Abstract
In this work, we studied the selective pressure and evolutionary analysis on the SARS-CoV-2 BF.7 and BQ.1.1 lineages circulating in Italy from July to December 2022. Two different datasets were constructed: the first comprised 694 SARS-CoV-2 BF.7 lineage sequences and the second comprised 734 BQ.1.1 sequences, available in the Italian COVID-19 Genomic (I-Co-Gen) platform and GISAID (last access date 15 December 2022). Alignments were performed with MAFFT v.7 under the Galaxy platform. The HYPHY software was used to study the selective pressure. Four positively selected sites (two in nsp3 and two in the spike) were identified in the BF.7 dataset, and two (one in ORF8 and one in the spike gene) were identified in the BQ.1.1 dataset. Mutation analysis revealed that R408S and N440K are very common in the spike of the BF.7 genomes, as well as L452R among BQ.1.1. N1329D and Q180H in nsp3 were found, respectively, at low and rare frequencies in BF.7, while I121L and I121T were found to be rare in ORF8 for BQ.1.1. The positively selected sites may have been driven by the selection for increased viral fitness, under circumstances of defined selective pressure, as well by host genetic factors.
1. Introduction
The SARS-CoV-2 virus evolved rapidly, with the emergence of new variants over time. The Omicron variant of SARS-CoV-2, first discovered in Botswana and South Africa on 11 November 2021, was subsequently identified worldwide. The World Health Organization designated Omicron as a variant of concern on 26 November 2021, as it became the leading variant [1].
The Omicron variant became dominant in Italy starting from January 2022 [2,3].
Omicron developed several sub-variants. In Italy, BA.5 showed a growing trend starting from July 2022, with a parallel decrease in the BA.2 sub-variant, reaching 81.7% of the sequences deposited on I-Co-Gen in the week 11–17 July 2022 [4] and a national prevalence of 90.8% at the end of August 2022 [5].
The BF.7 and BQ.1.1 sub-lineages of the Omicron variant BA.5 showed a growing trend in Italy starting from the end of November 2022 [6] in line with the international context [7,8].
The presence of the mutations K444T, N460K, and R346T in the spike of the sub-lineage BQ.1.1 and the R346T in the spike of BF.7 sub-lineage represents one of the main reasons to monitor these strains. In this regard, the receptor-binding domain (RBD) included position 346, whereas residue 658 is in close proximity to the S1/S2 cleavage [9].
Monitoring SARS-CoV-2 amino acid substitutions can help to explain viral behaviour and to detect potential alterations in transmissibility, infection severity, and immunity.
The amino-acid changes that result in reduced fitness are generally removed by negative selection, whereas changes that increase virus fitness are generally maintained by positive selection. The changes are considered neutral if they do not decrease or increase fitness.
In this work, we studied the selective pressure and evolutionary dynamics on SARS-CoV-2 BF.7 and BQ.1.1 lineages circulating in Italy from July to December 2022. The selective pressure allows us to estimate the nonsynonymous/synonymous rate (dN/dS, ω), considering a nonsynonymous rate higher than the synonymous as positive selection [10,11]. Since the selective pressure of SARS-CoV-2 BF.7 and BQ.1.1 lineages in Italy has not been investigated, this study might be useful to identify (i) the positive and negative selected sites and (ii) the recurrent and less frequent mutations.
2. Materials and Methods
2.1. Dataset and Sequence Alignment
All the high-quality (sequences containing <5% of ambiguous nucleotides N) SARS-CoV-2 genomes belonging to the BF.7 and BQ.1.1 lineages collected in Italy from July to December 2022, available in the national platform Italian COVID-19 Genomic I-Co-Gen (last access 15 December 2022) and GISAID [12,13], were downloaded.
The sequences (FASTA format) were downloaded from the I-Co-Gen platform and obtained through the open-source SARS-CoV-2 RECoVERY software v4.0 (developed by the ISS and available in the I-Co-Gen project), which automatically performs data quality control, the construction of genomes from NGS sequencing data (consensus) and other functions.
Therefore, two different datasets were investigated here. The first one is composed of 694 SARS-CoV-2 lineage BF.7 genomes; the second one included 734 lineage BQ.1.1 sequences. The GISAID Accession Numbers and the details of the sequences included in the first and second dataset were reported in Table S1.
In order to investigate the location of the Italian BF.7 and BQ.1.1 genomes in the phylogenetic trees with respect to those collected from other European countries, two additional datasets were created: the first one composed of a total of 2305 genomes (694 BF.7 Italian plus 1611 European) and the second one by 2459 genomes (734 BQ.1.1 Italian plus 1725 BQ.1.1 European). The lineage of the Italian sequences was assigned directly through the I-Co-Gen platform and the Pangolin 4.2 v1.17 software, and also confirmed uploading the sequences into the “Pangolin COVID-19 Lineage Assigner” online [14,15], last access 15 December 2022]. All the sequence alignments were obtained with the program MAFFT v.7 [16] under the Galaxy platform [17,18,19] and followed by manual editing through the Bioedit program [20].
2.2. Phylogenetic Analysis
The maximum likelihood phylogenetic trees on the two additional datasets were generated through IQ-TREE software using 1000 as the number of bootstrap replicates for branch support, SH-aLRT.
2.3. Selective Pressure Analysis
In this study, the following protein-coding gene sequence sub-sets (for both the first and second dataset) were created with the aim of investigating the SARS-CoV-2 amino acid substitutions, the evolutionary dynamics, and the positively and negatively selected sites: nsp1, nsp2, nsp3, nsp4, 3C-like proteinase (nsp5), nsp6, nsp7, nsp8, nsp9, nsp10, nsp11, nsp12, helicase (nsp13), 3′-to-5′-exonuclease (nsp14), endoRNAse (nsp15), 2′-O-ribosemethyltransferase (nsp16), S (surface glycoprotein), ORF3a, E, M, N, ORF6, ORF7a, ORF7b, ORF8, and ORF10.
A positive diversifying selection occurred when ω > 1 (ω, rate of nonsynonymous substitutions to that of synonymous), while purifying selection was inferred for ω < 1 [21].
The models Fast Unconstrained Bayesian AppRoximation (FUBAR) [22], Fixed Effects Likelihood (FEL), and Single-Likelihood Ancestor Counting (SLAC) [23] were used in HYPHY software v 2.2.4 [23] to identify selection.
The method FUBAR infers the nonsynonymous (dN) and synonymous (dS) substitution rates on a per-site basis in large datasets, based on the assumption that a pervasive selection pressure is constant in the entire phylogeny [22], and with this method, improved robustness is obtained for large datasets [22]. The FEL model uses an ML approach to infer dN and dS substitution rates on a per-site basis [23], assuming that the selection for each site is constant along the phylogeny. The SLAC model uses a combination of maximum-likelihood and counting approaches to infer dN and dS substitution rates on a per-site basis. Additionally, in this case, the selection for each site is constant along with the phylogeny [23]. The SLAC uses ML to infer the most likely ancestral sequence at each node, with a Suzuki–Gojobori counting method to directly count the total number of nonsynonymous and synonymous changes at each site [23].
Only the sites found by at least two models under significant selection (FUBAR, posterior probability ≥ 0.9; FEL, p ≤ 0.05 and SLAC p ≤ 0.1) were reported. The amino acid positions of the sites were referred with respect to the protein products obtained from the SARS-CoV-2 reference sequence Wuhan-Hu-1 (Accession Number: NC_045512.2). The frequency of each amino acid substitutions in the positive selected sites was calculated in order to classify the mutations as very common (frequency ≥ 83.7%–84%), common (frequency ≥ 64.0%), low-frequency (between 2.1% and 21.0%) or rare (frequency ≤ 2.0%).
The prediction of the possible impact on protein stability of the amino acid substitutions found in the positively selected sites was investigated through I-Mutant 2.0 and PolyPhen-2 (Polymorphism Phenotyping v2) tools, as previously reported [24].
3. Results
3.1. Phylogenetic Analysis
The BF.7 and BQ.1.1 Italian genomes appeared in relation to foreign European sequences, but, beyond a general intermixing in the tree, several internal supported subclades/subclusters composed only by Italian genomes were identified, indicating a certain genomic divergence.
3.2. Selective Pressure Analysis
Overall, the selective pressure analysis showed a variation in the SARS-CoV-2 protein-coding genes (Table S2).
The first dataset (lineage BF.7) revealed a total of four significant positively selected sites, two of which were localized in the nsp3 and two in the spike. The two sites (408, 440) identified in the spike of the lineage BF.7 dataset were located inside the RBD portion. Evidence of supported negative selection in the first dataset was detected for 29 sites, 5 of which (17.2%) were localized in nsp3, 5 (17.2%) in nsp 13, 4 (14.0%) in the spike, 3 (10.3%) in nsp14, 2 (6.9%) in nsp5, 2 (7.0%) in nsp16, 2 (7.0%) in N, and 1 (3.4%) in nsp2, nsp4, nsp6, nsp10, nsp12, and ORF7a. No positively or negatively selected sites were identified in nsp1, nsp7, nsp8, nsp9, nsp11, nsp15, ORF3a, E, M, ORF6, ORF7b, ORF8, ORF10 (Table S2).
The second dataset (SARS-CoV-2 lineage BQ.1.1) was characterized by two statistically significant positively selected sites, one in ORF8 and the other in the spike (Table S2). The positively selected site identified in the spike (lineage BQ.1.1 dataset) was located inside the RBD.
Negative selection for the second dataset was detected in 15 sites, 3 (20%) of which were localized in nsp3; 3 (20%) in nsp15; 2 (13.3%) in nsp4, nsp16, and spike; and 1 (6.7%) in nsp1, nsp9, and ORF3a. No positively or negatively selected sites were identified in nsp2, nsp5, nsp6, nsp7, nsp8, nsp10, nsp11, nsp12, nsp13, nsp14, E, M, N, ORF6, ORF7a, or ORF7b, ORF10 (Table S2).
3.3. Frequency of the Amino Acid Substitutions Harbored by the Italian SARS-CoV-2 Genomes and the Prediction of the Possible Impact of the Amino Acid Substitutions
The positively selected sites were investigated to track the frequency of each amino acid replacement in our datasets (Table 1a,b).

Table 1.
Panel (a): Frequency of amino acid substitutions harbored by the Italian BF.7 lineage genomes (first dataset, n = 694) and detected as positively selected sites. Panel (b): Frequency of amino acid substitutions harbored by the Italian BQ.1.1 lineage genomes (second dataset, n = 734) and detected as positively selected sites.
The amino acid substitution N1329D in nsp3 was identified rarely; the amino acid substitution Q180H in nsp3 was identified at low frequency and the R408S, N440K) in the spike were detected as very common in the Italian SARS-CoV-2 BF.7 genomes (Table 1 panel a).
Meanwhile, Table 1 panel b indicates one substitution as very common (L452R in the spike) and two as rare (I121L, I121T in ORF8) in the Italian BQ.1.1 lineage genomes (second dataset).
In particular, Q180H was located at the N-terminal of NSP3, in the nsp3a portion. Meanwhile, the N1329D was located in the nsp3e portion. All the amino acid substitutions identified as positively selected sites in the spike were located inside the RBD (Figure 1). Finally, the I121L and I121T were found in ORF8, which is one of the so-called accessory proteins, composed of 121-amino acids (Figure 1).
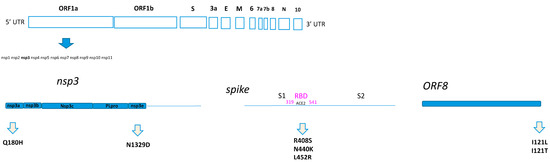
Figure 1.
Home-made schematic diagram relating to the location of the amino acid substitutions on the SARS-CoV-2 proteins.
Table 2 shows that 75% of the amino acid replacements identified as positively selected sites in the first dataset were predicted to decrease, while 25% were predicted to increase the stability of the proteins. All the amino acid replacements identified as positively selected sites for the second dataset (100%) were predicted to decrease the stability of the reported proteins (Table 2). Only two amino acid replacements (one in the first and the other in the second dataset) were predicted by PolyPhen-2 as probably or possibly damaging the protein structure (Table 2, score > 0.92). The remaining mutations were predicted with a benign effect on the protein structure (Table 2).
Table 2.
Results of the prediction obtained through I-Mutant 2.0 and PolyPhen-2.
4. Discussion
The SARS-CoV-2 virus evolved rapidly with the emergence of new variants over time.
Tracking the evolution of SARS-CoV-2 offers the opportunity to understand the viral genetic diversity, potentially predicting possible future evolutionary trajectories of the virus and offering routes for prevention and treatment.
Omicron’s BF.7 and BQ.1 variants have circulated widely in different parts of the world [25,26] and also in Italy [6]. Both revealed increased resistance to neutralization antibodies [25] and increased ACE2 binding affinity, infectivity, and fusogenicity [26].
This study provides a genomic and evolutionary analysis of SARS-CoV-2 BF.7 and BQ.1.1 lineages circulating in Italy from July to December 2022 with the aim to understand their fitness landscape, the selective pressure, and the amino acid changes with a selective advantage. Although these sublineages are actually bypassed by others, which emerged later, the selective pressure, the mutational profile, and the genetic diversity of the SARS-CoV-2 BF.7 and BQ.1.1 Italian genomes can contribute to expanding the data available from a large dataset, particularly on the amino acid substitutions identified. The positively selected sites identified here suggest that they may have been driven by selection for increased viral fitness under defined selective pressure circumstances, as well as by possible host genetic factors. Notably, the mutations R408S and N440K were identified in a higher frequency in the spike of the Italian BF.7 dataset. The accumulation of mutations can occur faster if some of them are considered advantageous. Some studies showed that the mutation R408S can lead to the escape of the antibodies and can induce smaller binding free energy changes in RBD-human ACE2 complexes [27], reducing the efficacy of many antibodies [28]. The N440K mutation has also been reported to escape antibody neutralization, which could increase infection risk [29] while also showing increased infectious fitness [30].
As regards the data on nsp3, the Q180H and N1329D mutations were identified at a low frequency (2.59%) in Italian genomes of the BF.7 lineage. In particular, literature data showed that the frequency of the Q180H mutation ranged from 0.05% to 1.18% in SARS-CoV-2 genomes collected in Africa, Asia, Australia, Europe, North America, and South America [31,32]. The same authors identified the mutation Q180H with a significant negative correlation with a case fatality ratio. The frequency of Q180H (nsp3) in the BF.7 lineage in specific European countries showed values ranging from 0.11% (France), 0.32% (Spain), 0.43% (Austria), 1.76% (Slovenia), and 1.67% (Switzerland), according to GISAID data [12, 13, last accessed 25 March 2024]. Local genomic evolution could partly explain our data.
The mutation N1329D identified in the nsp3 of the Italian BF.7 dataset in this study, if compared to BF.7 genomes from GISAID [12, 13, last accessed 25 March 2024], was reported in some Italian genomes and only in one foreign BF.7 genome collected in Austria (EPI_ISL_16377903). This mutation should be better evaluated in future studies since it falls within the nsp3e. Our data reinforce the concept that the substitutions that fall within the non-structural proteins, together with their prevalences, need to be monitored, in particular those occurring inside the nsp3e, which play an important role in the virus RNA synthesis and replication, suggesting that they may be useful targets for anti-viral drug discovery or therapeutic strategies [33].
In the SARS-CoV-2 BQ.1.1 dataset, the L452R (spike), was identified as a very common amino acid substitution, in line with what also occurred in other BQ.1.1 European genomes (data from GISAID). The L452R plays a key role, as it has been reported to increase SARS-CoV-2 fusogenicity and infectivity [34].
Even though I121L and I121T (ORF8, BQ.1.1 dataset) were identified at a low frequency in this study, they have to be closely monitored because they are localized in ORF8, a protein that has several important functions within SARS-CoV-2 pathogenesis [35].
Finally, to better understand the functional characteristics of the nonsynonymous mutations identified in the positively selected sites, the changes in the proteins were evaluated using PolyPhen-2, while I-Mutant 2.0 was used to evaluate their structural stability.
Some amino acid changes are expected to have a possible impact on the structure and stability of proteins, and for all these reasons, the need to be closely monitored. The evolution of SARS-CoV-2 will depend on the intensity and rate of fixation of further evolutionary changes and on the rate of emergence of distinct lineages. For this reason it is important to continue to monitor the selective pressure to identify relevant mutational “hot spots” and potentially useful to roll out and update vaccines and/or therapies.
Before drawing conclusions, it is worth mentioning the possible limitations of this study. In particular, the model depends on the availability of SARS-CoV-2 genomes and can to some extent influence the models used for the analysis.
5. Conclusions
In conclusion, our study provides an overview of the selective pressure profile in a dataset of SARS-CoV-2 BF.7 and BQ.1.1 lineages identified in Italy, highlighting how specific key mutations can become fixed in this viral population and the possible impact on the stability of the proteins. Furthermore, a specific key mutation (N1329D—nsp3) needs to be better evaluated in future studies to better clarify its role and impact.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/microorganisms12050908/s1. Table S1: (a) GISAID Accession Numbers of SARS-CoV-2 genomes belonging to BF.7 lineage (first dataset); (b) GISAID Accession Numbers of SARS-CoV-2 genomes belonging to BQ.1.1 lineage (second dataset). Table S2: Selective pressure analysis on SARS-CoV-2 lineages BF.7 and BQ.1.1.
Author Contributions
Conceptualization, A.L.P.; methodology, A.L.P.; formal analysis, A.L.P.; investigation, A.L.P., L.A. and A.D.M.; data curation, L.A., A.D.M., A.K. and L.D.S.; resources, the Italian genomic laboratory network; writing—original draft preparation, A.L.P.; writing—review and editing, A.L.P., L.A., A.D.M., A.K., L.D.S., G.V., I.D.B., S.M., A.T.P., P.S. and the Italian Genomic Laboratory Network; supervision, A.T.P. and P.S.; funding acquisition, A.T.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by EU funding within the NextGenerationEU-MUR PNRR Extended Partnership initiative on Emerging Infectious Diseases (Project no. PE00000007, INF-ACT).
Data Availability Statement
The sequences used in this study were available in GISAID (https://gisaid.org/ accessed on 15 December 2022).
Acknowledgments
We gratefully acknowledge all the authors and all the originating laboratories responsible for obtaining the specimens, as well as all the submitting laboratories where genetic sequence data were generated and shared via the GISAID Initiative, on which this research is based. The Italian Genomic Laboratory Network: Liborio Stuppia, Federico Anaclerio, Center for Advanced Studies and Technology (CAST), G. d’Annunzio University of Chieti-Pescara, Chieti, Italy; Giovanni Savini, Cesare Cammà, Luigi Possenti, Istituto Zooprofilattico Sperimentale dell’Abruzzo e del Molise “Giuseppe Caporale”, Teramo, Italy; Domenico Dell’Edera, Medical Genetics Unit, “Madonna delle Grazie” Hospital, Matera, Italy; Antonio Picerno, Teresa Lopizzo, Clinical Pathology and Microbiology Unit, AOR San Carlo, Potenza, Italy; Maria Teresa Fiorillo, Unit of Microbiology and Virology, North Health Center ASP 5, Reggio Calabria, Italy; Giuseppe Viglietto, CIS (Interdepartmental Center for Services and Research), Genomics and Molecular Pathology, “Magna Graecia” University, Catanzaro, Italy; Pasquale Minchella, Department of Microbiology and Virology, Pugliese Ciaccio Hospital, Catanzaro, Italy; Francesca Greco, Microbiology and Virology Unit, “Annunziata” Hospital of Cosenza, Cosenza, Italy; Antonio Limone, Giovanna Fusco, Istituto Zooprofilattico Sperimentale del Mezzogiorno, Portici, Naples, Italy; Claudia Tiberio, Luigi Atripaldi, Mariagrazia Coppola, UOC Microbiologia e Virologia, P.O. Cotugno A.O. dei Colli, Naples, Italy; Davide Cacchiarelli, Antonio Grimaldi, Telethon Institute of Genetics and Medicine (TIGEM), Pozzuoli, Napoli, Italy; Stefano Pongolini, Erika Scaltriti, Risk Analysis and Genomic Epidemiology Unit, Istituto Zooprofilattico Sperimentale della Lombardia e dell’Emilia-Romagna (IZSLER) “Bruno Ubertini”, Parma, Italy; Vittorio Sambri, Giorgio Dirani, Silvia Zannoli, Unit of Microbiology, The Greater Romagna Area Hub Laboratory, Cesena, Italy; Tiziana Lazzarotto, Giada Rossini, Microbiology Unit, IRCCS Azienda Ospedaliero-Universitaria di Bologna, Bologna, Italy; Federica Baldan, Sabrina Lombino, Department of Laboratory Medicine, Azienda Sanitaria Universitaria Friuli Centrale Udine, Italy; Pierlanfranco D’Agaro, Ludovica Segat, Hygiene and Preventive Medicine Clinical Operative Unit, Trieste University Hospital—ASUGI, Trieste, Italy; Fabio Barbone, Raffaella Koncan, Department of Medicine, Surgery and Health Sciences, University of Trieste, Italy; Antonio Battisti, Patricia Alba, Department of General Diagnostics, Istituto Zooprofilattico Sperimentale del Lazio e della Toscana (IZSLT), Rome, Italy; Maria Teresa Scicluna, Department of Virology, Istituto Zooprofilattico Sperimentale del Lazio e della Toscana M. Aleandri; Silvia Angeletti, Elisabetta Riva, Unità di ricerca Laboratorio Fondazione Policlinico Universitario Campus Bio- Medico, Roma, Italy; Fulvia Pimpinelli, UOSD Microbiology and Virology, IRCCS San Gallicano Dermatological Institute, IFO, Rome, Italy; Maurizio Fanciulli, SAFU Laboratory, IRCCS Regina Elena National Cancer Institute, IFO, Rome, Italy; Alice Massacci, Biostatistics, Bioinformatics and Clinical Trial Center, IRCCS Regina Elena National Cancer Institute, IFO, Rome, Italy; Maurizio Sanguinetti, Dipartimento di Scienze di Laboratorio e Infettivologiche, Fondazione Policlinico Universitario A. Gemelli IRCCS, Rome, Italy; Fabrizio Maggi, Maria Rosaria Capobianchi, Emanuela Giombini, Cesare Ernesto Maria Gruber, Laboratory of Virology, National Institute for Infectious Diseases “Lazzaro Spallanzani” (IRCCS), Rome, Italy; Maria Rosaria Capobianchi, Saint Camillus International University of Health Sciences, Via di Sant’Alessandro, 8, 00131 Rome, Italy; Department of Infectious Tropical Diseases and Microbiology, Sacro Cuore Don Calabria Hospital I.R.C.C.S., Via Don A. Sempreboni 5, 37024—Negrar di Valpolicella, Verona, Italy; Ombretta Turriziani, Department of Molecular Medicine, Sapienza University of Rome, Sapienza University Hospital “Policlinico Umberto I”, Rome, Italy; Carlo Federico Perno, Microbiology and Diagnostic Immunology, Bambino Gesù Children’s Hospital, IRCCS, Rome, Italy; Francesca Ceccherini-Silberstein, Maria Concetta Bellocchi, Department of Experimental Medicine, University of Rome Tor Vergata, Rome, Italy; Bianca Bruzzone, Giancarlo Icardi, Andrea Orsi, Hygiene Unit, San Martino Policlinico Hospital-IRCCS for Oncology and Neurosciences Genoa, Italy; Flavia Lillo, Laboratory of Clinical Pathology, ASL 2 Regione Liguria, Savona, Italy; Annapaola Callegaro, Rea Valaperta, ASST Bergamo Est, Italy; Maria Oggionni, ASST Bergamo Ovest, Bergamo, Italy; Sophie Testa, Fabio Sagradi, ASST Cremona, Cremona, Italy; Arnaldo Caruso, Department of Molecular and Translational Medicine University of Brescia Medical School, Brescia, Italy; Elisa Bonomi, Laboratory of Microbiology and Virology, ASST degli Spedali Civili Brescia, Brescia, Italy; Valerio Leoni, Claudia Siracusa, ASST della Brianza—Laboratory of Clinical Pathology and Toxichology, Hospital Pio XI of Desio, Italy; Diana Fanti, Alice Nava, S.C. Clinical Microbiology, ASST Grande Ospedale Metropolitano Niguarda Milan, Italy; Sergio Malandrin, Annalisa Cavallero, Microbiology and Virology Unit, Fondazione IRCCS San Gerardo dei Tintori, Monza, Italy; Claudio Farina, Marco Arosio, Microbiology and Virology Laboratory, ASST Papa Giovanni XXIII, Bergamo, Italy; Ferruccio Ceriotti, Sara Colonia Uceda Renteria, Stefania Paganini, Clinical Laboratory, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Milano, Italy; Anna Maria Di Blasio, Erminio Torresani, IRCCS Istituto Auxologico Italiano, Milan, Italy; Marina Noris, Istituto di Ricerche Farmacologiche Mario Negri IRCCS Ranica, Bergamo, Italy; Maria Beatrice Boniotti, Cristina Bertasio, Istituto Zooprofilattico Sperimentale della Lombardia ed Emilia Romagna, Brescia, Italy; Nicola Clementi, Laboratory of Microbiology and Virology, Vita-Salute San Raffaele University, Milan, Italy; Laboratory of Microbiology and Virology, IRCCS Ospedale San Raffaele, Milan, Italy; Enzo Boeri, Michela Sampaolo, Laboratory of Microbiology and Virology, IRCCS San Raffaele Hospital, Milan, Italy; Federica Novazzi, Nicasio Mancini, Laboratory of Microbiology, ASST Sette Laghi, Varese, Italy; Maria Rita Gismondo, Valeria Micheli, Laboratory of Clinical Microbiology, Virology and Bioemergencies, ASST Fatebenefratelli Sacco, Luigi Sacco Hospital, Milan, Italy; Laura Cardarelli, CERBA HealthCare Italia—RDI, Rete Diagnostica Italiana, Limena (PD), Italy; Flavia Maggiolini, CERBA HealthCare Italia—Centro Medico S. Nicola, Tradate (VA), Italy; Fausto Baldanti, Microbiology and Virology Department, Fondazione IRCCS Policlinico San Matteo, Pavia, Italy; Dpt. of Clinical, Surgical, Diagnostics and Pediatric Sciences, University of Pavia, Pavia, Italy; Antonio Piralla, Microbiology and Virology Department, Fondazione IRCCS Policlinico San Matteo, Pavia, Italy; Giulia Bassanini, Michela Musarra, Sofia Grandi, Claudia Cagioni, PTP Science Park S.c.a.r.l.—Laboratory SmeL, Lodi, Italy; Fabiana Cro, Cristina Lapucci, Cristina Kullmann, SYNLAB ITALIA SRL, Brescia, Italy; Elena Pariani, Cristina Galli, Laura Pellegrinelli, Department of Biomedical Sciences for Health, University of Milan, Milan, Italy; Stefano Menzo, Department of Biomedical Sciences and Public Health, Virology Unit, Polytechnic University of Marche, Ancona, Italy; Massimiliano Scutellà, Silvio Garofalo, U.O.C. Laboratory Medicine, Cardarelli Hospital and Department of Medicine and Health Sciences “V. Tiberio” (DiMeS), University of Molise, Campobasso (CB), Italy; Elisabetta Pagani, Irene Bianconi, Angela Maria Di Pierro, Laboratory of Microbiology and Virology, Hospital of Bolzano (SABES-ASDAA), Bolzano-Bozen, Italy; Lehr-Krankenhaus der Paracelsus Medizinischen Privatuniversität (PMU); Lucia Collini, Giovanni Lorenzin, Laboratory of Microbiology and Virology, Country Health Service APSS, S. Chiara Hospital, Trento, Italy; Valeria Ghisetti, Laboratory of Microbiology and Molecular Biology, Amedeo di Savoia Hospital, Turin, Italy; Anna Sapino, Silvia Brossa, Paola Marino, Antonino Sottile, Giorgia Migliardi, Candiolo Cancer Institute FPO-IRCCS, Candiolo, Turin, Italy; Simone Peletto, Giuseppe Ru, Pier Luigi Acutis, Elena Bozzetta, Istituto Zooprofilattico Sperimentale del Piemonte, Liguria e Valle d’Aosta, Turin, Italy; Maria Chironna, Daniela Loconsole, Hygiene Unit, Interdisciplinary Department of Medicine—DIM, University of Bari “Aldo Moro”, Bari, Italy; Antonio Parisi, Genetic and Molecular Epidemiology Laboratory, Experimental Zooprophylactic Institute of Apulia and Basilicata, Foggia, Italy; Rossella De Nittis, Microbiology and Virology, “Policlinico Riuniti”, University Hospital, Foggia, Italy; Florigio Romano Lista, Scientific Department, Army Medical Center, Rome, Italy; Ferdinando Coghe, Laboratorio Generale (HUB) Analisi Chimico Cliniche e Microbiologia, PO “Duilio Casula”, Azienda Ospedaliera Universitaria di Cagliari, Cagliari, Italy; Sergio Uzzau, Salvatore Rubino, Flavia Angioj, Gabriele Ibba, Caterina Serra, Department of Biomedical Sciences, S.C. Microbiology and Virology, Azienda Ospedaliera Universitaria di Sassari, Sassari, Italy; Giovanna Piras, Giuseppe Mameli, Rosanna Asproni, Laboratorio Specialistico, UOC Ematologia e CTMO, P.O. “San Francesco,” ASL Nuoro, Nuoro, Italy; Francesca Di Gaudio, Department of Health Promotion, Mother and Child Care, Internal Medicine and medical Specialties “G. D’Alessandro”, University of Palermo, Palermo, Italy; Stefano Vullo, Stefano Reale, Istituto Zooprofilattico Sperimentale della Sicilia, Palermo, Italy; Teresa Pollicino, Division of Advanced Diagnostic Laboratories, University Hospital “G. Martino” Messina, Italy; Francesco Vitale, Fabio Tramuto, Clinical Epidemiology Unit and Regional Reference Laboratory, University Hospital “P. Giaccone”, Palermo, Italy; Department of Health Promotion, Mother and Child Care, Internal Medicine and Medical Specialties “G. D’Alessandro”, University of Palermo, Italy; Stefania Stefani, Clinical Virology Laboratory, “G. Rodolico—S. Marco” Hospital, Catania, Italy; Department of Biomedical and Biotechnological Sciences, University of Catania, Catania, Italy; Guido Scalia, A.O.U. Policlinico “G. Rodolico- S. Marco”, U.O.C. Laboratory Analysis, Virology Section, and Department of Biomedical and Biotechnological Sciences, University of Catania, Catania, Italy; Concetta Ilenia Palermo, A.O.U. Policlinico “G. Rodolico- S. Marco”, U.O.C. Laboratory Analysis, Virology Section, Catania, Italy; Giuseppe Mancuso, UOC Microbiology, University Hospital “G. Martino”, Messina, Italy; Vincenzo Bramanti, Carmelo Fidone, Giuseppe Barrano, U.O.C. Laboratory Analysis, ASP Ragusa, Ragusa, Italy; Mauro Pistello, Department of Translational Research, University of Pisa; Virology Unit, Pisa University Hospital, Pisa, Italy; Gian Maria Rossolini, Department of Experimental and Clinical Medicine, University of Florence, Florence, Italy; Microbiology and Virology Unit, Florence Careggi University Hospital, Florence, Italy; Francesca Malentacchi, Microbiology and Virology Unit, Careggi University Hospital, Florence, Italy; Maria Grazia Cusi, Virology Unit, Department of Medical Biotechnologies, University of Siena, Siena, Italy; Antonella Mencacci, Barbara Camilloni, Microbiology and Clinical Microbiology, Department of Medicine and Surgery, University of Perugia, Santa Maria della Misericordia Hospital, Perugia, Italy; Calogero Terregino, Alice Fusaro, Isabella Monne, Angela Salomoni, Division of Comparative Biomedical Sciences, Istituto Zooprofilattico Sperimentale delle Venezie, Legnaro, Padova, Italy; Davide Gibellini, Department of Diagnostic and Public Health, Verona University, Verona, Italy; Laura Squarzon, Mosè Favarato, Molecular Diagnostics and Genetics, AULSS 3 Serenissima, Venice, Italy.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- World Health Organization (WHO). Classification of Omicron (B.1.1.529): SARS-CoV-2 Variant of Concern. 26 November 2021. Available online: https://www.who.int/news/item/26-11-2021-classification-of-omicron-(b.1.1.529)-sars-cov-2-variant-of-concern (accessed on 24 April 2023).
- Epicentro. Indagine del 17/01/2022. Stima Della Prevalenza Delle Varianti VOC (Variant of Concern) e di Altre Varianti di SARS-CoV-2 in Italia. January 2022. Available online: https://www.epicentro.iss.it/coronavirus/pdf/sars-cov-2-monitoraggio-varianti-indagini-rapide-17-gennaio-2022.pdf (accessed on 24 April 2023).
- Epicentro. Monitoraggio Delle Varianti del Virus SARS-CoV-2 di Interesse in Sanità Pubblica in Italia Prevalenza e Distribuzione Delle Varianti di SARS-CoV-2 di Interesse per la Sanità Pubblica in Italia—Rapporto n. 17 del 18 Febbraio 2022. February 2022. Available online: https://www.epicentro.iss.it/coronavirus/pdf/sars-cov-2-monitoraggio-varianti-rapporti-periodici-18-febbraio-2022.pdf (accessed on 24 April 2023).
- Epicentro. Monitoraggio Delle Varianti del Virus SARS-CoV-2 di Interesse in Sanità Pubblica in Italia Prevalenza e Distribuzione Delle Varianti di SARS-CoV-2 di interesse per la sanità pubblica in Italia—Rapporto n. 22 del 28 luglio 2022. July 2022. Available online: https://www.epicentro.iss.it/coronavirus/pdf/sars-cov-2-monitoraggio-varianti-rapporti-periodici-28-luglio-2022.pdf (accessed on 24 April 2023).
- Epicentro. Monitoraggio Delle Varianti del Virus SARS-CoV-2 di Interesse in Sanità Pubblica in Italia Prevalenza e Distribuzione Delle Varianti di SARS-CoV-2 di Interesse per la Sanità Pubblica in Italia—Rapporto n. 23 del 2 Settembre 2022. September 2022. Available online: https://www.epicentro.iss.it/coronavirus/pdf/sars-cov-2-monitoraggio-varianti-rapporti-periodici-2-settembre-2022.pdf (accessed on 24 April 2023).
- Epicentro. Monitoraggio Delle Varianti del Virus SARS-CoV-2 di Interesse in Sanità Pubblica in Italia Prevalenza e Distribuzione Delle Varianti di SARS-CoV-2 di Interesse per la Sanità Pubblica in Italia—Rapporto n. 26 del 2 Dicembre 2022. December 2022. Available online: https://www.epicentro.iss.it/coronavirus/pdf/sars-cov-2-monitoraggio-varianti-rapporti-periodici-2-dicembre-2022.pdf (accessed on 24 April 2023).
- GISAID. Genomic Epidemiology of SARS-CoV-2 with Subsampling Focused Globally over the Past 6 Months. 2023. Available online: https://gisaid.org/phylodynamics/global/nextstrain/ (accessed on 24 April 2023).
- Velavan, T.P.; Ntoumi, F.; Kremsner, P.G.; Lee, S.S.; Meyer, C.G. Emergence and geographic dominance of Omicron subvariants XBB/XBB.1.5 and BF.7—The public health challenges. Int. J. Infect. Dis. 2023, 128, 307–309. [Google Scholar] [CrossRef]
- Hachmann, N.P.; Miller, J.; Collier, A.Y.; Barouch, D.H. Neutralization escape by SARS-CoV-2 Omicron subvariant BA.4.6. N. Engl. J. Med. 2022, 387, 1904–1906. [Google Scholar] [CrossRef]
- Nielsen, R.; Yang, Z. Likelihood models for detecting positive selected amino acid sites and applications to the HIV-1 envelope gene. Genetics 1998, 148, 929–936. [Google Scholar] [CrossRef]
- Kosakovsky Pond, S.L.; Frost, S.D.W. Not So Different After All: A Comparison of Methods for Detecting Amino Acid Sites Under Selection. Mol. Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef]
- Khare, S.; Gurry, C.; Freitas, L.; Schultz, M.B.; Bach, G.; Diallo, A.; Akite, N.; Ho, J.; Lee, R.T.C.; Yeo, W.; et al. GISAID’s Role in Pandemic Response. China CDC Wkly. 2021, 3, 1049–1051. [Google Scholar] [CrossRef] [PubMed Central]
- GISAID. Available online: https://gisaid.org/ (accessed on 15 December 2022).
- O’Toole, A.; Scher, E.; Underwood, A.; Jackson, B.; Hill, V.; McCrone, J.T.; Colquhoun, R.; Ruis, C.; Abu-Dahab, K.; Taylor, B.; et al. Assignment of epidemiological lineages in an emerging pandemic using the pangolin tool. Virus Evol. 2021, 7, veab064. [Google Scholar] [CrossRef] [PubMed]
- Pangolin. Available online: https://pangolin.cog-uk.io/ (accessed on 20 December 2022).
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Čech, M. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef] [PubMed]
- The Galaxy Community. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2022 update. Nucleic Acids Res. 2022, 50, W345–W351. [Google Scholar] [CrossRef]
- Galaxy Platform. Available online: https://usegalaxy.org/ (accessed on 20 December 2022).
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Zhang, J.; Nielsen, R.; Yang, Z. Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol. Biol. Evol. 2005, 22, 2472–2479. [Google Scholar] [CrossRef] [PubMed]
- Murrell, B.; Moola, S.; Mabona, A.; Weighill, T.; Sheward, D.; Kosakovsky Pond, S.L.; Scheffler, K. FUBAR: A Fast, Unconstrained Bayesian AppRoximation for Inferring Selection. Mol. Biol. Evol. 2013, 30, 1196–1205. [Google Scholar] [CrossRef] [PubMed]
- Kosakovsky Pond, S.L.; Frost, S.D.W.; Muse Spencer, V. HyPhy: Hypothesis testing using phylogenies. Bioinformatics 2005, 21, 676–679. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, N.; Nandi, S.; Saha, I. Phylogenetic analysis of 17271 Indian SARS-CoV-2 genomes to identify temporal and spatial hotspot mutations. PLoS ONE 2022, 17, e0265579. [Google Scholar] [CrossRef] [PubMed]
- Qu, P.; Evans, J.P.; Faraone, J.N.; Zheng, Y.; Carlin, C.; Anghelina, M.; Stevens, P.; Fernandez, S.; Jones, D.; Lozanski, G.; et al. Enhanced neutralization resistance of SARS-CoV-2 Omicron subvariants BQ.1, BQ.1.1, BA.4.6, BF.7, and BA.2.75.2. Cell Host Microbe 2023, 31, 9–17.e3. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). Weekly Epidemiological Update on COVID-19. 19 January 2023. Available online: https://www.who.int/publications/m/item/weekly-epidemiological-update-on-covid-19---19-january-2023 (accessed on 18 July 2023).
- Ito, J.; Suzuki, R.; Uriu, K.; Itakura, Y.; Zahradnik, J.; Kimura, K.T.; Deguchi, S.; Wang, L.; Lytras, S.; Tamura, T.; et al. Convergent evolution of SARS-CoV-2 Omicron subvariants leading to the emergence of BQ.1.1 variant. Nat. Commun. 2023, 14, 2671. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, R.; Hozumi, Y.; Liu, G.; Qiu, Y.; Wei, X.; Wei, G.W. Emerging dominant SARS-CoV-2 variants. Version 1. arXiv 2022, arXiv:2210.09485v1. [Google Scholar] [PubMed]
- Chen, J.; Wei, G.W. Omicron BA.2 (B.1.1.529.2): High potential to becoming the next dominating variant. arXiv 2022, arXiv:2202.05031v1. [Google Scholar] [PubMed] [PubMed Central]
- Kullappan, M.; Mary, U.; Ambrose, J.M.; Veeraraghavan, V.P.; Surapaneni, K.M. Elucidating the role of N440K mutation in SARS-CoV-2 spike—ACE-2 binding affinity and COVID-19 severity by virtual screening, molecular docking and dynamics approach. J. Biomol. Struct. Dyn. 2023, 41, 912–929. [Google Scholar] [CrossRef]
- Tandel, D.; Gupta, D.; Sah, V.; Harshan, K.H. N440K variant of SARS-CoV-2 has Higher Infectious Fitness. bioRxiv 2021. [Google Scholar] [CrossRef]
- Al-Awaida, W.J.; Al Hourani, B.J.; Swedan, S.; Nimer, R.; Alzoughool, F.; Al-Ameer, H.J.; Al Tamam, S.E.; Alashqar, R.; Al Bawareed, O.; Gushchina, Y.; et al. Correlates of SARS-CoV-2 Variants on Deaths, Case Incidence and Case Fatality Ratio among the Continents for the Period of 1 December 2020 to 15 March 2021. Genes 2021, 12, 1061. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X.; et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B 2020, 10, 766–788. [Google Scholar] [CrossRef]
- Motozono, C.; Toyoda, M.; Zahradnik, J.; Saito, A.; Nasser, H.; Tan, T.S.; Ngare, I.; Kimura, I.; Uriu, K.; Kosugi, Y.; et al. SARS-CoV-2 spike L452R variant evades cellular immunity and increases infectivity. Cell Host Microbe 2021, 29, 1124–1136.e11. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 Protein Interaction Map Reveals Targets for Drug Repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).