
Improving Bacterial Metagenomic Research through Long-Read Sequencing

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
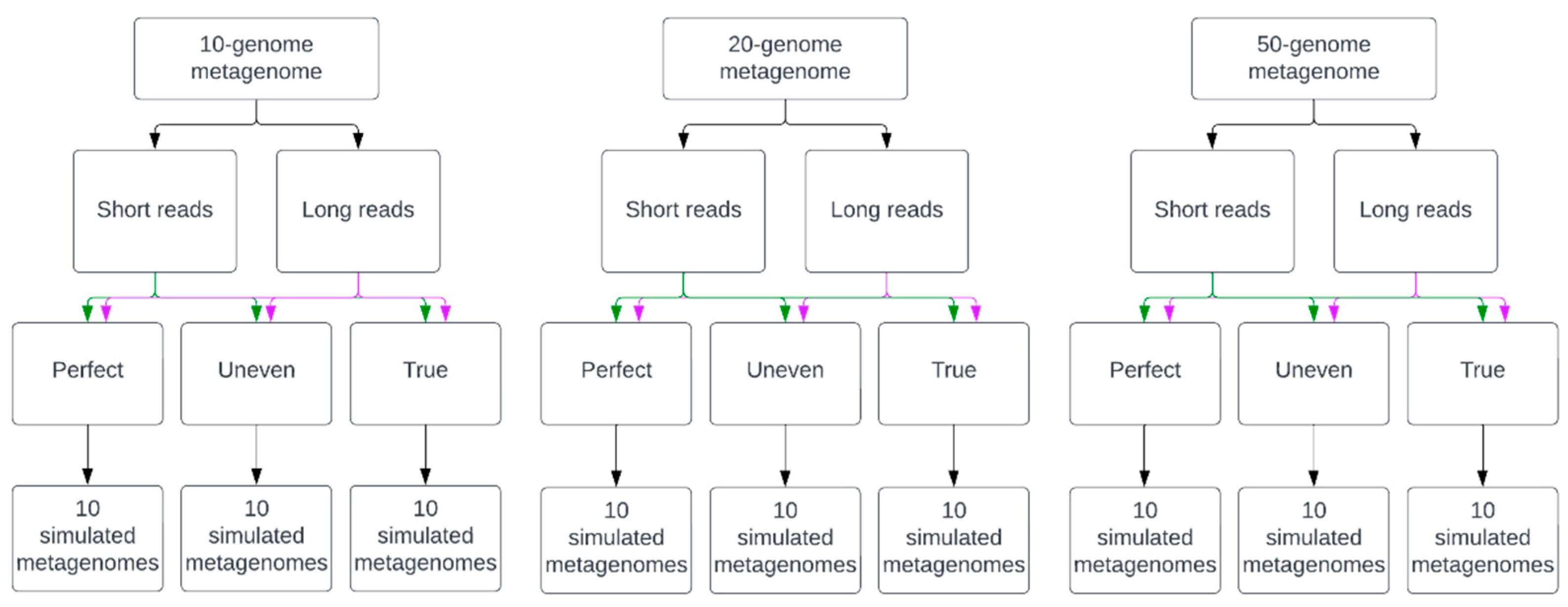
2.1. Simulated Metagenomes of Synthetic Microbial Communities
2.2. Metagenome Assembly and Quality Assessment
2.3. Metagenome-Assembled Genome Recovery and Taxonomic Classification
2.4. Result Evaluation
2.5. Comparison of Experimental Metagenomic Data
3. Results
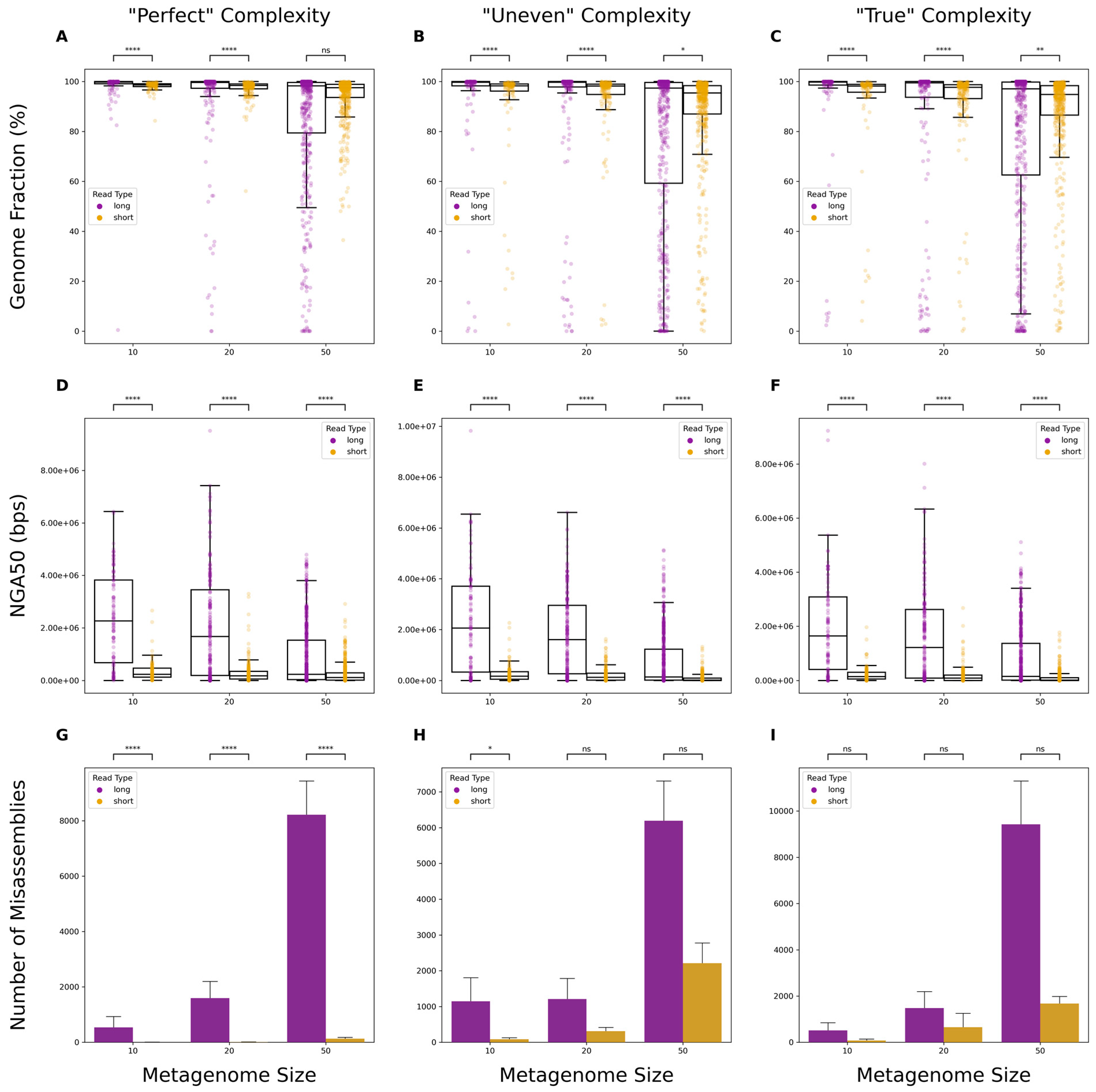
3.1. Comparison of Metagenomic Assembly Completeness
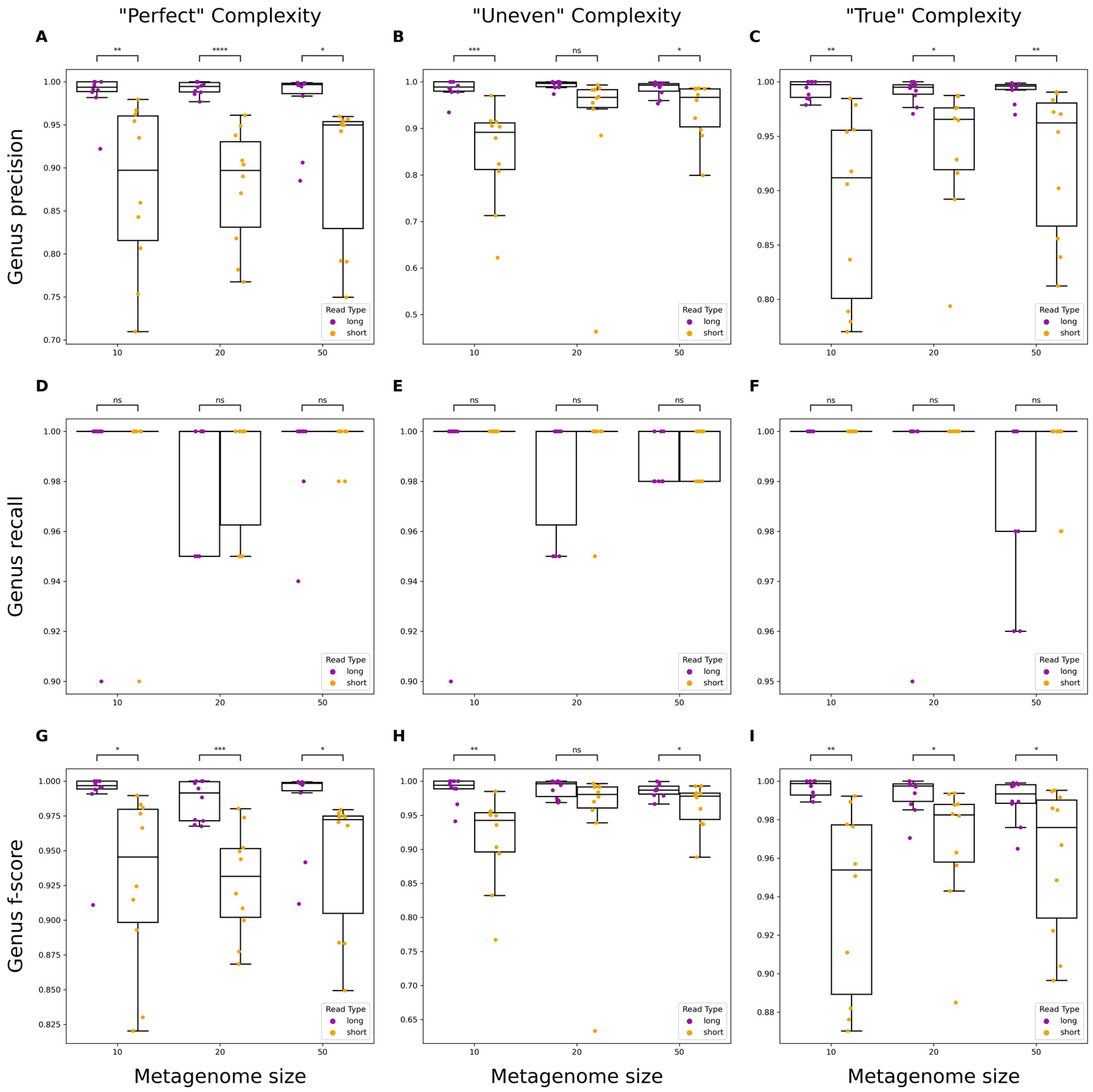
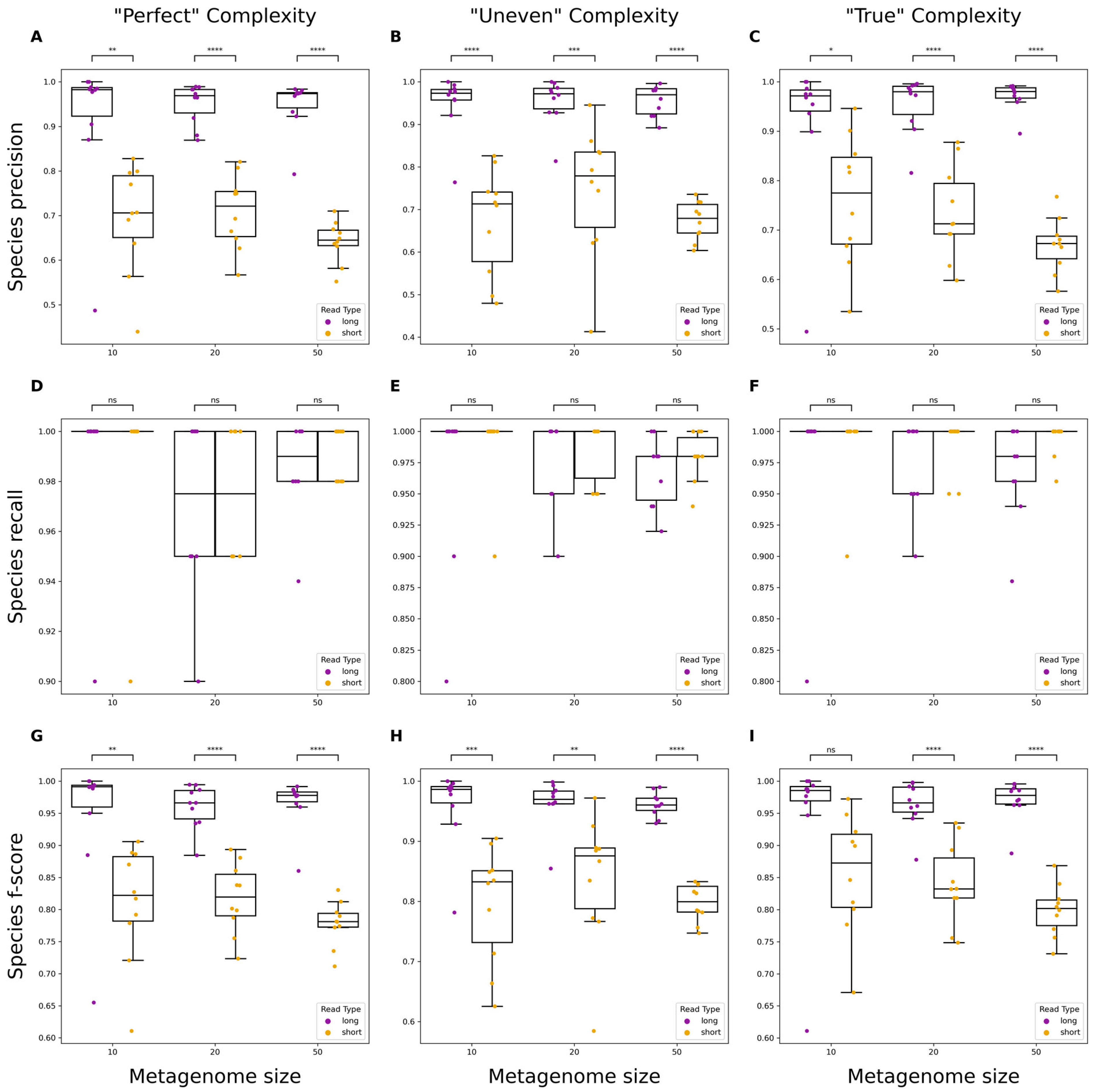
3.2. Evaluating Taxonomic Classification
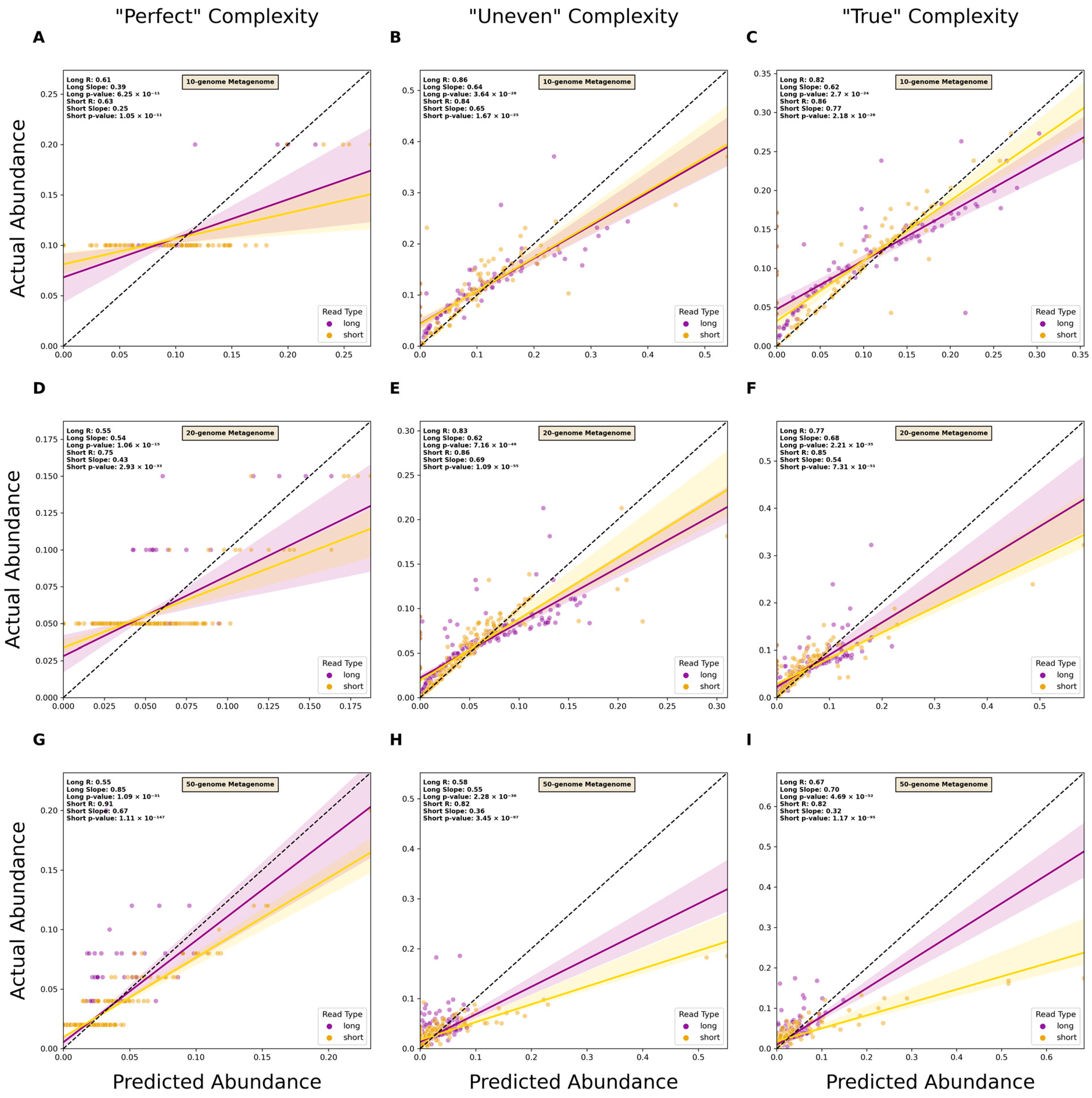
3.3. Estimating Relative Abundance Using Short- and Long-Read Data
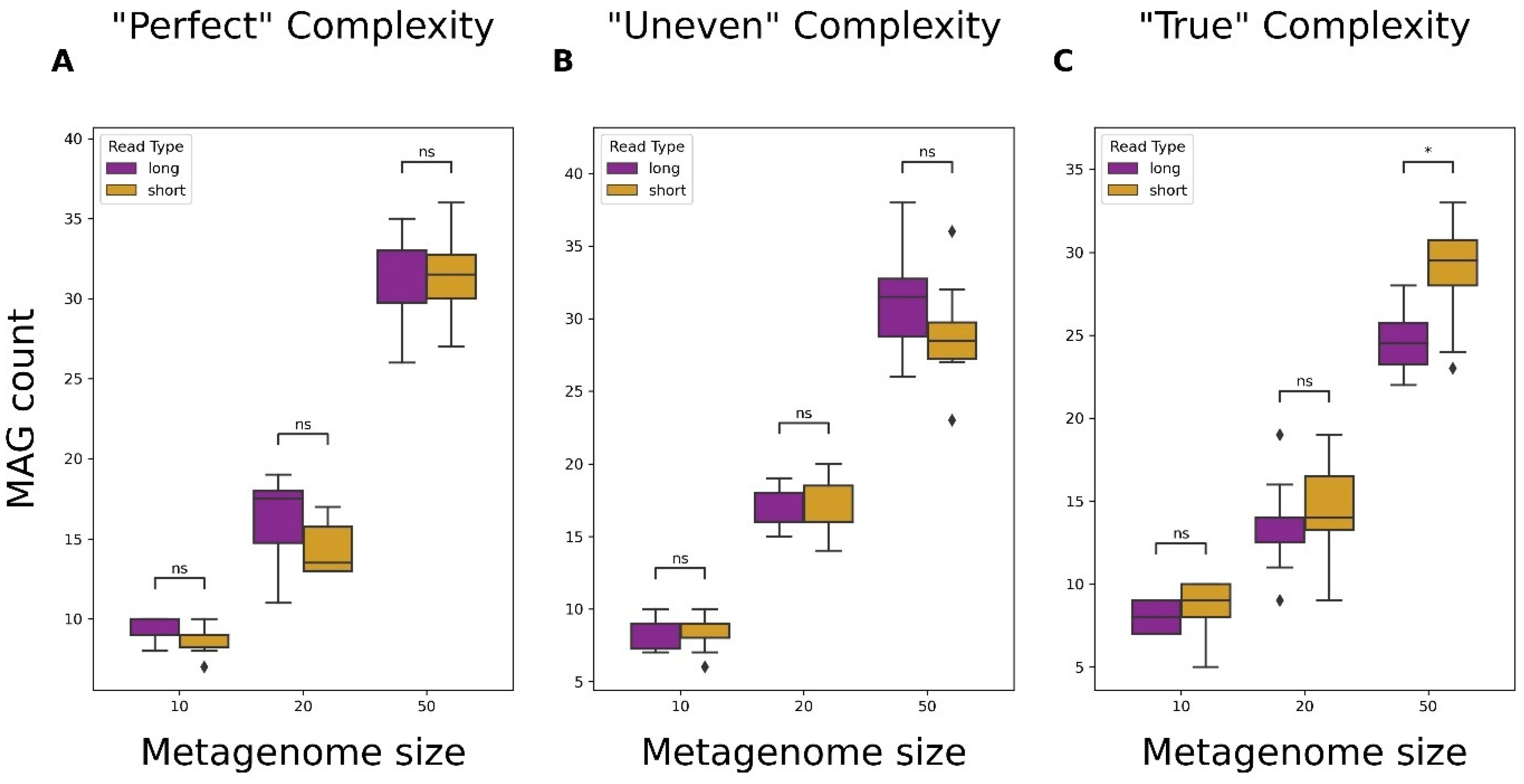
3.4. Metagenome-Assembled Genome (MAG) Recovery from Short- and Long-Read Data
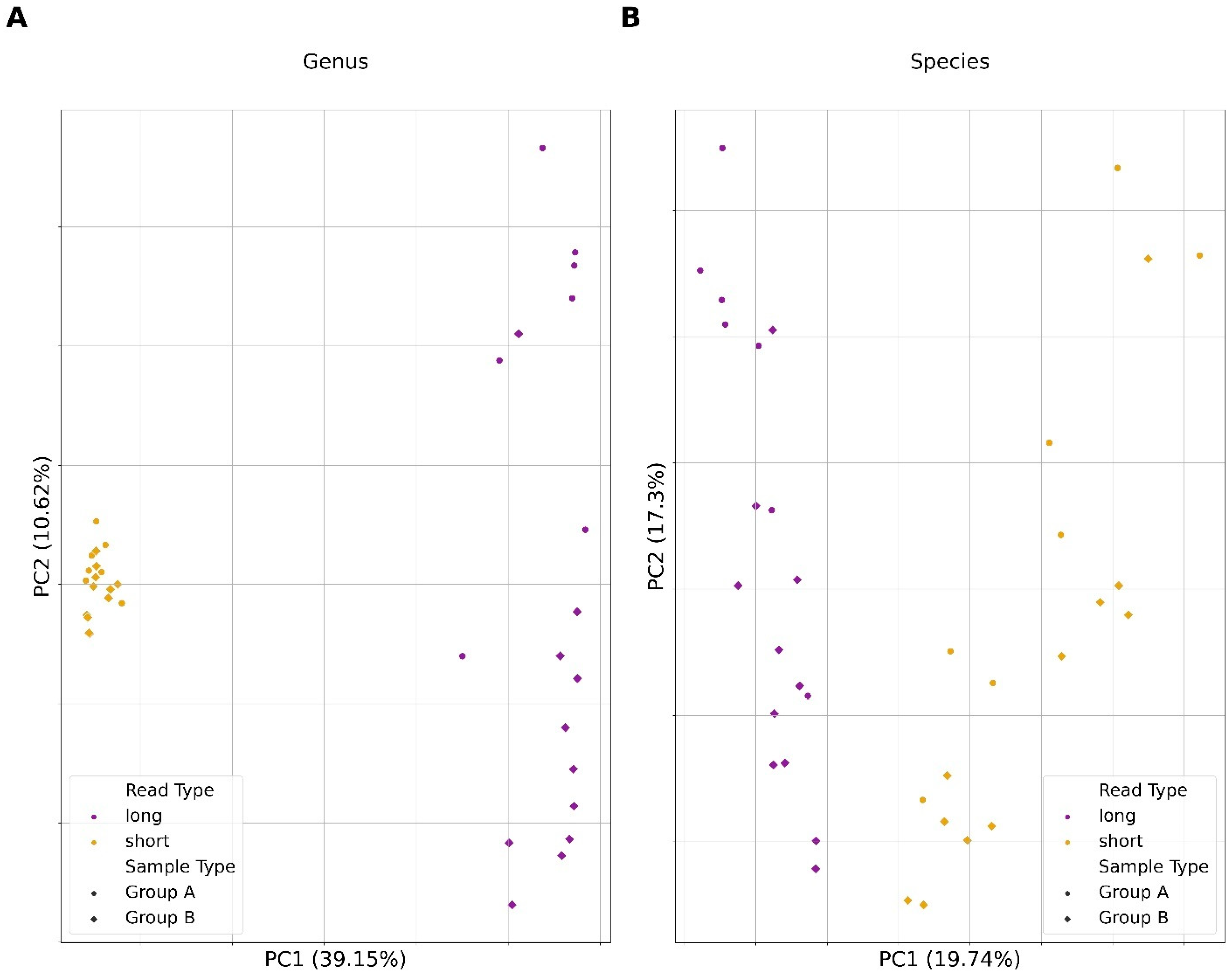
3.5. Microbial Compositional Differences between Short and Long Reads
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, K.; Pachter, L. Bioinformatics for Whole-Genome Shotgun Sequencing of Microbial Communities. PLoS Comput. Biol. 2005, 1, e24. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Hu, H.; Fang, W.; Shi, D.; Liang, C.; Sun, Y.; Gao, G.; Wang, H.; Zhang, Q.; Wang, L.; et al. Detection of Pathogens from Resected Heart Valves of Patients with Infective Endocarditis by Next-Generation Sequencing. Int. J. Infect. Dis. 2019, 83, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Song, R.; Sun, Y.; Li, X.; Ding, C.; Huang, Y.; Du, X.; Wang, J. Biodegradable Microplastics Induced the Dissemination of Antibiotic Resistance Genes and Virulence Factors in Soil: A Metagenomic Perspective. Sci. Total. Environ. 2022, 828, 154596. [Google Scholar] [CrossRef] [PubMed]
- Suttner, B.; Johnston, E.R.; Orellana, L.H.; Rodriguez-R, L.M.; Hatt, J.K.; Carychao, D.; Carter, M.Q.; Cooley, M.B.; Konstantinidis, K.T. Metagenomics as a Public Health Risk Assessment Tool in a Study of Natural Creek Sediments Influenced by Agricultural and Livestock Runoff: Potential and Limitations. Appl. Environ. Microbiol. 2020, 86, e02525-19. [Google Scholar] [CrossRef] [PubMed]
- Wallen, Z.D.; Demirkan, A.; Twa, G.; Cohen, G.; Dean, M.N.; Standaert, D.G.; Sampson, T.R.; Payami, H. Metagenomics of Parkinson’s Disease Implicates the Gut Microbiome in Multiple Disease Mechanisms. Nat. Commun. 2022, 13, 6958. [Google Scholar] [CrossRef] [PubMed]
- Latorre-Pérez, A.; Villalba-Bermell, P.; Pascual, J.; Vilanova, C. Assembly Methods for Nanopore-Based Metagenomic Sequencing: A Comparative Study. Sci. Rep. 2020, 10, 13588. [Google Scholar] [CrossRef] [PubMed]
- Begmatov, S.; Dorofeev, A.G.; Kadnikov, V.V.; Beletsky, A.V.; Pimenov, N.V.; Ravin, N.V.; Mardanov, A.V. The Structure of Microbial Communities of Activated Sludge of Large-Scale Wastewater Treatment Plants in the City of Moscow. Sci. Rep. 2022, 12, 3458. [Google Scholar] [CrossRef]
- Oyserman, B.O.; Flores, S.S.; Griffioen, T.; Pan, X.; van der Wijk, E.; Pronk, L.; Lokhorst, W.; Nurfikari, A.; Paulson, J.N.; Movassagh, M.; et al. Disentangling the Genetic Basis of Rhizosphere Microbiome Assembly in Tomato. Nat. Commun. 2022, 13, 3228. [Google Scholar] [CrossRef]
- Sun, X.; Cai, Y.; Dai, W.; Jiang, W.; Tang, W. The Difference of Gut Microbiome in Different Biliary Diseases in Infant before Operation and the Changes after Operation. BMC Pediatr. 2022, 22, 502. [Google Scholar] [CrossRef]
- Gupta, S.; Mortensen, M.S.; Schjørring, S.; Trivedi, U.; Vestergaard, G.; Stokholm, J.; Bisgaard, H.; Krogfelt, K.A.; Sørensen, S.J. Amplicon Sequencing Provides More Accurate Microbiome Information in Healthy Children Compared to Culturing. Commun. Biol. 2019, 2, 291. [Google Scholar] [CrossRef]
- Berglund, F.; Österlund, T.; Boulund, F.; Marathe, N.P.; Larsson, D.G.J.; Kristiansson, E. Identification and reconstruction of novel antibiotic resistance genes from metagenomes. Microbiome 2019, 7, 52. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, N.D.; Swann, J.; Barker, L.; Kavanagh, J.; Hoosdally, S.; Crook, D.; Street, T.L.; Eyre, D.W.; The GonFast Investigators Group. High Precision Neisseria gonorrhoeae Variant and Antimicrobial Resistance Calling from Metagenomic Nanopore Sequencing. Genome Res. 2020, 30, 1354–1363. [Google Scholar] [CrossRef] [PubMed]
- Charalampous, T.; Kay, G.L.; Richardson, H.; Aydin, A.; Baldan, R.; Jeanes, C.; Rae, D.; Grundy, S.; Turner, D.J.; Wain, J.; et al. Nanopore Metagenomics Enables Rapid Clinical Diagnosis of Bacterial Lower Respiratory Infection. Nat. Biotechnol. 2019, 37, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Petersen, L.M.; Martin, I.W.; Moschetti, W.E.; Kershaw, C.M.; Tsongalis, G.J. Third-Generation Sequencing in the Clinical Laboratory: Exploring the Advantages and Challenges of Nanopore Sequencing. J. Clin. Microbiol. 2019, 58, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Phillippy, A.M. One Chromosome, One Contig: Complete Microbial Genomes from Long-Read Sequencing and Assembly. Curr. Opin. Microbiol. 2015, 23, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Singleton, C.M.; Petriglieri, F.; Kristensen, J.M.; Kirkegaard, R.H.; Michaelsen, T.Y.; Andersen, M.H.; Kondrotaite, Z.; Karst, S.M.; Dueholm, M.S.; Nielsen, P.H.; et al. Connecting Structure to Function with the Recovery of Over 1000 High-Quality Metagenome-Assembled Genomes from Activated Sludge Using Long-Read Sequencing. Nat. Commun. 2021, 12, 2009. [Google Scholar] [CrossRef] [PubMed]
- Amarasinghe, S.L.; Su, S.; Dong, X.; Zappia, L.; Ritchie, M.E.; Gouil, Q. Opportunities and Challenges in Long-Read Sequencing Data Analysis. Genome Biol. 2020, 21, 30. [Google Scholar] [CrossRef] [PubMed]
- Sereika, M.; Kirkegaard, R.H.; Karst, S.M.; Michaelsen, T.Y.; Sørensen, E.A.; Wollenberg, R.D.; Albertsen, M. Oxford Nanopore R10.4 Long-Read Sequencing Enables the Generation of Near-Finished Bacterial Genomes from Pure Cultures and Metagenomes without Short-Read or Reference Polishing. Nat. Methods 2022, 19, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Amarasinghe, S.L.; Ritchie, M.E.; Gouil, Q. long-read-tools.org: An interactive catalogue of analysis methods for long-read sequencing data. GigaScience 2021, 10, giab003. [Google Scholar] [CrossRef]
- Amarasinghe, S.L.; Ritchie, M.E.; Gouil, Q. Long-Read-Tools. Long Read Tools. 2023. Available online: https://long-read-tools.org/analysis.html (accessed on 10 September 2023).
- Govender, K.N.; Street, T.L.; Sanderson, N.D.; Eyre, D.W. Metagenomic Sequencing as a Pathogen-Agnostic Clinical Diagnostic Tool for Infectious Diseases: A Systematic Review and Meta-analysis of Diagnostic Test Accuracy Studies. J. Clin. Microbiol. 2021, 59, 10–1128. [Google Scholar] [CrossRef]
- Yang, C.; Lo, T.; Nip, K.M.; Hafezqorani, S.; Warren, R.L.; Birol, I. Meta-NanoSim_2021.11.19.469328v1.Full. BioRxiv 2021. [Google Scholar] [CrossRef]
- Wick, R. Filtlong. 2021. Available online: https://github.com/rrwick/Filtlong (accessed on 30 June 2021).
- Bushnell, B. BBTools. Walnut Creek: Sourceforge.net/projects/bbmap/. 2020. Available online: https://sourceforge.net/projects/bbmap/ (accessed on 13 June 2020).
- Schirmer, M.; D’amore, R.; Ijaz, U.Z.; Hall, N.; Quince, C. Illumina Error Profiles: Resolving Fine-Scale Variation in Metagenomic Sequencing Data. BMC Bioinform. 2016, 17, 125. [Google Scholar] [CrossRef] [PubMed]
- Kolmogorov, M.; Bickhart, D.M.; Behsaz, B.; Gurevich, A.; Rayko, M.; Shin, S.B.; Kuhn, K.; Yuan, J.; Polevikov, E.; Smith, T.P.L.; et al. metaFlye: Scalable Long-Read Metagenome Assembly Using Repeat Graphs. Nat. Methods 2020, 17, 1103–1110. [Google Scholar] [CrossRef] [PubMed]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. metaSPAdes: A new versatile metagenomic assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef] [PubMed]
- Mikheenko, A.; Saveliev, V.; Gurevich, A. MetaQUAST: Evaluation of metagenome assemblies. Bioinformatics 2016, 32, 1088–1090. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Lh3/SEQTK: Toolkit for Processing Sequences in FASTA/Q Formats. GitHub. Available online: https://github.com/lh3/seqtk (accessed on 19 May 2023).
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Woodcroft, B. CoverM. Available online: https://github.com/wwood/ (accessed on 24 February 2021).
- Kang, D.D.; Li, F.; Kirton, E.; Thomas, A.; Egan, R.; An, H.; Wang, Z. MetaBAT 2: An Adaptive Binning Algorithm for Robust and Efficient Genome Reconstruction from Metagenome Assemblies. PeerJ 2019, 7, e7359. [Google Scholar] [CrossRef] [PubMed]
- Chklovski, A.; Parks, D.H.; Woodcroft, B.J.; Tyson, G.W. CheckM2: A Rapid, Scalable and Accurate Tool for Assessing Microbial Genome Quality Using Machine Learning. BioRxiv 2022. [Google Scholar] [CrossRef]
- Wood, D.E.; Lu, J.; Langmead, B. Improved Metagenomic Analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef]
- Wick, R.R.; Schultz, M.B.; Zobel, J.; Holt, K.E. Bandage: Interactive Visualization of de novo Genome Assemblies. Bioinformatics 2015, 31, 3350–3352. [Google Scholar] [CrossRef] [PubMed]
- Charlier, F.; Weber, M.; Izak, D.; Harkin, E.; Magnus, M.; Lalli, J.; Fresnais, L.; Chan, M.; Markov, N.; Amsalem, O.; et al. Statannotations (0.5). 2022. Available online: https://doi.org/10.5281/zenodo.7213391 (accessed on 16 October 2022).
- Lu, J.; Breitwieser, F.P.; Thielen, P.; Salzberg, S.L. Bracken: Estimating species abundance in metagenomics data. PeerJ Comput. Sci. 2017, 3, e104. [Google Scholar] [CrossRef]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0 Fundamental Algorithms for Scientific Computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Eren, A.E. Assessing Completion and Contamination of Metagenome-Assembled Genomes. 2016. Available online: https://merenlab.org/2016/06/09/assessing-completion-and-contamination-of-MAGs/ (accessed on 21 August 2022).
- Chiou, K.L.; Bergey, C.M. Methylation-based enrichment facilitates low-cost, noninvasive genomic scale sequencing of populations from feces. Sci. Rep. 2018, 8, 1975. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Park, J.; Park, S.; Baek, I.; Chun, J. Introducing murine microbiome database (MMDB): A curated database with taxonomic profiling of the healthy mouse gastrointestinal microbiome. Microorganisms 2019, 7, 480. [Google Scholar] [CrossRef] [PubMed]
- Kieser, S.; Zdobnov, E.M.; Trajkovski, M. Comprehensive mouse microbiota genome catalog reveals major difference to its human counterpart. PLoS Comput. Biol. 2022, 18, e1009947. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Rincon, N.; Wood, D.E.; Breitwieser, F.P.; Pockrandt, C.; Langmead, B.; Salzberg, S.L.; Steinegger, M. Metagenome analysis using the Kraken software suite. Nat. Protoc. 2022, 17, 2815–2839. [Google Scholar] [CrossRef] [PubMed]
- Aitchison, J. The Statistical Analysis of Compositional Data; Chapman and Hall: London, UK, 1986. [Google Scholar]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Watson, C.M.; Crinnion, L.A.; Lindsay, H.; Mitchell, R.; Camm, N.; Robinson, R.; Joyce, C.; Tanteles, G.A.; Halloran, D.J.O.; Pena, S.D.J.; et al. Assessing the Utility of Long-Read Nanopore Sequencing for Rapid and Efficient Characterization of Mobile Element Insertions. Mod. Pathol. 2020, 101, 442–449. [Google Scholar] [CrossRef]
- Hu, T.; Chitnis, N.; Monos, D.; Dinh, A. Next-generation sequencing technologies: An overview. Hum. Immunol. 2021, 82, 801–811. [Google Scholar] [CrossRef]
- Hahn, A.; Sanyal, A.; Perez, G.F.; Colberg-Poley, A.M.; Campos, J.; Rose, M.C.; Pérez-Losada, M. Different Next Generation Sequencing Platforms Produce Different Microbial Profiles and Diversity in Cystic Fibrosis Sputum. J. Microbiol. Methods 2016, 130, 95–99. [Google Scholar] [CrossRef]
- Bastida, F.; Eldridge, D.J.; García, C.; Png, G.K.; Bardgett, R.D.; Delgado-Baquerizo, M. Soil Microbial Diversity–Biomass Relationships Are Driven by Soil Carbon Content across Global Biomes. ISME J. 2021, 15, 2081–2091. [Google Scholar] [CrossRef] [PubMed]
- Hoang, M.T.V.; Irinyi, L.; Hu, Y.; Schwessinger, B.; Meyer, W. Long-Reads-Based Metagenomics in Clinical Diagnosis With a Special Focus on Fungal Infections. Front. Microbiol. 2022, 12, 708550. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Jennison, A.; Whiley, D.; McMahon, J.; Hewitson, G.; Graham, R.; De Jong, A.; Warrilow, D. Illumina sequencing of clinical samples for virus detection in a public health laboratory. Sci. Rep. 2019, 9, 5409. [Google Scholar] [CrossRef]
- Verma, S.K.; Sharma, P.C. NGS-based characterization of microbial diversity and functional profiling of solid tannery waste metagenomes. Genomics 2020, 112, 2903–2913. [Google Scholar] [CrossRef]
- Warwick-Dugdale, J.; Solonenko, N.; Moore, K.; Chittick, L.; Gregory, A.C.; Allen, M.J.; Sullivan, M.B.; Temperton, B. Long-read viral metagenomics captures abundant and microdiverse viral populations and their niche-defining Genomic Islands. PeerJ 2019, 7, e6800. [Google Scholar] [CrossRef]
- Wen, C.; Zheng, Z.; Shao, T.; Liu, L.; Xie, Z.; Le Chatelier, E.; He, Z.; Zhong, W.; Fan, Y.; Zhang, L.; et al. Quantitative metagenomics reveals unique gut microbiome biomarkers in ankylosing spondylitis. Genome Biol. 2017, 18, 142. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Rinke, C.; Chuvochina, M.; Chaumeil, P.-A.; Woodcroft, B.J.; Evans, P.N.; Hugenholtz, P.; Tyson, G.W. Recovery of Nearly 8,000 Metagenome-Assembled Genomes Substantially Expands the Tree of Life. Nat. Microbiol. 2017, 2, 1533–1542. [Google Scholar] [CrossRef]
- Stevens, B.M.; Creed, T.B.; Reardon, C.L.; Manter, D.K. Comparison of Oxford Nanopore Technologies and Illumina MiSeq sequencing with mock communities and agricultural soil. Sci. Rep. 2023, 13, 9323. [Google Scholar] [CrossRef]
- Wickramarachchi, A.; Lin, Y. Binning long reads in metagenomics datasets using composition and coverage information. Algorithms Mol. Biol. 2022, 17, 14. [Google Scholar] [CrossRef]
- Bertrand, D.; Shaw, J.; Kalathiyappan, M.; Ng, A.H.Q.; Kumar, M.S.; Li, C.; Dvornicic, M.; Soldo, J.P.; Koh, J.Y.; Tong, C.; et al. Hybrid metagenomic assembly enables high-resolution analysis of resistance determinants and mobile elements in human microbiomes. Nat. Biotechnol. 2019, 37, 937–944. [Google Scholar] [CrossRef] [PubMed]
- Wick, R.R.; Holt, K.E. Polypolish: Short-read polishing of long-read bacterial genome assemblies. PLoS Comput. Biol. 2022, 18, e1009802. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Greenman, N.; Hassouneh, S.A.-D.; Abdelli, L.S.; Johnston, C.; Azarian, T. Improving Bacterial Metagenomic Research through Long-Read Sequencing. Microorganisms 2024, 12, 935. https://doi.org/10.3390/microorganisms12050935
Greenman N, Hassouneh SA-D, Abdelli LS, Johnston C, Azarian T. Improving Bacterial Metagenomic Research through Long-Read Sequencing. Microorganisms. 2024; 12(5):935. https://doi.org/10.3390/microorganisms12050935
Chicago/Turabian StyleGreenman, Noah, Sayf Al-Deen Hassouneh, Latifa S. Abdelli, Catherine Johnston, and Taj Azarian. 2024. "Improving Bacterial Metagenomic Research through Long-Read Sequencing" Microorganisms 12, no. 5: 935. https://doi.org/10.3390/microorganisms12050935
APA StyleGreenman, N., Hassouneh, S. A.-D., Abdelli, L. S., Johnston, C., & Azarian, T. (2024). Improving Bacterial Metagenomic Research through Long-Read Sequencing. Microorganisms, 12(5), 935. https://doi.org/10.3390/microorganisms12050935