|---|---|---|---|
| Fundamentum divisions | Differences in living environment | 18 ≤ Age ≤ 39, Young; Age > 39, Middle aged and elderly people. | ---- | BMI > 28, Obesity; BMI < 28, Health. | ||||
| Group name | XN | XW | Young | Mid–eld | Male | Female | Obesity | Health |
| Number (n) | 48 | 50 | 85 | 13 | 43 | 55 | 18 | 80 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, S.; Wu, S.; Zhao, F.; Zhao, Z.; Shen, X.; Yu, X.; Zhang, M.; Wen, F.; Sun, Z.; Menghe, B. Diversity Analysis of Intestinal Bifidobacteria in the Hohhot Population. Microorganisms 2024, 12, 756. https://doi.org/10.3390/microorganisms12040756
Yang S, Wu S, Zhao F, Zhao Z, Shen X, Yu X, Zhang M, Wen F, Sun Z, Menghe B. Diversity Analysis of Intestinal Bifidobacteria in the Hohhot Population. Microorganisms. 2024; 12(4):756. https://doi.org/10.3390/microorganisms12040756
Chicago/Turabian StyleYang, Shuying, Su Wu, Feiyan Zhao, Zhixin Zhao, Xin Shen, Xia Yu, Meng Zhang, Fang Wen, Zhihong Sun, and Bilige Menghe. 2024. "Diversity Analysis of Intestinal Bifidobacteria in the Hohhot Population" Microorganisms 12, no. 4: 756. https://doi.org/10.3390/microorganisms12040756