
Ecological Trait-Based Digital Categorization of Microbial Genomes for Denitrification Potential

Abstract
:1. Introduction
2. Materials and Methods
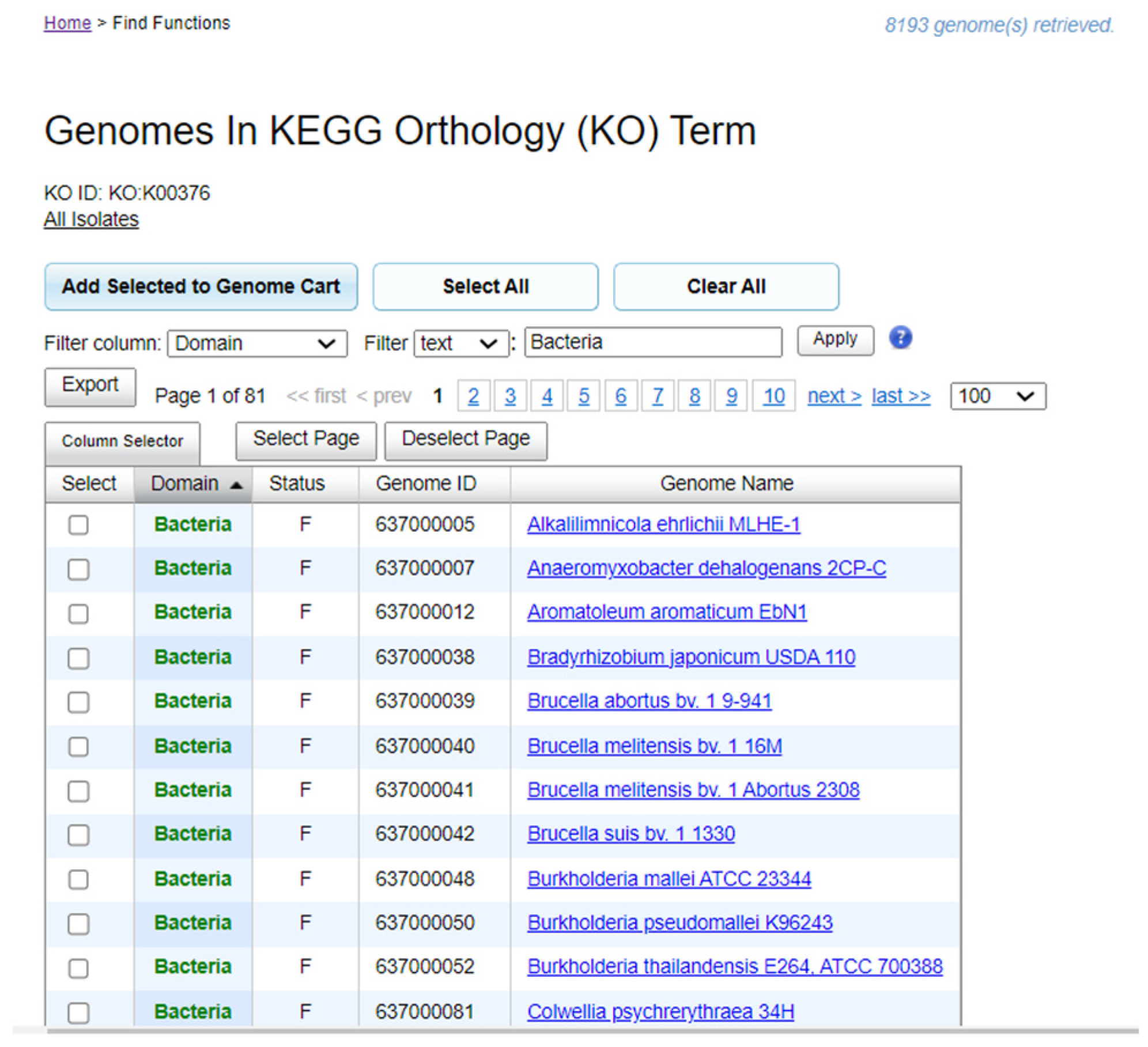
2.1. Data Sources for Functional Annotation Identifiers of Denitrification Enzyme Genes
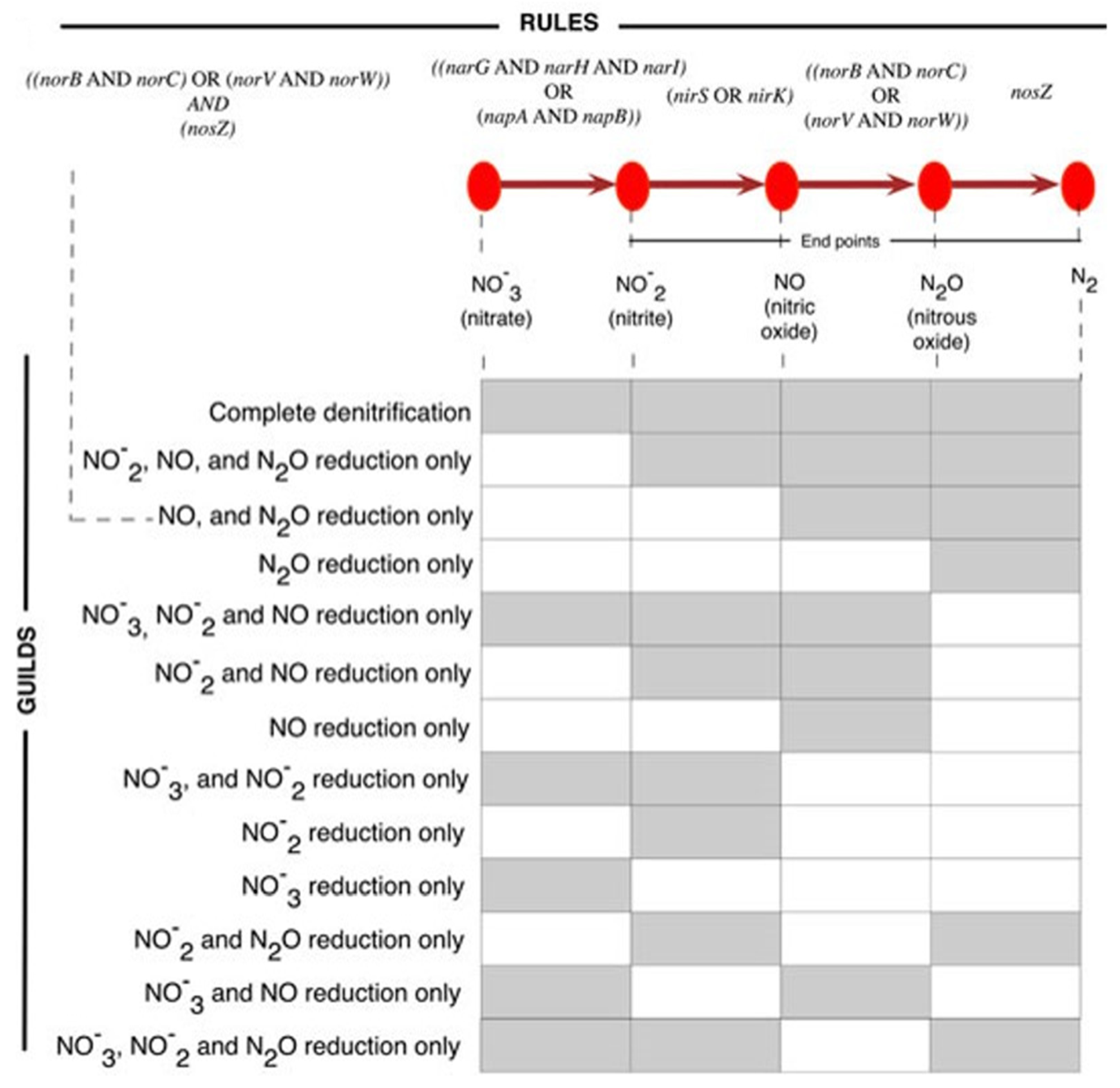
2.2. Construction of a Denitrification Potential of Archaeal and Bacterial Genomes Dataset
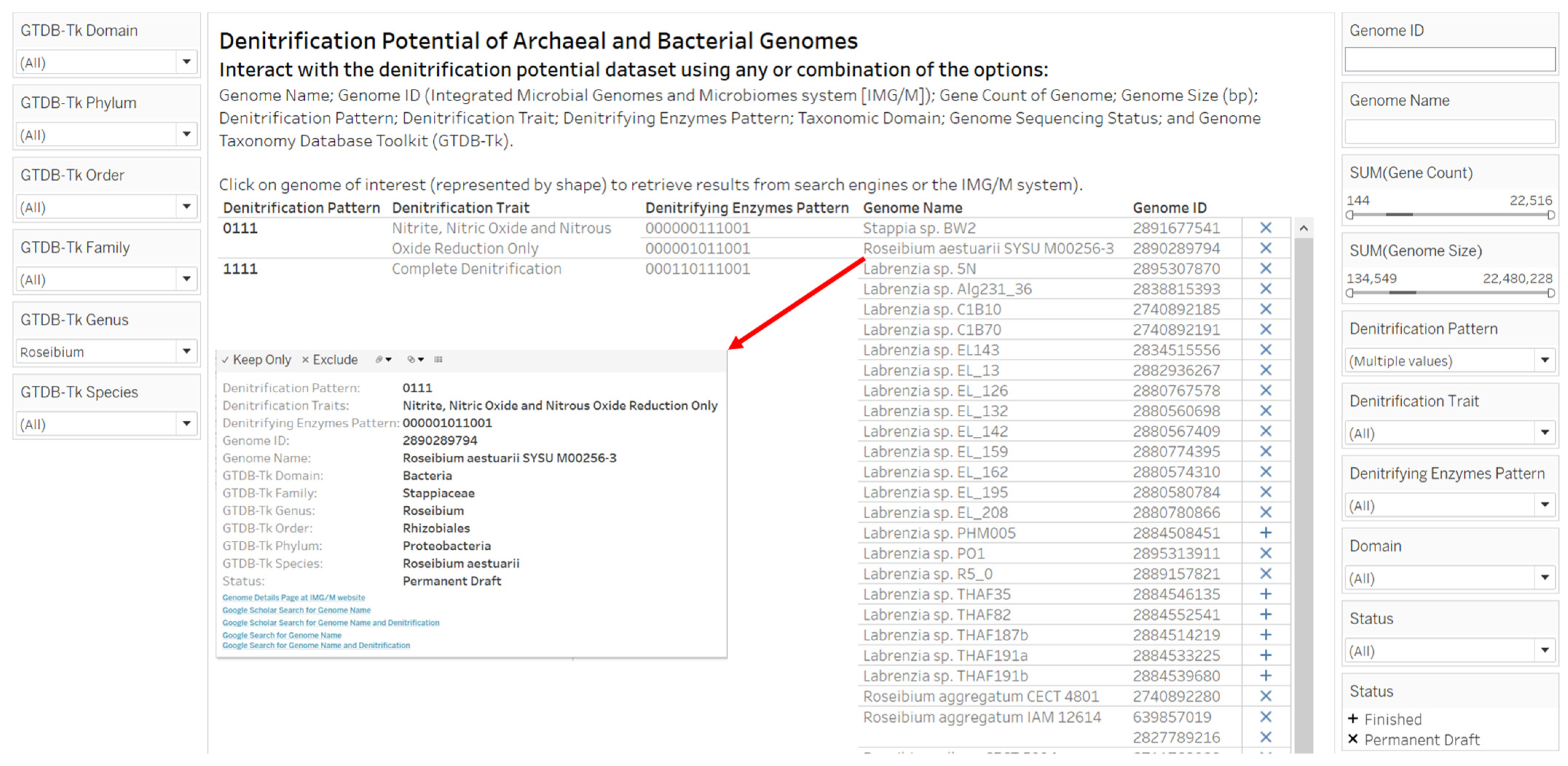
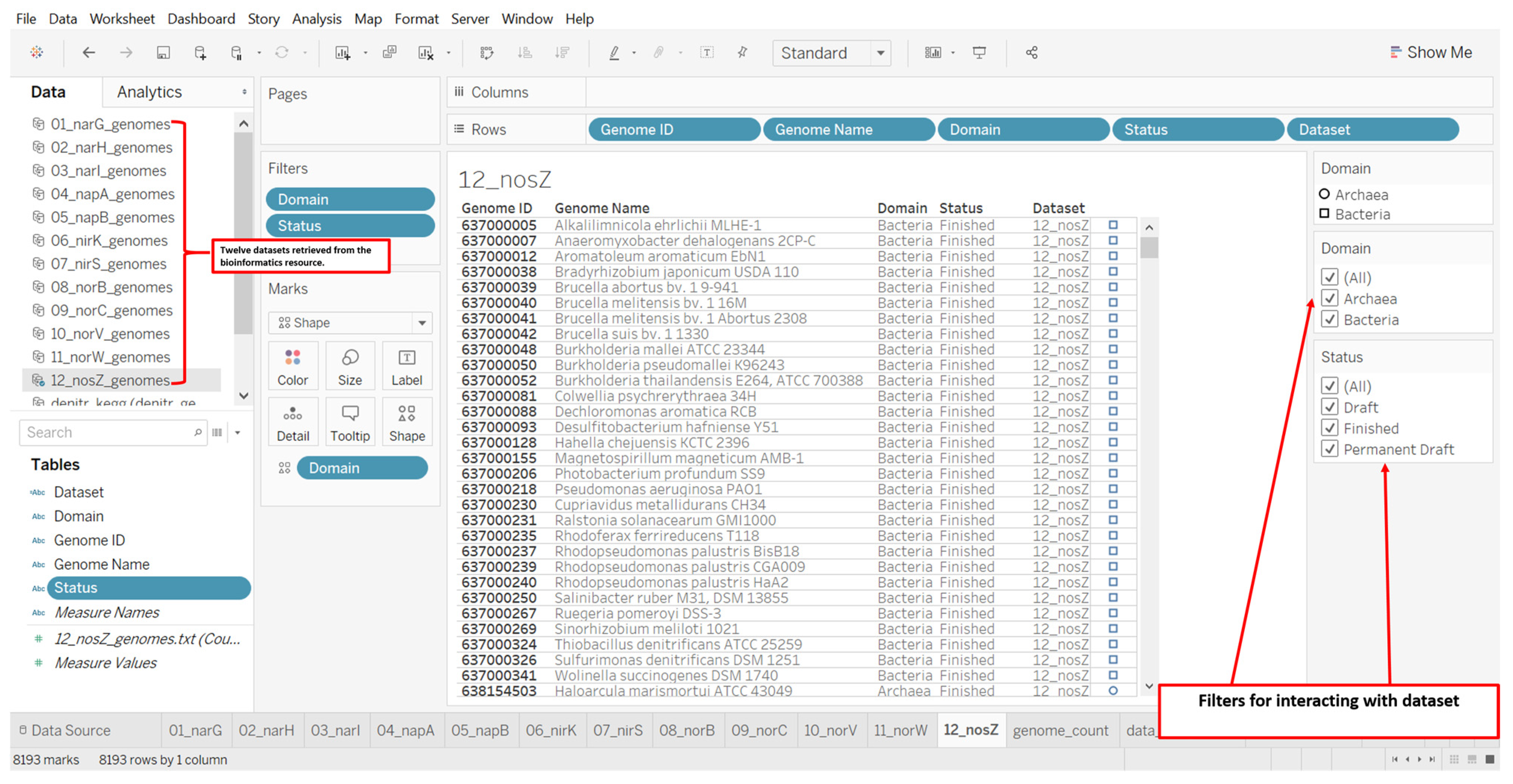
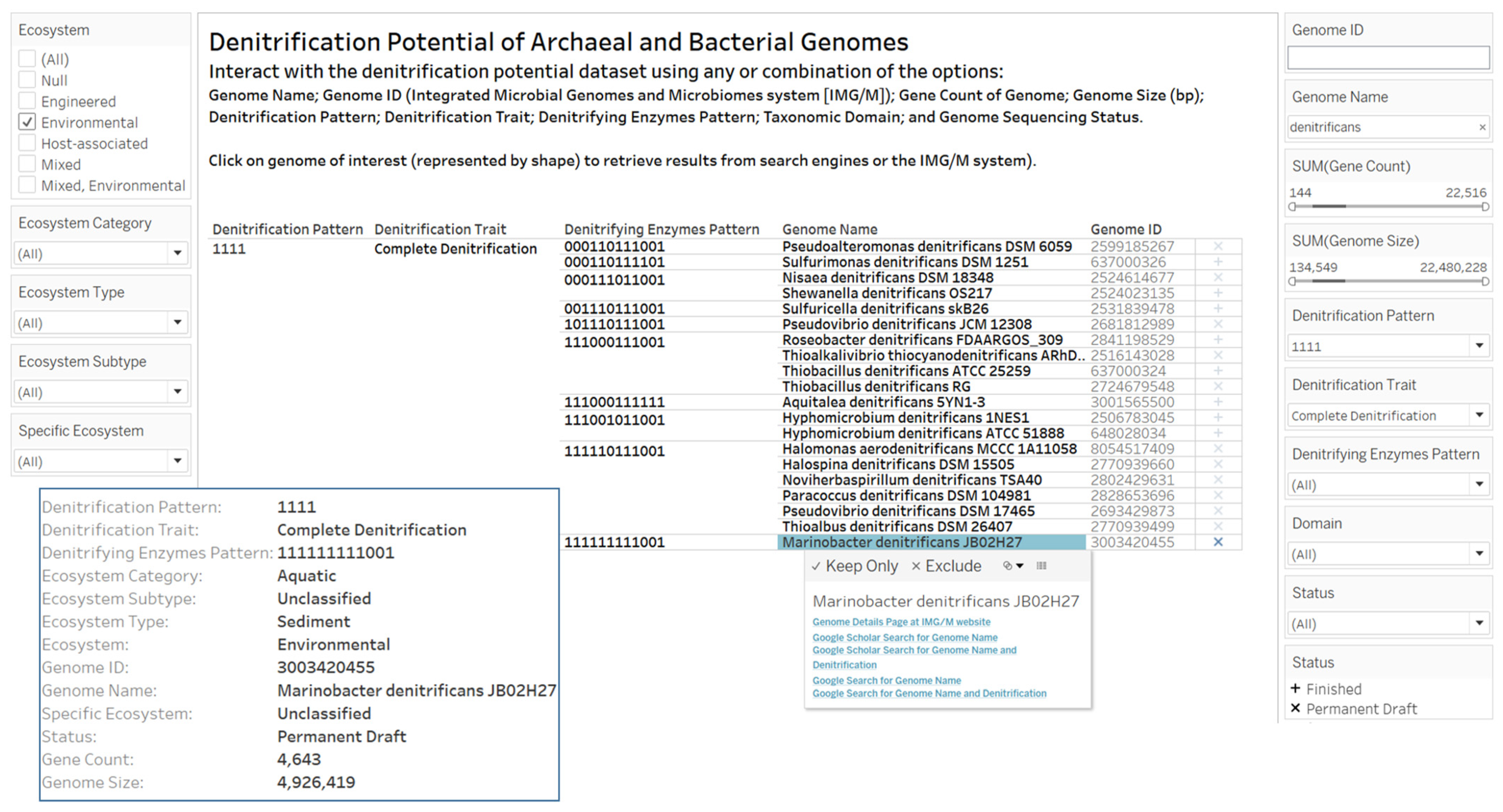
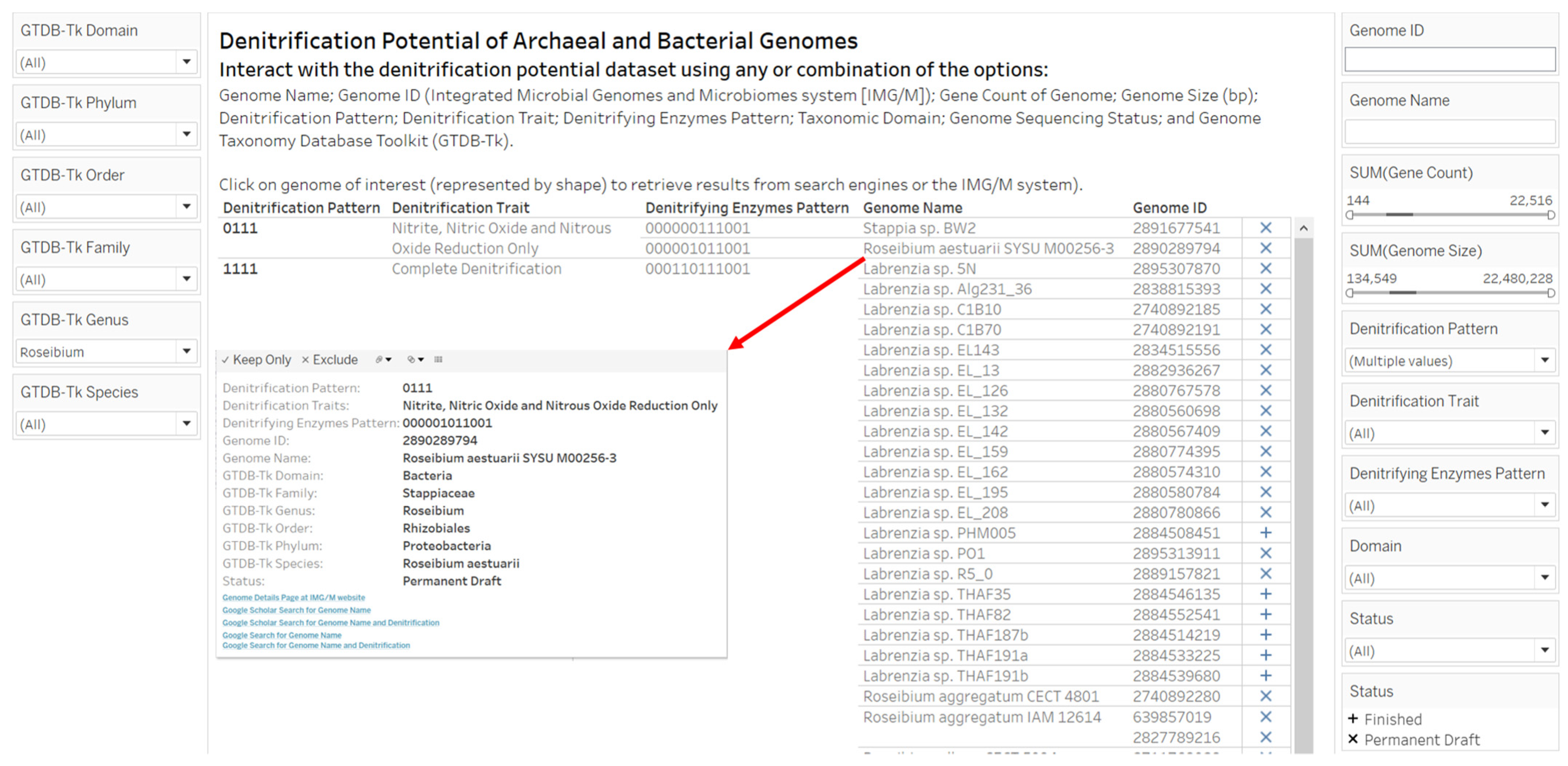
2.3. Designs and Implementations of Visual Analytics Resources to Support Human Interaction with the Dataset on the Denitrification Potential of Microbial Genomes
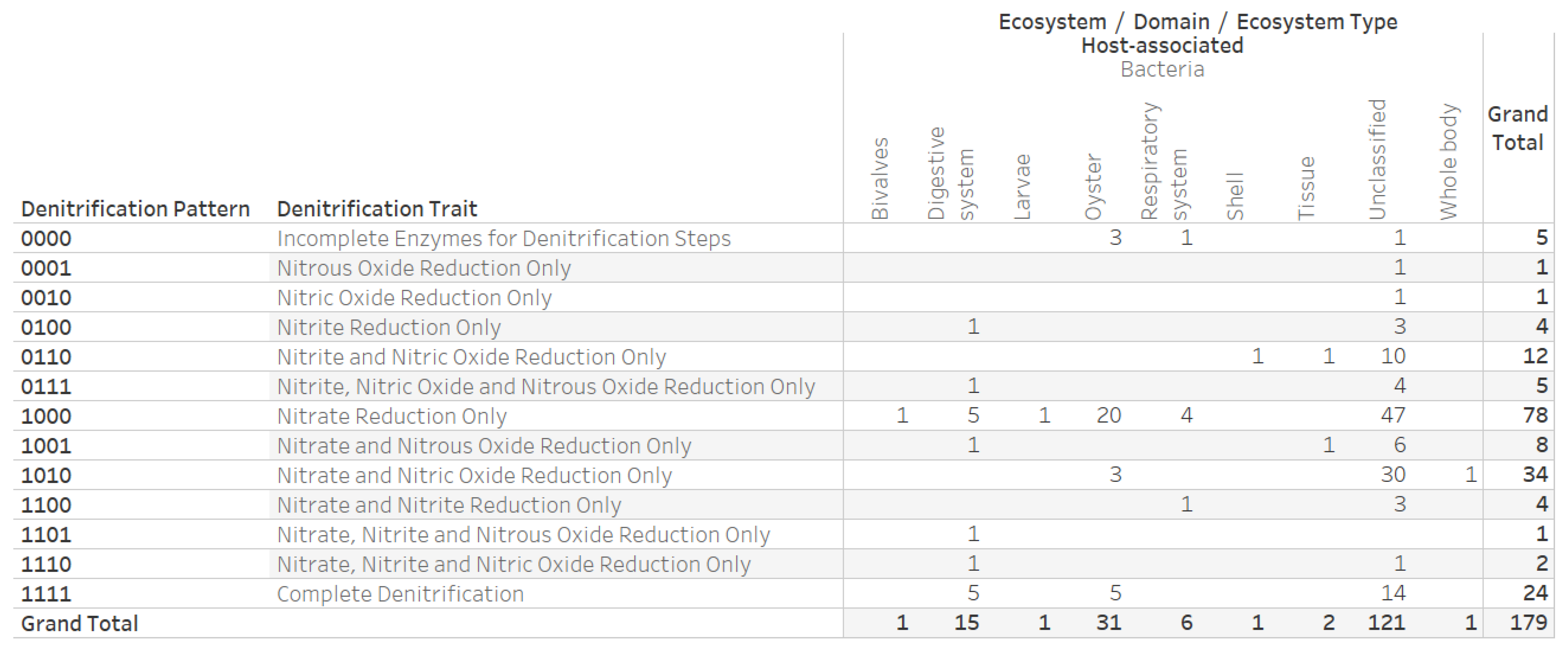
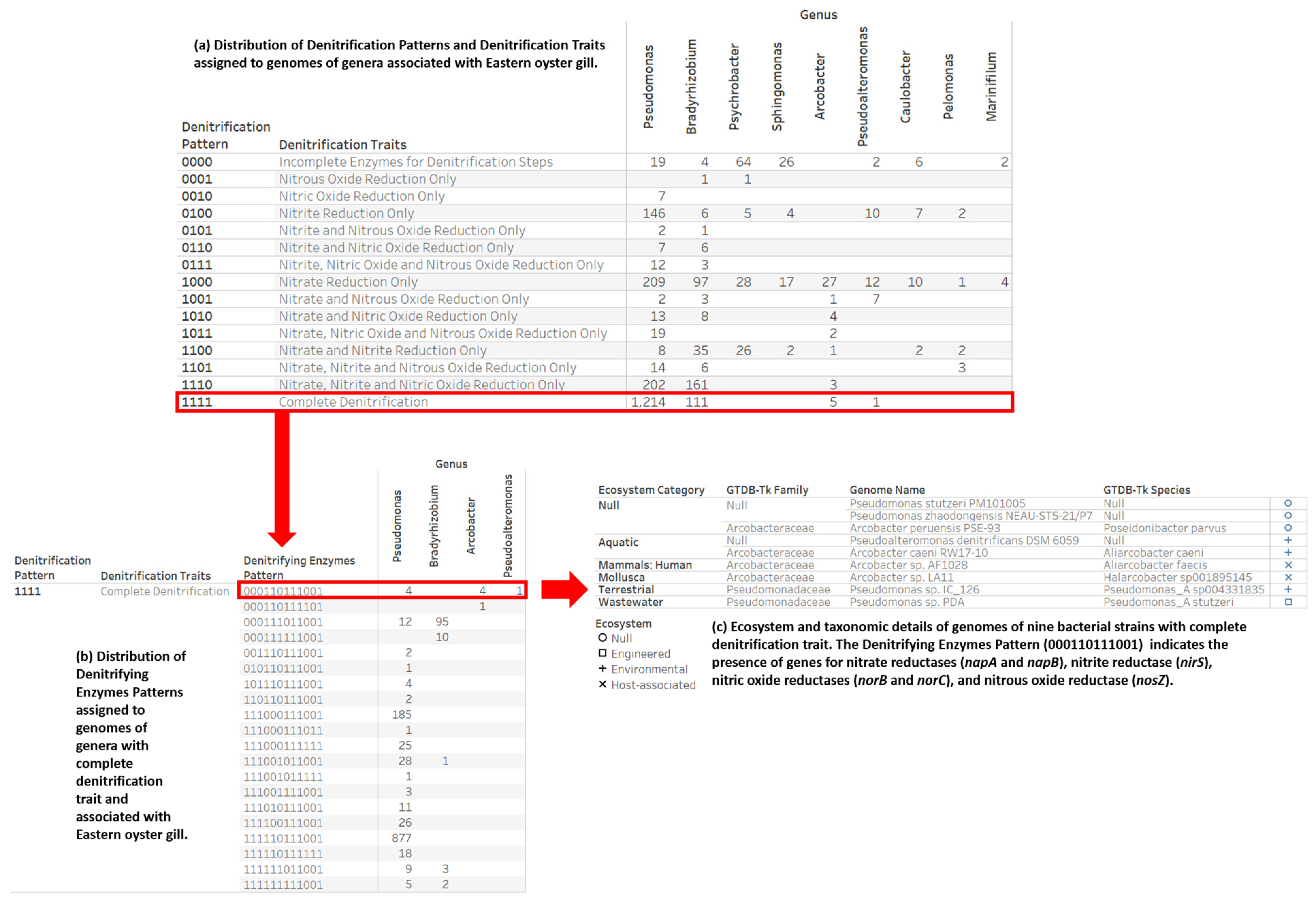
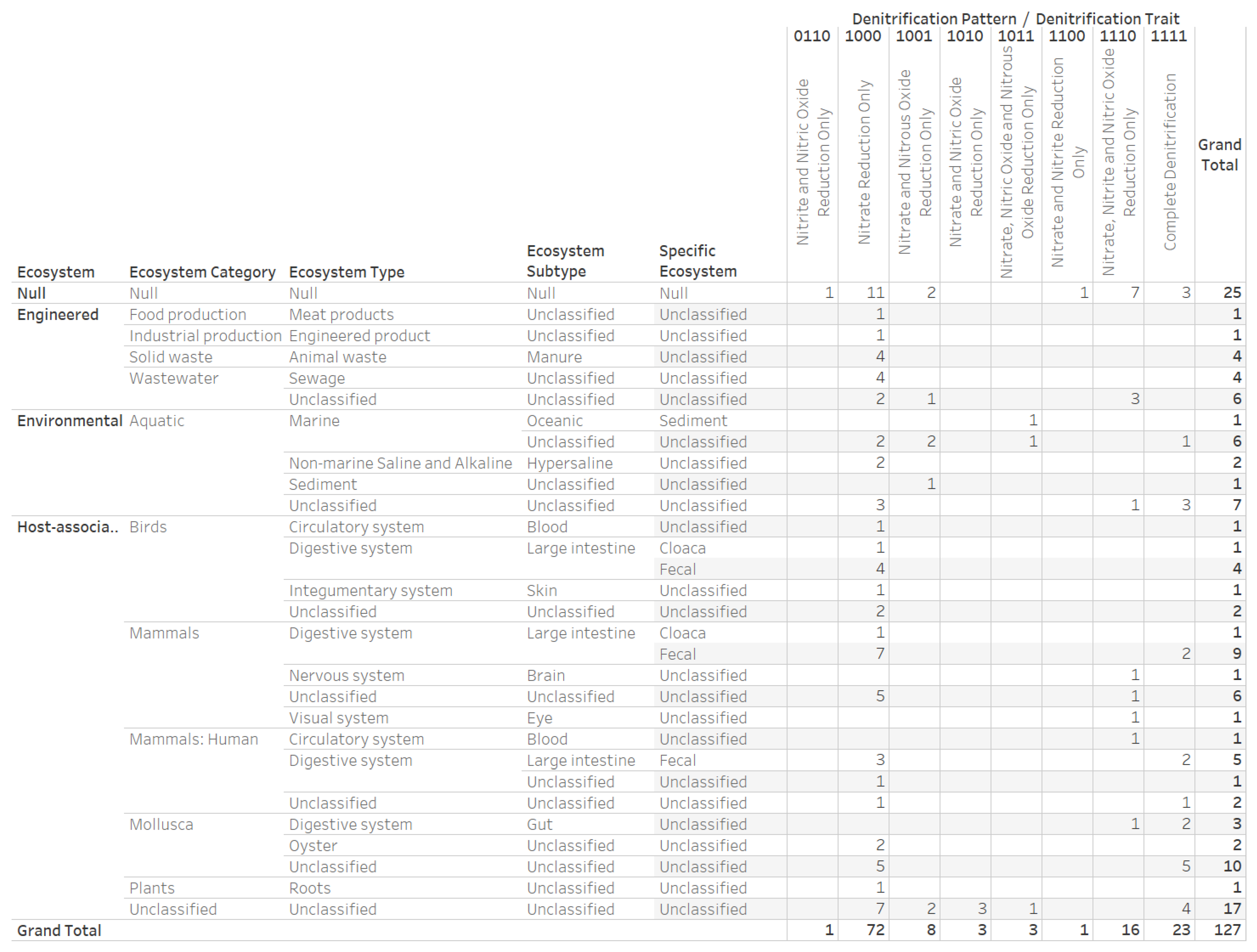
2.4. Denitrification Potential Categorization of Bacterial Genera Associated with Eastern Oyster (Crassostrea virginica)
3. Results
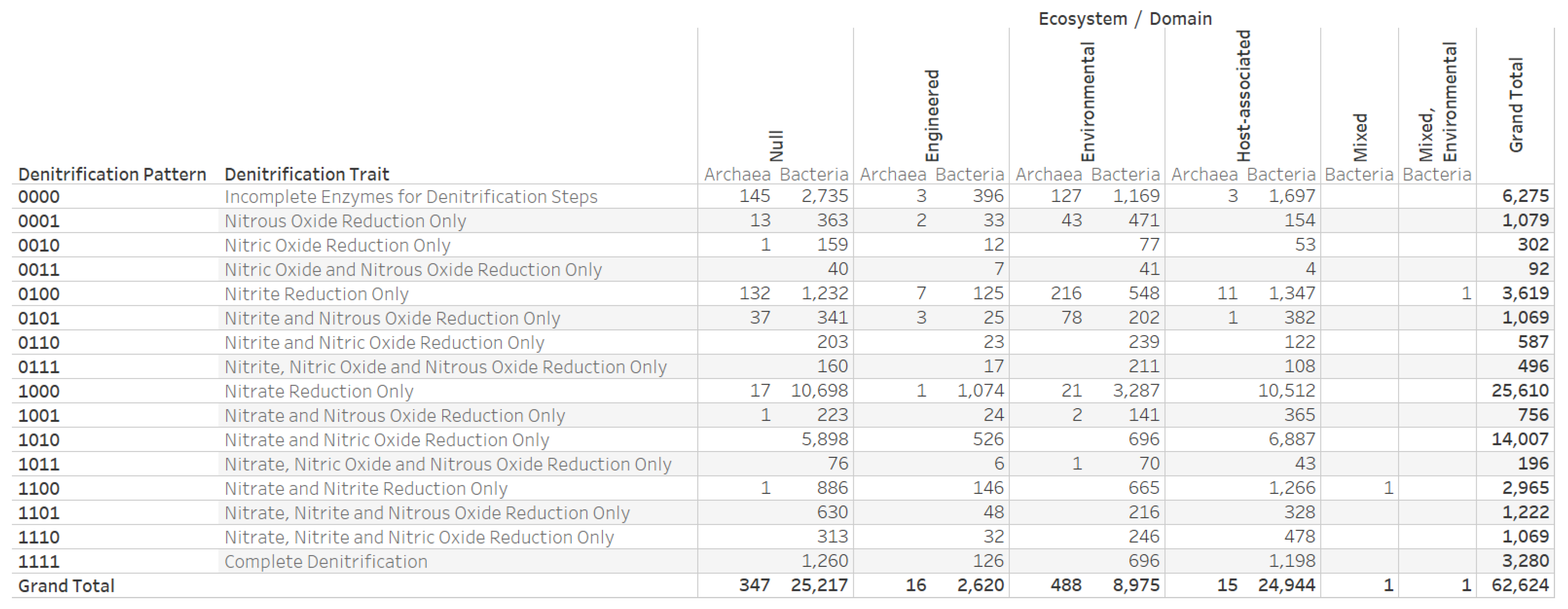
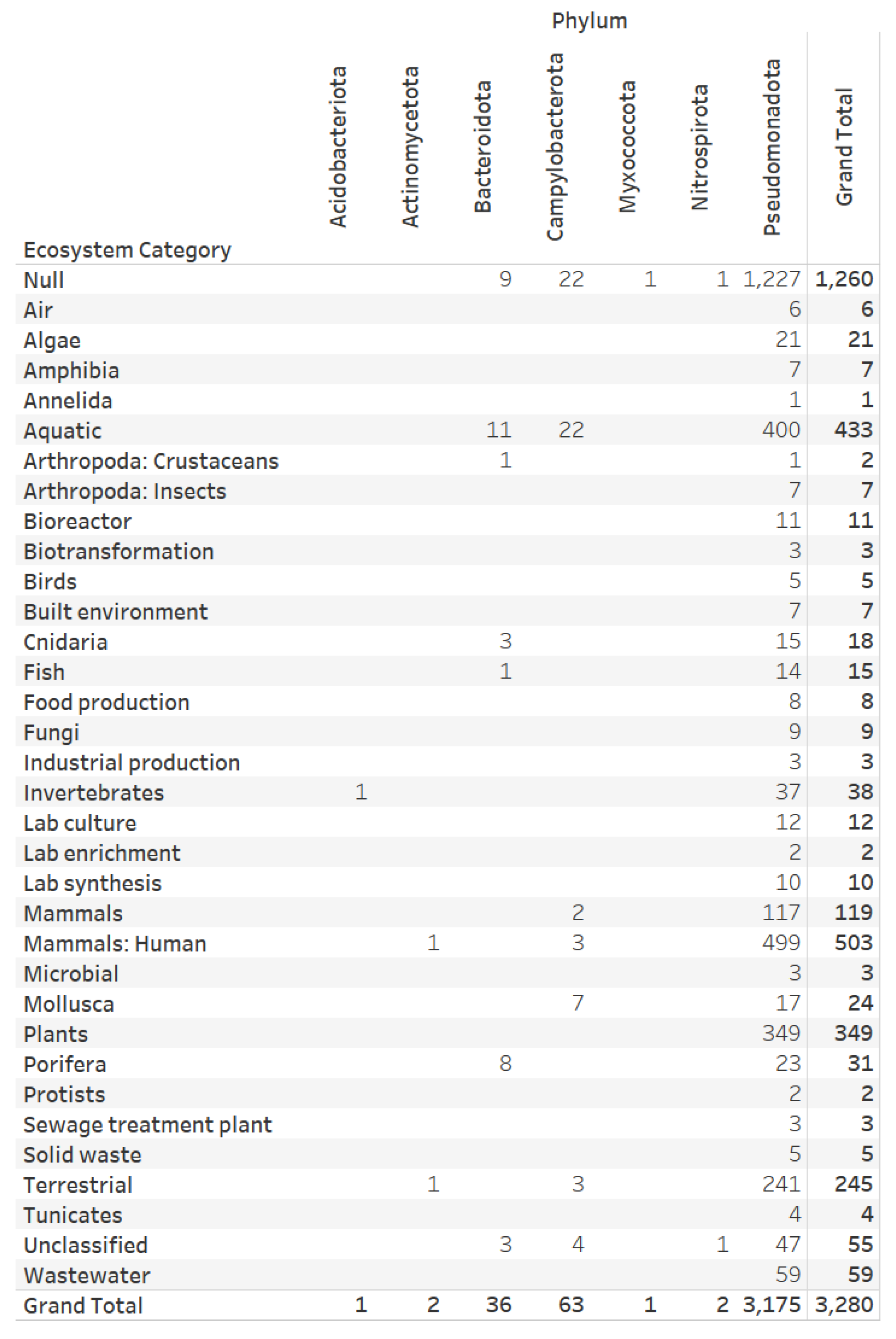
3.1. Dataset on the Denitrification Potential of Microbial Genomes
3.2. Designs and Implementations of Visual Analytics Resources to Support Interaction with the Dataset on the Denitrification Potential of Microbial Genomes
3.3. Denitrification Potential Categorization of Bacterial Genera Associated with Eastern Oyster (Crassostrea virginica)
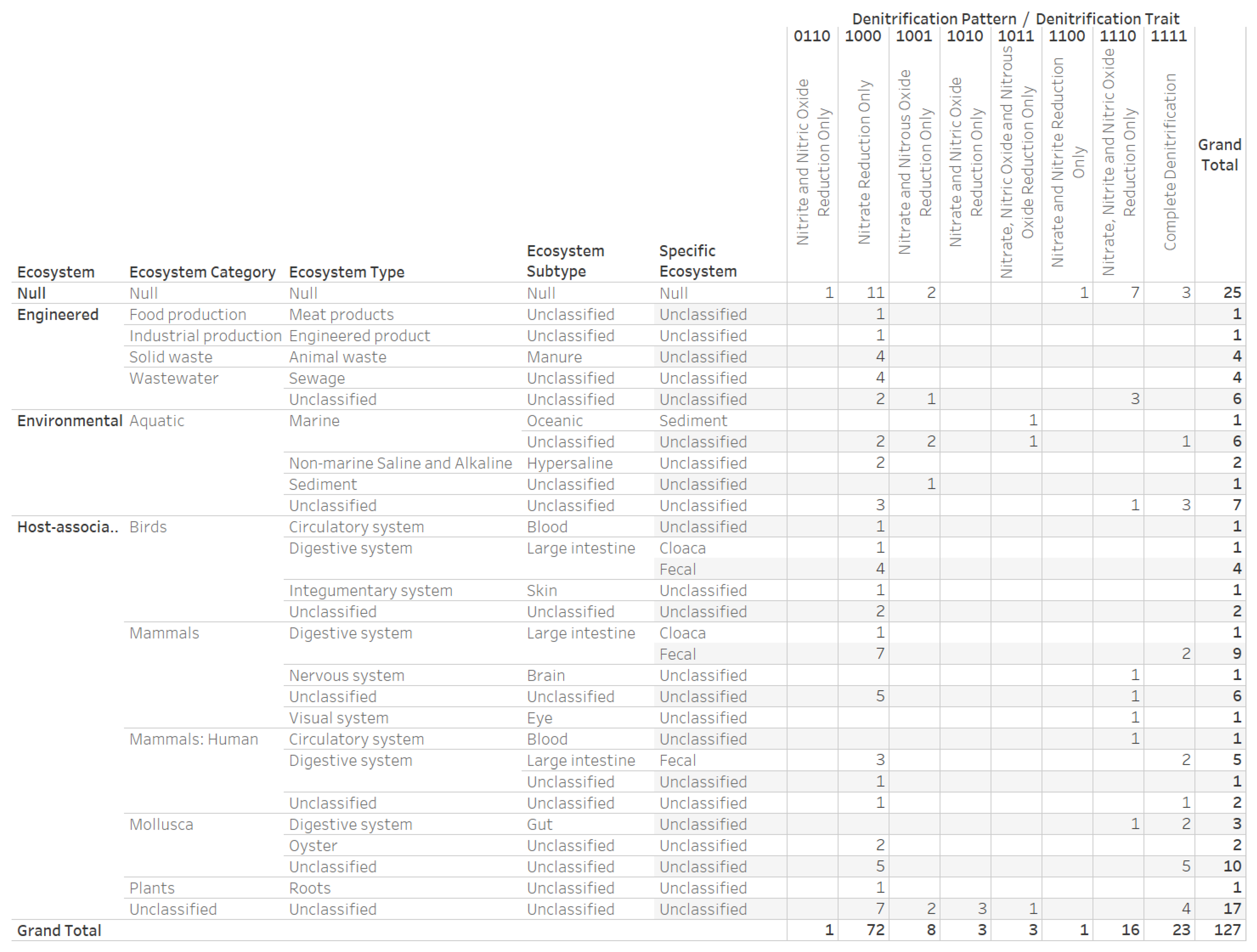
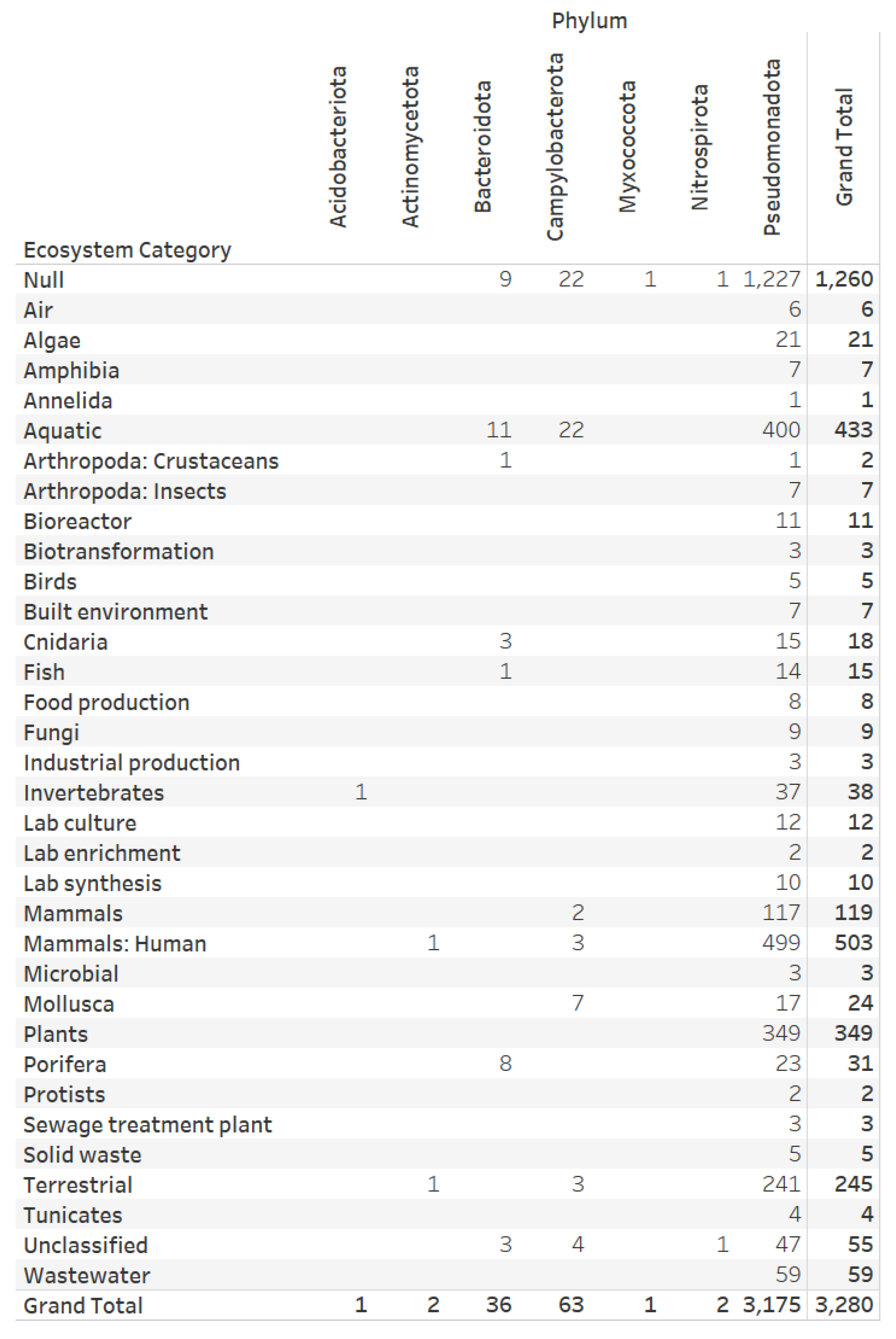
3.4. Nitrogen Assimilation, Taxonomic, and Ecosystems Annotations for Genomes with a Complete Denitrification Pattern
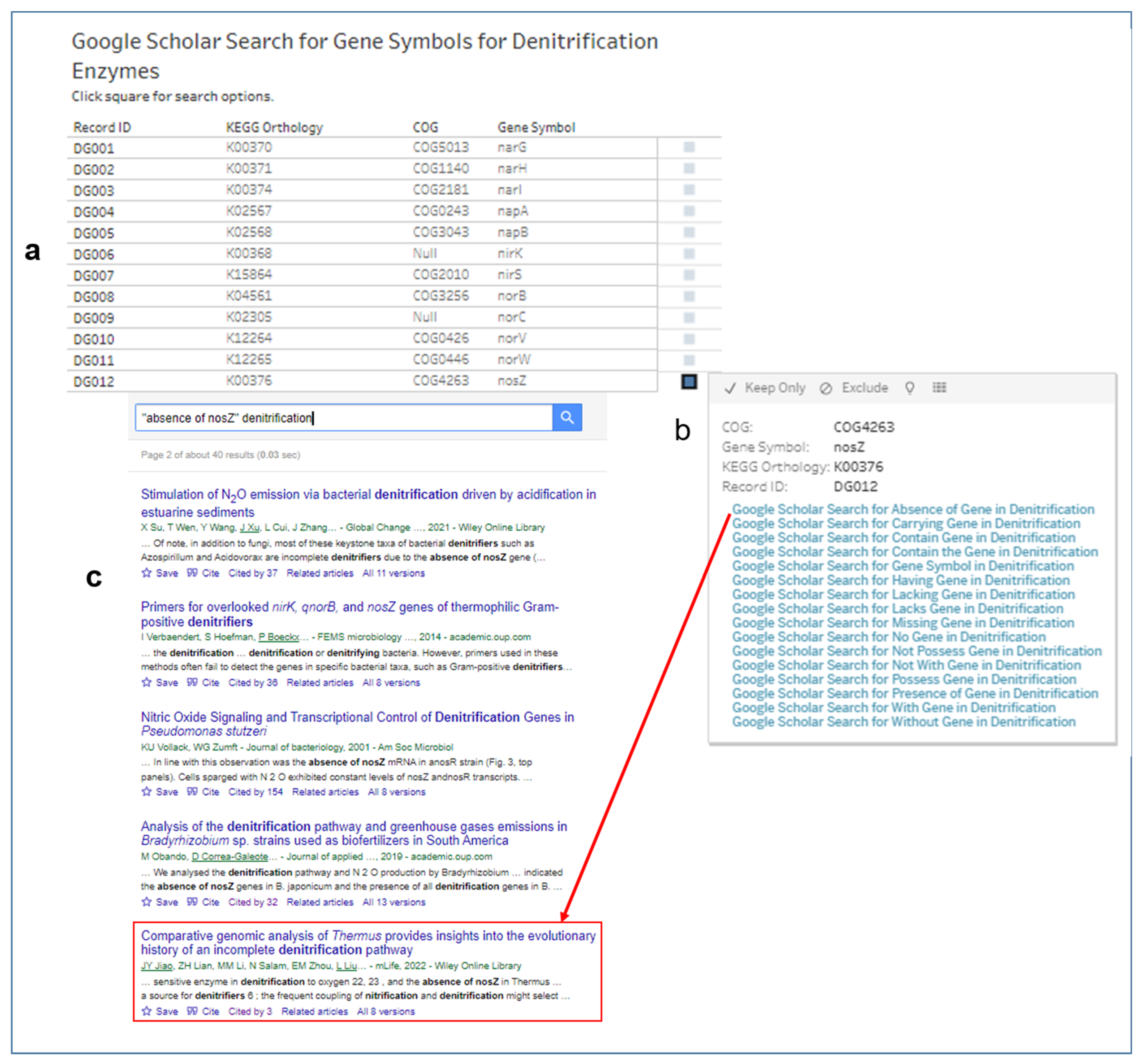
3.5. Searches for Scholarly Articles with Gene Symbols of Enzymes for Denitrification
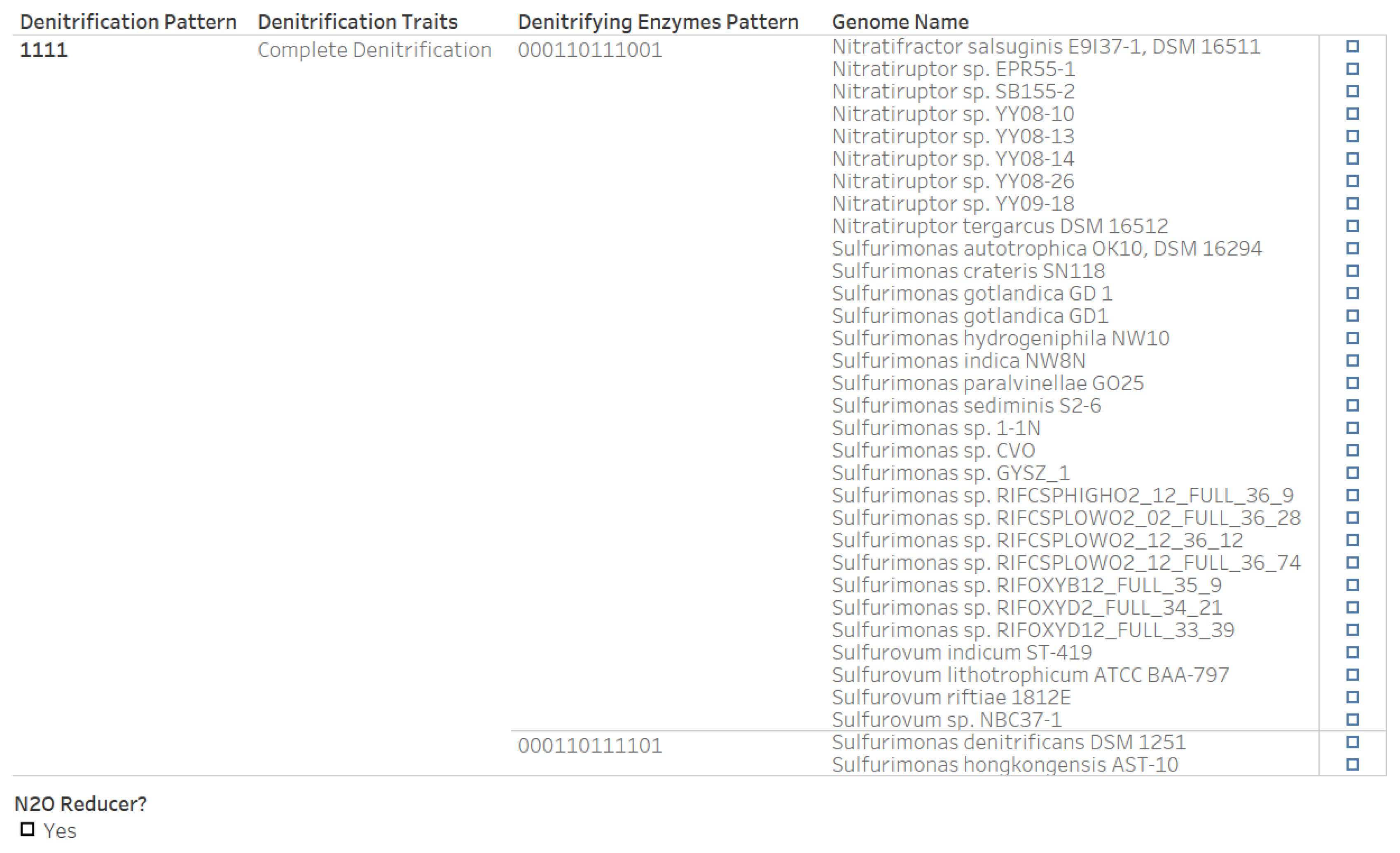
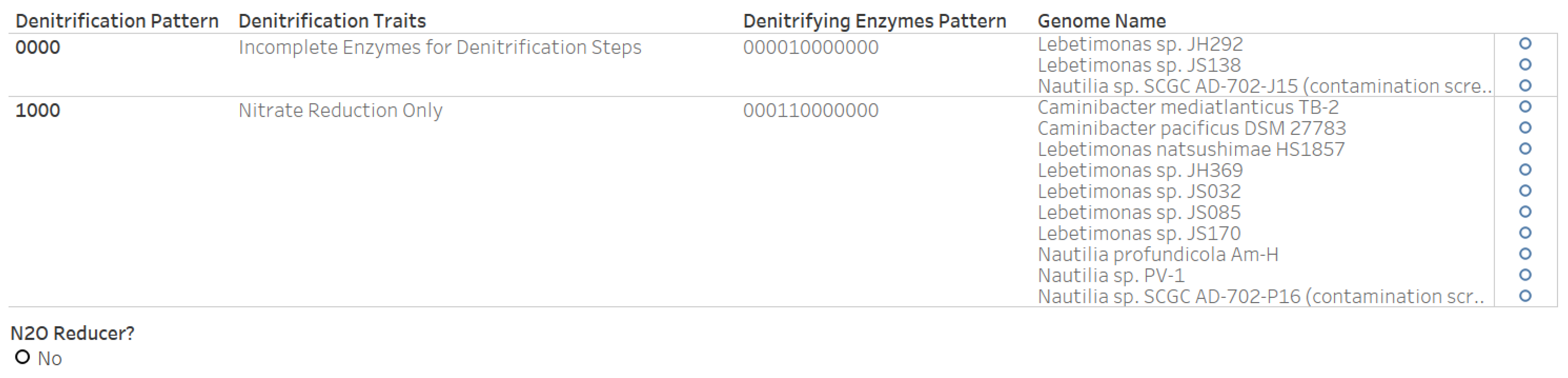
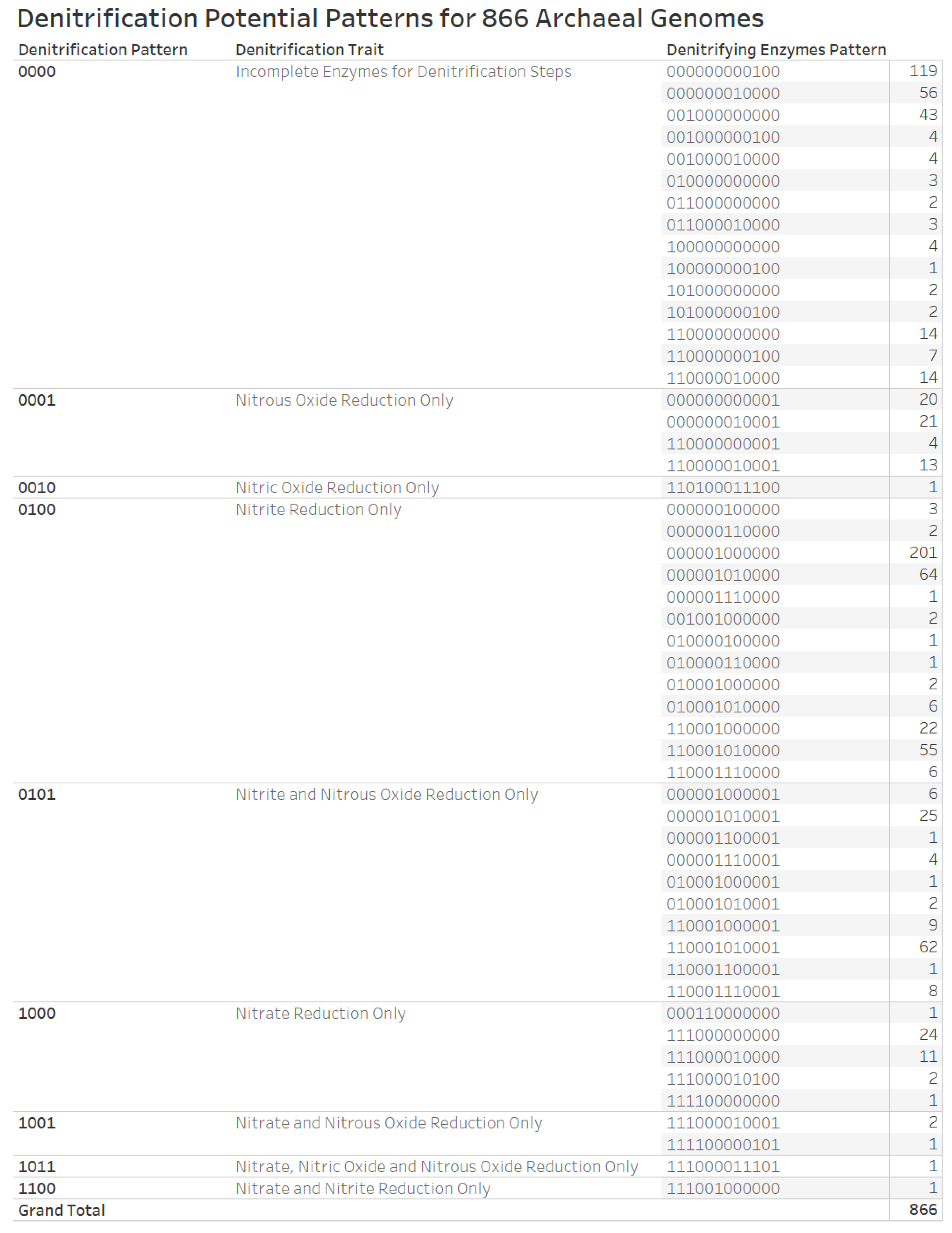
3.6. Denitrification Patterns of Archaeal Genomes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
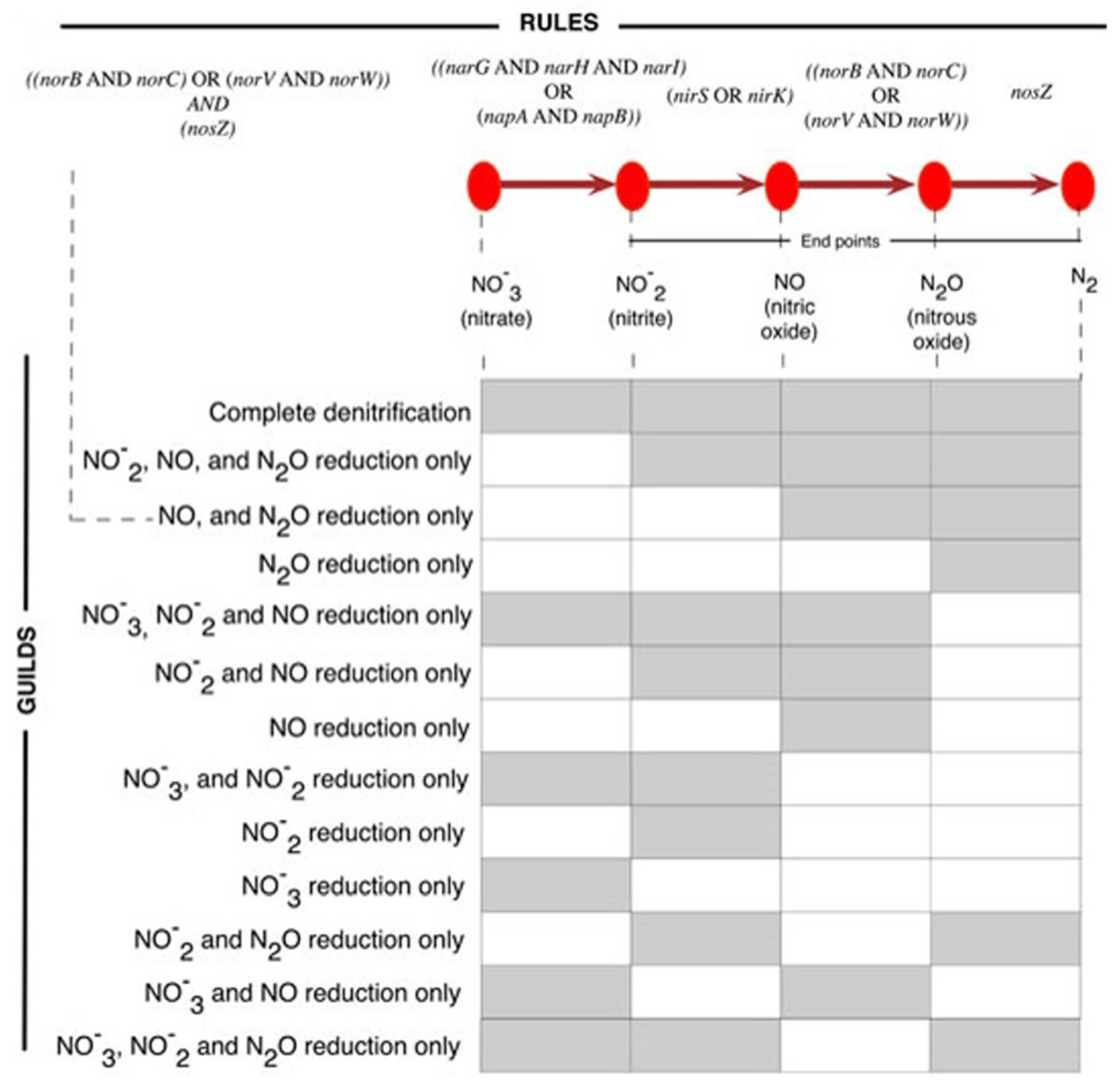
Appendix B

Appendix C
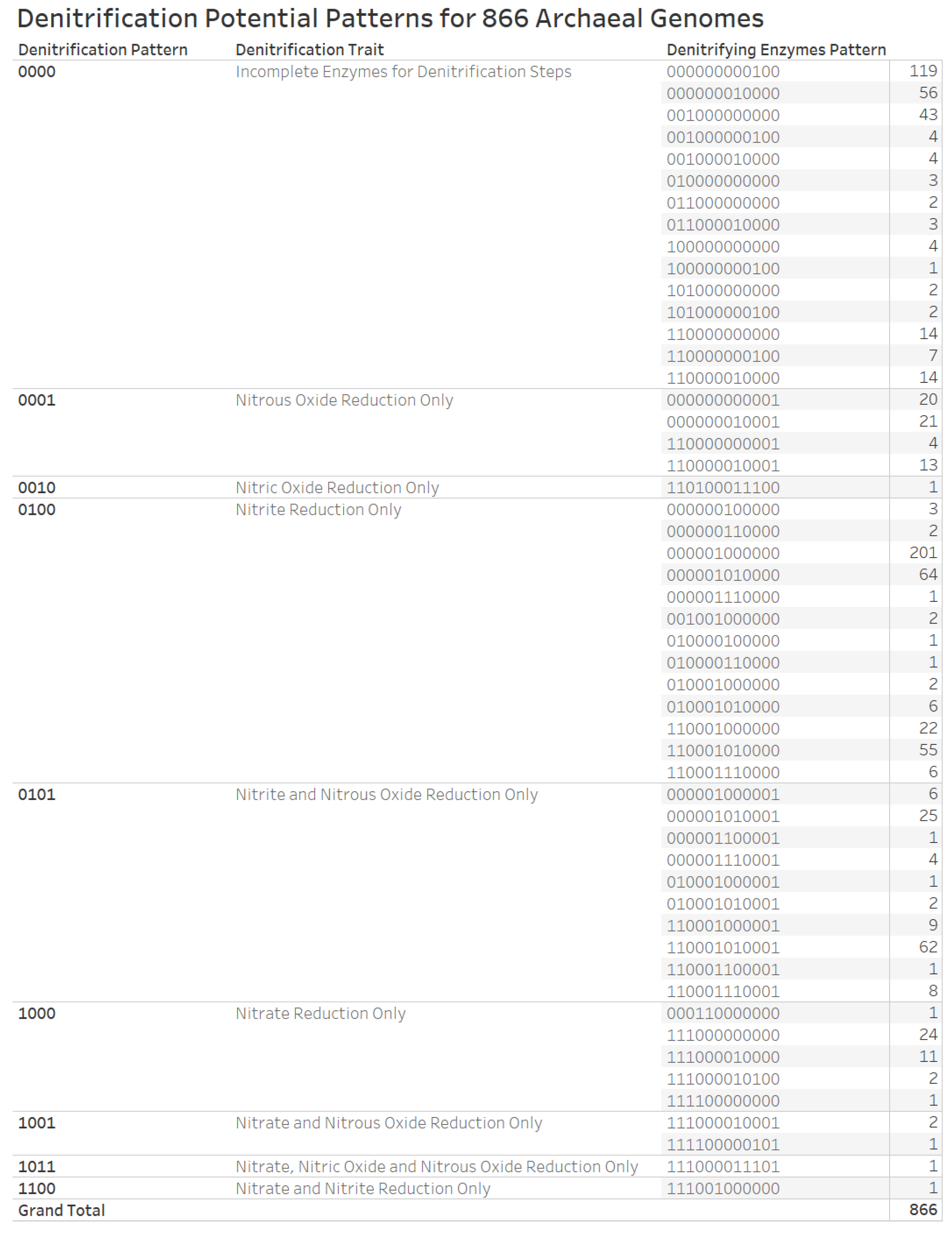
References
- Stein, L.Y.; Klotz, M.G. The nitrogen cycle. Curr. Biol. 2016, 26, R94–R98. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ward, B.B.; Sigman, D.M. Global nitrogen cycle: Critical enzymes, organisms, and processes for nitrogen budgets and dynamics. Chem. Rev. 2020, 120, 5308–5351. [Google Scholar] [CrossRef] [PubMed]
- Bernhard, A. The nitrogen cycle: Processes, players, and human impact. Nat. Educ. Knowl. 2010, 3, 25. [Google Scholar]
- Robertson, G.P.; Groffman, P. Nitrogen transformations. In Soil Microbiology, Ecology and Biochemistry; Elsevier: Amsterdam, The Netherlands, 2015; pp. 407–438. [Google Scholar]
- Albright, M.B.; Timalsina, B.; Martiny, J.B.; Dunbar, J. Comparative genomics of nitrogen cycling pathways in bacteria and archaea. Microb. Ecol. 2019, 77, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Mosley, O.E.; Gios, E.; Close, M.; Weaver, L.; Daughney, C.; Handley, K.M. Nitrogen cycling and microbial cooperation in the terrestrial subsurface. ISME J. 2022, 16, 2561–2573. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Guan, X.; Deng, T.; Yang, X.; Li, J.; Zhou, M.; Wang, C.; Wang, S.; Yan, Q.; Shu, L. Synthetic denitrifying communities reveal a positive and dynamic biodiversity-ecosystem functioning relationship during experimental evolution. Microbiol. Spectr. 2023, 11, e04528-22. [Google Scholar] [CrossRef] [PubMed]
- Arat, S.; Bullerjahn, G.S.; Laubenbacher, R. A network biology approach to denitrification in Pseudomonas aeruginosa. PLoS ONE 2015, 10, e0118235. [Google Scholar] [CrossRef] [PubMed]
- Enwall, K.; Philippot, L.; Hallin, S. Activity and composition of the denitrifying bacterial community respond differently to long-term fertilization. Appl. Environ. Microbiol. 2005, 71, 8335–8343. [Google Scholar] [CrossRef] [PubMed]
- Karaoz, U.; Brodie, E.L. microTrait: A toolset for a trait-based representation of microbial genomes. Front. Bioinform. 2022, 2, 918853. [Google Scholar] [CrossRef]
- Verbaendert, I.; Boon, N.; De Vos, P.; Heylen, K. Denitrification is a common feature among members of the genus Bacillus. Syst. Appl. Microbiol. 2011, 34, 385–391. [Google Scholar] [CrossRef]
- Schneider, F.D.; Fichtmueller, D.; Gossner, M.M.; Güntsch, A.; Jochum, M.; König-Ries, B.; Le Provost, G.; Manning, P.; Ostrowski, A.; Penone, C. Towards an ecological trait-data standard. Methods Ecol. Evol. 2019, 10, 2006–2019. [Google Scholar] [CrossRef]
- Blake, J.A.; Bult, C.J. Beyond the data deluge: Data integration and bio-ontologies. J. Biomed. Inform. 2006, 39, 314–320. [Google Scholar] [CrossRef]
- Karp, P.D.; Ivanova, N.; Krummenacker, M.; Kyrpides, N.; Latendresse, M.; Midford, P.; Ong, W.K.; Paley, S.; Seshadri, R. A comparison of microbial genome web portals. Front. Microbiol. 2019, 10, 208. [Google Scholar] [CrossRef]
- Chen, I.-M.A.; Chu, K.; Palaniappan, K.; Ratner, A.; Huang, J.; Huntemann, M.; Hajek, P.; Ritter, S.J.; Webb, C.; Wu, D. The IMG/M data management and analysis system v. 7: Content updates and new features. Nucleic Acids Res. 2023, 51, D723–D732. [Google Scholar] [CrossRef]
- Kang, I.; Kim, S.; Islam, M.R.; Cho, J.-C. The first complete genome sequences of the acI lineage, the most abundant freshwater Actinobacteria, obtained by whole-genome-amplification of dilution-to-extinction cultures. Sci. Rep. 2017, 7, 42252. [Google Scholar] [CrossRef]
- Isokpehi, R.D.; Udensi, U.K.; Simmons, S.S.; Hollman, A.L.; Cain, A.E.; Olofinsae, S.A.; Hassan, O.A.; Kashim, Z.A.; Enejoh, O.A.; Fasesan, D.E. Evaluative profiling of arsenic sensing and regulatory systems in the human microbiome project genomes. Microbiol. Insights 2014, 7, 25–34. [Google Scholar] [CrossRef]
- Tu, Q.; Lin, L.; Cheng, L.; Deng, Y.; He, Z. NCycDB: A curated integrative database for fast and accurate metagenomic profiling of nitrogen cycling genes. Bioinformatics 2019, 35, 1040–1048. [Google Scholar] [CrossRef]
- Gadegaonkar, S.S.; Mander, Ü.; Espenberg, M. A state-of-the-art review and guidelines for enhancing nitrate removal in bio-electrochemical systems (BES). J. Water Process Eng. 2023, 53, 103788. [Google Scholar] [CrossRef]
- Zumft, W.G. Cell biology and molecular basis of denitrification. Microbiol. Mol. Biol. Rev. 1997, 61, 533–616. [Google Scholar] [PubMed]
- Knowles, R. Denitrification. Microbiol. Rev. 1982, 46, 43–70. [Google Scholar] [CrossRef]
- Gineyts, R.; Niboyet, A. Nitrification, denitrification, and related functional genes under elevated CO2: A meta-analysis in terrestrial ecosystems. Glob. Chang. Biol. 2023, 29, 1839–1853. [Google Scholar] [CrossRef]
- Sadaiappan, B.; PrasannaKumar, C.; Nambiar, V.U.; Subramanian, M.; Gauns, M.U. Meta-analysis cum machine learning approaches address the structure and biogeochemical potential of marine copepod associated bacteriobiomes. Sci. Rep. 2021, 11, 3312. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- Djemiel, C.; Maron, P.-A.; Terrat, S.; Dequiedt, S.; Cottin, A.; Ranjard, L. Inferring microbiota functions from taxonomic genes: A review. Gigascience 2022, 11, giab090. [Google Scholar] [CrossRef]
- Das, B.K.; Ishii, S.; Antony, L.; Smart, A.J.; Scaria, J.; Brözel, V.S. The microbial nitrogen cycling, bacterial community composition, and functional potential in a natural grassland are stable from breaking dormancy to being dormant again. Microorganisms 2022, 10, 923. [Google Scholar] [CrossRef]
- Read-Daily, B.; Maamar, S.B.; Sabba, F.; Green, S.; Nerenberg, R. Effect of nitrous oxide (N2O) on the structure and function of nitrogen-oxide reducing microbial communities. Chemosphere 2022, 307, 135819. [Google Scholar] [CrossRef]
- Zhang, S.; Tang, Z.; Xia, S.; Jiang, Y.; Li, M.; Wang, B. The intrinsic relevance of nitrogen removal pathway to varying nitrate loading rate in a polycaprolactone-supported denitrification system. Biodegradation 2022, 33, 317–331. [Google Scholar] [CrossRef]
- Sabdaningsih, A.; Adyasari, D.; Suryanti, S.; Febrianto, S.; Eshananda, Y. Environmental legacy of aquaculture and industrial activities in mangrove ecosystems. J. Sea Res. 2023, 196, 102454. [Google Scholar] [CrossRef]
- Bowman, J.S.; Ducklow, H.W. Microbial communities can be described by metabolic structure: A general framework and application to a seasonally variable, depth-stratified microbial community from the coastal West Antarctic Peninsula. PLoS ONE 2015, 10, e0135868. [Google Scholar] [CrossRef]
- Wemheuer, F.; Taylor, J.A.; Daniel, R.; Johnston, E.; Meinicke, P.; Thomas, T.; Wemheuer, B. Tax4Fun2: Prediction of habitat-specific functional profiles and functional redundancy based on 16S rRNA gene sequences. Environ. Microbiome 2020, 15, 11. [Google Scholar] [CrossRef]
- Ye, R.W.; Averill, B.A.; Tiedje, J.M. Denitrification: Production and consumption of nitric oxide. Appl. Environ. Microbiol. 1994, 60, 1053–1058. [Google Scholar] [CrossRef]
- Sanchez, A.; Bajic, D.; Diaz-Colunga, J.; Skwara, A.; Vila, J.C.; Kuehn, S. The community-function landscape of microbial consortia. Cell Syst. 2023, 14, 122–134. [Google Scholar] [CrossRef]
- Zhou, M.; Guan, X.; Deng, T.; Hu, R.; Qian, L.; Yang, X.; Wu, B.; Li, J.; He, Q.; Shu, L. Synthetic phylogenetically diverse communities promote denitrification and stability. Environ. Res. 2023, 231, 116184. [Google Scholar] [CrossRef]
- De Souza, R.S.C.; Armanhi, J.S.L.; Arruda, P. From microbiome to traits: Designing synthetic microbial communities for improved crop resiliency. Front. Plant Sci. 2020, 11, 553605. [Google Scholar] [CrossRef]
- Svenningsen, N.B.; Heisterkamp, I.M.; Sigby-Clausen, M.; Larsen, L.H.; Nielsen, L.P.; Stief, P.; Schramm, A. Shell biofilm nitrification and gut denitrification contribute to emission of nitrous oxide by the invasive freshwater mussel Dreissena polymorpha (zebra mussel). Appl. Environ. Microbiol. 2012, 78, 4505–4509. [Google Scholar] [CrossRef]
- Arfken, A.; Song, B.; Bowman, J.S.; Piehler, M. Denitrification potential of the eastern oyster microbiome using a 16S rRNA gene based metabolic inference approach. PLoS ONE 2017, 12, e0185071. [Google Scholar] [CrossRef]
- Ray, N.E.; Maguire, T.J.; Al-Haj, A.N.; Henning, M.C.; Fulweiler, R.W. Low greenhouse gas emissions from oyster aquaculture. Environ. Sci. Technol. 2019, 53, 9118–9127. [Google Scholar] [CrossRef]
- Chauhan, A.; Wafula, D.; Lewis, D.E.; Pathak, A. Metagenomic assessment of the eastern oyster-associated microbiota. Genome Announc. 2014, 2, e01083-14. [Google Scholar] [CrossRef]
- Horodesky, A.; Castilho-Westphal, G.G.; Pont, G.D.; Faoro, H.; Balsanelli, E.; Tadra-Sfeir, M.Z.; Cozer, N.; Pie, M.R.; Ostrensky, A. Metagenomic analysis of the bacterial microbiota associated with cultured oysters (Crassostrea sp.) in estuarine environments. An. Acad. Bras. Ciências 2020, 92, e20180432. [Google Scholar] [CrossRef]
- Pimentel, Z.T.; Dufault-Thompson, K.; Russo, K.T.; Scro, A.K.; Smolowitz, R.M.; Gomez-Chiarri, M.; Zhang, Y. Microbiome analysis reveals diversity and function of mollicutes associated with the Eastern oyster, Crassostrea virginica. Msphere 2021, 6, e00227-21. [Google Scholar] [CrossRef]
- Pathak, A.; Marquez, M.; Stothard, P.; Chukwujindu, C.; Su, J.-Q.; Zhou, Y.; Zhou, X.-Y.; Jagoe, C.H.; Chauhan, A. Microbiome analysis of the Eastern oyster as a function of ploidy and seasons. bioRxiv 2023. [Google Scholar] [CrossRef]
- Pathak, A.; Stothard, P.; Chauhan, A. Comparative genomic analysis of three Pseudomonas species isolated from the Eastern Oyster (Crassostrea virginica) tissues, mantle fluid, and the overlying estuarine water column. Microorganisms 2021, 9, 490. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef]
- Valk, L.C.; Peces, M.; Singleton, C.M.; Laursen, M.D.; Andersen, M.H.; Mielczarek, A.T.; Nielsen, P.H. Exploring the microbial influence on seasonal nitrous oxide concentration in a full-scale wastewater treatment plant using metagenome assembled genomes. Water Res. 2022, 219, 118563. [Google Scholar] [CrossRef]
- Isokpehi, R.D.; Simmons, S.S.; Johnson, M.O.; Payton, M. Genomic evidence for bacterial determinants influencing obesity development. Int. J. Environ. Res. Public Health 2017, 14, 345. [Google Scholar] [CrossRef]
- Beard, L.; Aghassibake, N. Tableau (version 2020.3). J. Med. Libr. Assoc. 2021, 109, 159. [Google Scholar] [CrossRef]
- Sedig, K.; Parsons, P. Interaction design for complex cognitive activities with visual representations: A pattern-based approach. AIS Trans. Hum.-Comput. Interact. 2013, 5, 84–133. [Google Scholar] [CrossRef]
- Ola, O.; Sedig, K. Discourse with visual health data: Design of human-data interaction. Multimodal Technol. Interact. 2018, 2, 10. [Google Scholar] [CrossRef]
- Unzueta-Martínez, A.; Welch, H.; Bowen, J.L. Determining the composition of resident and transient members of the oyster microbiome. Front. Microbiol. 2022, 12, 828692. [Google Scholar] [CrossRef]
- Singh, P.; Williams, D.; Velez, F.J.; Nagpal, R. Comparison of the gill microbiome of retail oysters from two geographical locations exhibited distinct microbial signatures: A pilot study for potential future applications for monitoring authenticity of their origins. Appl. Microbiol. 2022, 3, 1–10. [Google Scholar] [CrossRef]
- Chen, H.; Wang, M.; Yang, C.; Wan, X.; Ding, H.H.; Shi, Y.; Zhao, C. Bacterial spoilage profiles in the gills of Pacific oysters (Crassostrea gigas) and Eastern oysters (C. virginica) during refrigerated storage. Food Microbiol. 2019, 82, 209–217. [Google Scholar] [CrossRef]
- Kojima, H.; Fukui, M. Sulfuricella denitrificans gen. nov., sp. nov., a sulfur-oxidizing autotroph isolated from a freshwater lake. Int. J. Syst. Evol. Microbiol. 2010, 60, 2862–2866. [Google Scholar] [CrossRef]
- Zhang, M.-X.; Li, A.-Z.; Wu, Q.; Yao, Q.; Zhu, H.-H. Marinobacter denitrificans sp. nov., isolated from marine sediment of southern Scott Coast, Antarctica. Int. J. Syst. Evol. Microbiol. 2020, 70, 2918–2924. [Google Scholar] [CrossRef]
- Duan, L.; Li, J.-l.; Li, X.; Dong, L.; Fang, B.-Z.; Xiao, M.; Mou, X.; Li, W.-J. Roseibium aestuarii sp. nov., isolated from Pearl River Estuary. Int. J. Syst. Evol. Microbiol. 2020, 70, 2896–2900. [Google Scholar] [CrossRef]
- Mizutani, Y.; Tanaka, R. Genome sequence of Arcobacter sp. strain LA11, isolated from the abalone Haliotis discus. Genome Announc. 2017, 5, e00032-17. [Google Scholar] [CrossRef]
- Huang, S.; Fu, Y.; Zhang, H.; Wang, C.; Zou, C.; Lu, X. Research progress of novel bio-denitrification technology in deep wastewater treatment. Front. Microbiol. 2023, 14, 1284369. [Google Scholar] [CrossRef]
- Dos Santos, P.C.; Fang, Z.; Mason, S.W.; Setubal, J.C.; Dixon, R. Distribution of nitrogen fixation and nitrogenase-like sequences amongst microbial genomes. BMC Genom. 2012, 13, 162. [Google Scholar] [CrossRef]
- Pi, H.-W.; Lin, J.-J.; Chen, C.-A.; Wang, P.-H.; Chiang, Y.-R.; Huang, C.-C.; Young, C.-C.; Li, W.-H. Origin and evolution of nitrogen fixation in prokaryotes. Mol. Biol. Evol. 2022, 39, msac181. [Google Scholar] [CrossRef]
- Caffrey, J.M.; Hollibaugh, J.T.; Mortazavi, B. Living oysters and their shells as sites of nitrification and denitrification. Mar. Pollut. Bull. 2016, 112, 86–90. [Google Scholar] [CrossRef]
- Ayvazian, S.; Mulvaney, K.; Zarnoch, C.; Palta, M.; Reichert-Nguyen, J.; McNally, S.; Pilaro, M.; Jones, A.; Terry, C.; Fulweiler, R.W. Beyond bioextraction: The role of oyster-mediated denitrification in nutrient management. Environ. Sci. Technol. 2021, 55, 14457–14465. [Google Scholar] [CrossRef]
- Fukushi, M.; Mino, S.; Tanaka, H.; Nakagawa, S.; Takai, K.; Sawabe, T. Biogeochemical implications of N2O-Reducing thermophilic Campylobacteria in Deep-Sea vent fields, and the description of Nitratiruptor labii sp. nov. IScience 2020, 23, 101462. [Google Scholar] [CrossRef]
- Jiao, J.Y.; Lian, Z.H.; Li, M.M.; Salam, N.; Zhou, E.M.; Liu, L.; Ming, H.; Nie, G.; Shu, W.; Zhao, G. Comparative genomic analysis of Thermus provides insights into the evolutionary history of an incomplete denitrification pathway. mLife 2022, 1, 198–209. [Google Scholar] [CrossRef]
- Anderson, I.; Risso, C.; Holmes, D.; Lucas, S.; Copeland, A.; Lapidus, A.; Cheng, J.-F.; Bruce, D.; Goodwin, L.; Pitluck, S. Complete genome sequence of Ferroglobus placidus AEDII12DO. Stand. Genom. Sci. 2011, 5, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Cabello, P.; Roldan, M.D.; Moreno-Vivian, C. Nitrate reduction and the nitrogen cycle in archaea. Microbiology 2004, 150, 3527–3546. [Google Scholar] [CrossRef] [PubMed]
- Petri, R.; Imhoff, J.F. The relationship of nitrate reducing bacteria on the basis of narH gene sequences and comparison of narH and 16S rDNA based phylogeny. Syst. Appl. Microbiol. 2000, 23, 47–57. [Google Scholar] [CrossRef]
- Torregrosa-Crespo, J.; González-Torres, P.; Bautista, V.; Esclapez, J.M.; Pire, C.; Camacho, M.; Bonete, M.J.; Richardson, D.J.; Watmough, N.J.; Martínez-Espinosa, R.M. Analysis of multiple haloarchaeal genomes suggests that the quinone-dependent respiratory nitric oxide reductase is an important source of nitrous oxide in hypersaline environments. Environ. Microbiol. Rep. 2017, 9, 788–796. [Google Scholar] [CrossRef]
- Zhou, Z.; Liu, Y.; Xu, W.; Pan, J.; Luo, Z.-H.; Li, M. Genome-and community-level interaction insights into carbon utilization and element cycling functions of Hydrothermarchaeota in hydrothermal sediment. MSystems 2020, 5, e00795-19. [Google Scholar] [CrossRef]
- Torregrosa-Crespo, J.; Pire, C.; Martínez-Espinosa, R.M.; Bergaust, L. Denitrifying haloarchaea within the genus Haloferax display divergent respiratory phenotypes, with implications for their release of nitrogenous gases. Environ. Microbiol. 2019, 21, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Torregrosa-Crespo, J.; Pire, C.; Bergaust, L.; Martínez-Espinosa, R.M. Haloferax mediterranei, an archaeal model for denitrification in saline systems, characterized through integrated physiological and transcriptional analyses. Front. Microbiol. 2020, 11, 533614. [Google Scholar] [CrossRef]
- Cozen, A.E.; Weirauch, M.T.; Pollard, K.S.; Bernick, D.L.; Stuart, J.M.; Lowe, T.M. Transcriptional map of respiratory versatility in the hyperthermophilic crenarchaeon Pyrobaculum aerophilum. J. Bacteriol. 2009, 191, 782–794. [Google Scholar] [CrossRef]
- Vorholt, J.A.; Hafenbradl, D.; Stetter, K.O.; Thauer, R.K. Pathways of autotrophic CO2 fixation and of dissimilatory nitrate reduction to N2O in Ferroglobus placidus. Arch. Microbiol. 1997, 167, 19–23. [Google Scholar] [CrossRef]
- Baker, B.; Langwig, M.; De Anda, V.; Sneed, S.; Seitz, K.; Rasmussen, A.; Lee, J.; Anantharaman, K.; Francis, C. Metabolic capacity is maintained despite shifts in microbial diversity in estuary sediments. Res. Sq. 2023. [Google Scholar] [CrossRef]
- Robertson, G.P. Denitrification and the challenge of scaling microsite knowledge to the globe. mLife 2023, 2, 229–238. [Google Scholar] [CrossRef]
- Conthe, M.; Lycus, P.; Arntzen, M.Ø.; da Silva, A.R.; Frostegård, Å.; Bakken, L.R.; Kleerebezem, R.; van Loosdrecht, M.C. Denitrification as an N2O sink. Water Res. 2019, 151, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Ravishankara, A.; Daniel, J.S.; Portmann, R.W. Nitrous oxide (N2O): The dominant ozone-depleting substance emitted in the 21st century. Science 2009, 326, 123–125. [Google Scholar] [CrossRef] [PubMed]
- Goyal, A.; Qanungo, K. Nitrous oxide as a greenhouse gas: A state of art. AIP Conf. Proc. 2023, 2535, 040009. [Google Scholar]
- Grossart, H.-P.; Dziallas, C.; Leunert, F.; Tang, K.W. Bacteria dispersal by hitchhiking on zooplankton. Proc. Natl. Acad. Sci. USA 2010, 107, 11959–11964. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Jiang, S. A review on nirS-type and nirK-type denitrifiers via a scientometric approach coupled with case studies. Environ. Sci. Process. Impacts 2022, 24, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Abada, A.; Beiralas, R.; Narvaez, D.; Sperfeld, M.; Duchin-Rapp, Y.; Lipsman, V.; Yuda, L.; Cohen, B.; Carmieli, R.; Ben-Dor, S. Aerobic bacteria produce nitric oxide via denitrification and promote algal population collapse. ISME J. 2023, 17, 1167–1183. [Google Scholar] [CrossRef]
- Chen, J.; Liu, L.; Wang, W.; Gao, H. Nitric oxide, nitric oxide formers and their physiological impacts in bacteria. Int. J. Mol. Sci. 2022, 23, 10778. [Google Scholar] [CrossRef]
- Kobayashi, S.; Hira, D.; Yoshida, K.; Toyofuku, M.; Shida, Y.; Ogasawara, W.; Yamaguchi, T.; Araki, N.; Oshiki, M. Nitric oxide production from nitrite reduction and hydroxylamine oxidation by copper-containing dissimilatory nitrite reductase (NirK) from the aerobic ammonia-oxidizing archaeon, Nitrososphaera viennensis. Microbes Environ. 2018, 33, 428–434. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, H.; Takaki, Y.; Abe, M.; Imachi, H.; Ikuta, T.; Miyazaki, J.; Tasumi, E.; Uematsu, K.; Tame, A.; Tsuda, M. Multispecies populations of methanotrophic Methyloprofundus and cultivation of a likely dominant species from the Iheya North deep-sea hydrothermal field. Appl. Environ. Microbiol. 2022, 88, e0075821. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, C.; Minamisawa, K. Redundant roles of Bradyrhizobium oligotrophicum Cu-type (NirK) and cd 1-type (NirS) nitrite reductase genes under denitrifying conditions. FEMS Microbiol. Lett. 2018, 365, fny015. [Google Scholar] [CrossRef] [PubMed]
- Torregrosa-Crespo, J.; Miralles-Robledillo, J.M.; Bernabeu, E.; Pire, C.; Martínez-Espinosa, R.M. Denitrification in hypersaline and coastal environments. FEMS Microbiol. Lett. 2023, 370, fnad066. [Google Scholar] [CrossRef] [PubMed]
- Pold, G.; Bonilla-Rosso, G.; Saghaï, A.; Strous, M.; Jones, C.M.; Hallin, S. Phylogenetics and environmental distribution of nitric oxide forming nitrite reductases reveals their distinct functional and ecological roles. ISME Commun. 2024, 4, ycae020. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Mazzei, M.; Di Gregorio, S.; Niccolini, L.; Vitiello, V.; Ye, Y.; Guo, B.; Yan, X.; Buttino, I. Marine Copepods as a Microbiome Hotspot: Revealing Their Interactions and Biotechnological Applications. Water 2023, 15, 4203. [Google Scholar] [CrossRef]
- Zoccarato, L.; Grossart, H.P. Relationship between lifestyle and structure of bacterial communities and their functionality in aquatic systems. In The Structure and Function of Aquatic Microbial Communities; Springer: Berlin/Heidelberg, Germany, 2019; pp. 13–52. [Google Scholar]
- Ma, L.-c.; Zhao, H.-q.; Wu, L.B.; Liu, C. Impacts of the microbiome on human, animal, and environmental health from a One Health perspective. Sci. One Health 2023, 2, 100037. [Google Scholar] [CrossRef]
- Hernández-del Amo, E.; Menció, A.; Gich, F.; Mas-Pla, J.; Bañeras, L. Isotope and microbiome data provide complementary information to identify natural nitrate attenuation processes in groundwater. Sci. Total Environ. 2018, 613, 579–591. [Google Scholar] [CrossRef]
- Zhang, L.; Ma, X.; Li, Q.; Cui, H.; Shi, K.; Wang, H.; Zhang, Y.; Gao, S.; Li, Z.; Wang, A.-J. Complementary biotransformation of antimicrobial triclocarban obviously mitigates nitrous oxide emission toward sustainable microbial denitrification. Environ. Sci. Technol. 2023, 57, 7490–7502. [Google Scholar] [CrossRef]
- On, S.L.; Miller, W.G.; Biggs, P.J.; Cornelius, A.J.; Vandamme, P. A critical rebuttal of the proposed division of the genus Arcobacter into six genera using comparative genomic, phylogenetic, and phenotypic criteria. Syst. Appl. Microbiol. 2020, 43, 126108. [Google Scholar] [CrossRef]
- Pérez-Cataluña, A.; Salas-Massó, N.; Diéguez, A.L.; Balboa, S.; Lema, A.; Romalde, J.L.; Figueras, M.J. Revisiting the taxonomy of the genus Arcobacter: Getting order from the chaos. Front. Microbiol. 2018, 9, 2077. [Google Scholar] [CrossRef] [PubMed]
- Lobiuc, A.; Pavăl, N.-E.; Dimian, M.; Covașă, M. Nanopore sequencing assessment of bacterial pathogens and associated antibiotic resistance genes in environmental samples. Microorganisms 2023, 11, 2834. [Google Scholar] [CrossRef] [PubMed]
- Venâncio, I.; Luís, Â.; Domingues, F.; Oleastro, M.; Pereira, L.; Ferreira, S. The prevalence of Arcobacteraceae in aquatic environments: A systematic review and meta-analysis. Pathogens 2022, 11, 244. [Google Scholar] [CrossRef] [PubMed]
- Buzzanca, D.; Kerkhof, P.-J.; Alessandria, V.; Rantsiou, K.; Houf, K. Arcobacteraceae comparative genome analysis demonstrates genome heterogeneity and reduction in species isolated from animals and associated with human illness. Heliyon 2023, 9, e17652. [Google Scholar] [CrossRef]
- Gaimster, H.; Hews, C.L.; Griffiths, R.; Soriano-Laguna, M.J.; Alston, M.; Richardson, D.J.; Gates, A.J.; Rowley, G. A central small RNA regulatory circuit controlling bacterial denitrification and N2O emissions. MBio 2019, 10, e01165-19. [Google Scholar] [CrossRef]


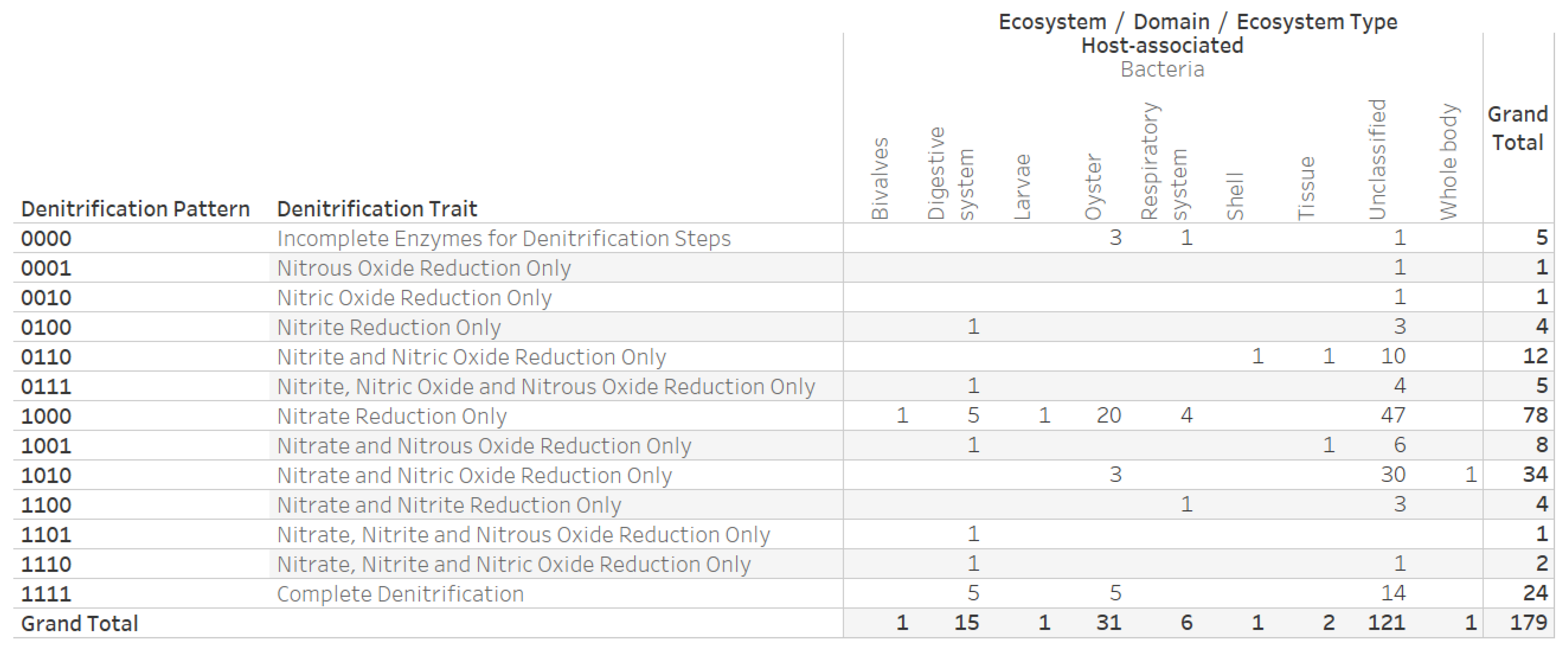





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset Column Category 1 | Dataset Columns |
|---|---|
| Genome | Domain, Gene Count, Genome ID, Genome Name, Genome Size, Genus, GOLD Sequencing Project ID, Sequencing Center, Sequencing Status, Status |
| Ecosystem | Ecosystem, Ecosystem Category, Ecosystem Type, Ecosystem Subtype, Specific Ecosystem |
| Lineage | GTDB-Tk Domain, GTDB-Tk Family, GTDB-Tk Genus, GTDB-Tk Order, GTDB-Tk Phylum, GTDB-Tk Species |
| Denitrifying Enzymes | E01_narG, E02_narH, E03_narI, E04_napA, E05_napB, E06_nirK, E07_nirS, E08_norB, E09_norC, E10_norV, E11_norW, E12_nosZ |
| Denitrification Potential | Denitrification Pattern, Denitrification Traits, Denitrifying enzymes pattern |
| KEGG Orthology (KO) Identifier 1 | KEGG Gene Name | Gene Symbol 2 | Genome Count 3 | ||
|---|---|---|---|---|---|
| Archaea | Bacteria | Total | |||
| K00370 | nitrate reductase/nitrite oxidoreductase, alpha subunit | narG | 268 | 39,961 | 40,229 |
| K00371 | nitrate reductase/nitrite oxidoreductase, beta subunit | narH | 280 | 39,985 | 40,265 |
| K00374 | nitrate reductase gamma subunit | narI | 105 | 41,172 | 41,277 |
| K02567 | nitrate reductase (cytochrome) | napA | 4 | 21,911 | 21,915 |
| K02568 | nitrate reductase (cytochrome), electron transfer subunit | napB | 1 | 21,754 | 21,755 |
| K00368 | nitrite reductase (NO-forming) | nirK | 479 | 10,873 | 11,352 |
| K15864 | nitrite reductase (NO-forming)/hydroxylamine reductase | nirS | 28 | 3205 | 3233 |
| K04561 | nitric oxide reductase subunit B | norB | 364 | 15,680 | 16,044 |
| K02305 | nitric oxide reductase subunit C | norC | 2 | 6163 | 6165 |
| K12264 | anaerobic nitric oxide reductase flavorubredoxin | norV | 138 | 17,164 | 17,302 |
| K12265 | nitric oxide reductase FlRd-NAD(+) reductase | norW | 14,224 | 14,224 | |
| K00376 | nitrous-oxide reductase | nosZ | 181 | 8009 | 8190 |
| Denitrification Trait | Genome Name (Mollusca Host) 1 |
|---|---|
| Complete Denitrification | Arcobacter sp. LA11 (abalone) Halarcobacter mediterraneus F156-34 (mussel) Malaciobacter mytili CECT 7386 (mussel) Malaciobacter mytili F2075 (mussel) Poseidonibacter parvus LPB0137 (squid) Poseidonibacter sp. SJOD-M-5 (oyster) Poseidonibacter sp. SJOD-M-33 (oyster) |
| Nitrate Reduction Only | Arcobacter ellisii LMG 26155 (mussel) Arcobacter venerupis CECT7836 (clam) Malaciobacter canalis F138-33 (oyster) Malaciobacter canalis LMG 29148 (oyster) Malaciobacter molluscorum CECT 7696 (mussel) Malaciobacter molluscorum F98-3 (mussel) |
| Nitrite and Nitric Oxide Reduction Only | Poseidonibacter ostreae JOD-M-6 (oyster) |
| Nitrogen Assimilation Pathway | KEGG Entry and Name 1 | Gene Symbol | Genome Count | Example Genome |
|---|---|---|---|---|
| Nitrogen Fixation | K02588 nitrogenase iron protein | nifH | 369 | Arcobacter acticola KCTC 52212 |
| Assimilatory Nitrate Reduction | K00372 assimilatory nitrate reductase catalytic subunit [EC:1.7.99.-] | nasA | 2294 | Shewanella denitrificans OS217 |
| Assimilatory Nitrate Reduction | K00360 assimilatory nitrate reductase electron transfer subunit [EC:1.7.99.-] | nasB | 2 | Halomonas icarae D1-1 |
| Assimilatory Nitrate Reduction | K00367 ferredoxin-nitrate reductase [EC:1.7.7.2] | narB | 47 | Sulfuricella denitrificans skB26 |
| Assimilatory Nitrite Reduction | K00366 ferredoxin-nitrite reductase [EC:1.7.7.1] | nirA | 170 | Arcobacter peruensis PSE-93 |
| Ammonia Assimilation to Glutamine | K01915 glutamine synthetase [EC:6.3.1.2] | glnA | 3164 | Aliiroseovarius crassostreae DSM 16950 |
| Denitrification Pattern 1 | Denitrification Trait (Count of Denitrifying Enzymes Pattern) 2 | Genome Count | Example Genome | Denitrifying Enzymes Pattern of the Example Genome 3 | Reference for the Denitrification Pattern |
|---|---|---|---|---|---|
| 0000 | Incomplete Enzymes for Denitrification Steps (15) | 278 | Aeropyrum pernix K1 | 110000000000 | [65,66] |
| 0001 | Nitrous Oxide Reduction Only (4) | 58 | Haloarcula japonica DSM 6131 | 110000010001 | [67] |
| 0010 | Nitric Oxide Reduction Only (1) | 1 | Candidatus Hydrothermarchaeota archaeon JdFR-18 | 110100011100 | [68] |
| 0100 | Nitrite Reduction Only (13) | 366 | Haloferax volcanii DS2 | 110001010000 | [69] |
| 0101 | Nitrite and Nitrous Oxide Reduction Only (10) | 119 | Haloferax mediterranei R-4 | 110001010001 | [70] |
| 1000 | Nitrate Reduction Only (5) | 39 | Pyrobaculum aerophilum IM2 | 111000010000 | [71] |
| 1001 | Nitrate and Nitrous Oxide Reduction Only (2) | 3 | Pyrobaculum calidifontis JCM 11548 | 111000010001 | [71] |
| 1011 | Nitrate, Nitric Oxide and Nitrous Oxide Reduction Only (1) | 1 | Ferroglobus placidus AEDII12DO, DSM 10642 | 111000011101 | [64,72] |
| 1100 | Nitrate and Nitrite Reduction Only (1) | 1 | Candidatus Heimdallarchaeota archaeon LC_3 | 111001000000 | [73] |
| Genome Name | Genome ID 1 | Denitrifying Enzymes Pattern 2 | Ecosystem Category |
|---|---|---|---|
| Halobiforma haloterrestris DSM 13078 | 2693429869 | 110001110001 | Terrestrial |
| Halobiforma lacisalsi AJ5, JCM 12983 | 2529293100 | 110001110001 | Aquatic |
| Halobiforma lacisalsi AJ5, JCM 12983 | 2806310686 | 110001110001 | Aquatic |
| Halobiforma nitratireducens JCM 10879 | 2554235466 | 110001100001 | Aquatic |
| Halorubrum amylolyticum ZC67 | 2881047951 | 110001110001 | Terrestrial |
| Halorubrum salipaludis WN019 | 2995789858 | 000001110001 | Terrestrial |
| Halosolutus halophilus LT55 | 8055007790 | 000001110001 | Terrestrial |
| Haloterrigena longa ABH32 | 8065811630 | 000001110001 | Aquatic |
| Haloterrigena sp. LL2A | 2639762614 | 000001110001 | Aquatic |
| Natrinema altunense AJ2 | 2585427993 | 110001110001 | Aquatic |
| Natrinema altunense JCM 12890 | 2554235488 | 110001110001 | Aquatic |
| Natrinema amylolyticum LT61 | 8056733939 | 110001110001 | Terrestrial |
| Natrinema pallidum BOL6-1 | 8058325716 | 110001110000 | Terrestrial |
| Natrinema pellirubrum 157 | 2509601048 | 110001110000 | Fish |
| Natrinema pellirubrum 157 | 2537562080 | 110001110000 | Fish |
| Natrinema sp. J7-2 | 2517093029 | 000001110000 | Terrestrial |
| Natrinema thermotolerans A29 | 2582580504 | 110001110000 | Food production |
| Natrinema thermotolerans A29 | 2914868299 | 110001110000 | Food production |
| Natrinema thermotolerans DSM 11552 | 2534681901 | 110001110000 | Aquatic |
| Natronomonas sp. LN261 | 3001203943 | 000001100001 | Terrestrial |
| Salinilacihabitans rarus AD-4 | 8054413294 | 110001110001 | Aquatic |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Isokpehi, R.D.; Kim, Y.; Krejci, S.E.; Trivedi, V.D. Ecological Trait-Based Digital Categorization of Microbial Genomes for Denitrification Potential. Microorganisms 2024, 12, 791. https://doi.org/10.3390/microorganisms12040791
Isokpehi RD, Kim Y, Krejci SE, Trivedi VD. Ecological Trait-Based Digital Categorization of Microbial Genomes for Denitrification Potential. Microorganisms. 2024; 12(4):791. https://doi.org/10.3390/microorganisms12040791
Chicago/Turabian StyleIsokpehi, Raphael D., Yungkul Kim, Sarah E. Krejci, and Vishwa D. Trivedi. 2024. "Ecological Trait-Based Digital Categorization of Microbial Genomes for Denitrification Potential" Microorganisms 12, no. 4: 791. https://doi.org/10.3390/microorganisms12040791