
Contribution of pks+ Escherichia coli (E. coli) to Colon Carcinogenesis




Abstract
:1. Introduction
2. E. coli: A Versatile Bacterium
2.1. Diverse Population and Phylogeny
2.2. Pathogenic Strains and Diseases
2.3. Environmental Adaptation
2.4. Biofilm Formation and Resistance to Antibiotics
2.5. Protective Role of E. coli in Gut Microflora against Enteropathogens
3. Role of E. coli in CRC Development
3.1. Epidemiological Evidence
3.2. Cyclomodulins: DNA-Damaging Compounds Produced by E. coli
3.3. Cyclomodulins Promoting Cell Proliferation and Tissue Dedifferentiation
E. coli Cytotoxic Necrotizing Factor 1 (CNF1)

3.4. Cyclomodulins Inhibiting Cellular Proliferation
3.4.1. Cytolethal-Distending Toxin
3.4.2. Cell Cycle-Inhibiting Factor (Cif)
3.5. Colibactin
3.5.1. pks and Its Role in Synthesizing Various Compounds

3.5.2. Colibactin-Producing E. coli and CRC Mutations
3.5.3. Activation of Colibactin Biosynthesis Pathway
3.6. Effects of Colibactin
3.6.1. Genotoxic Effects
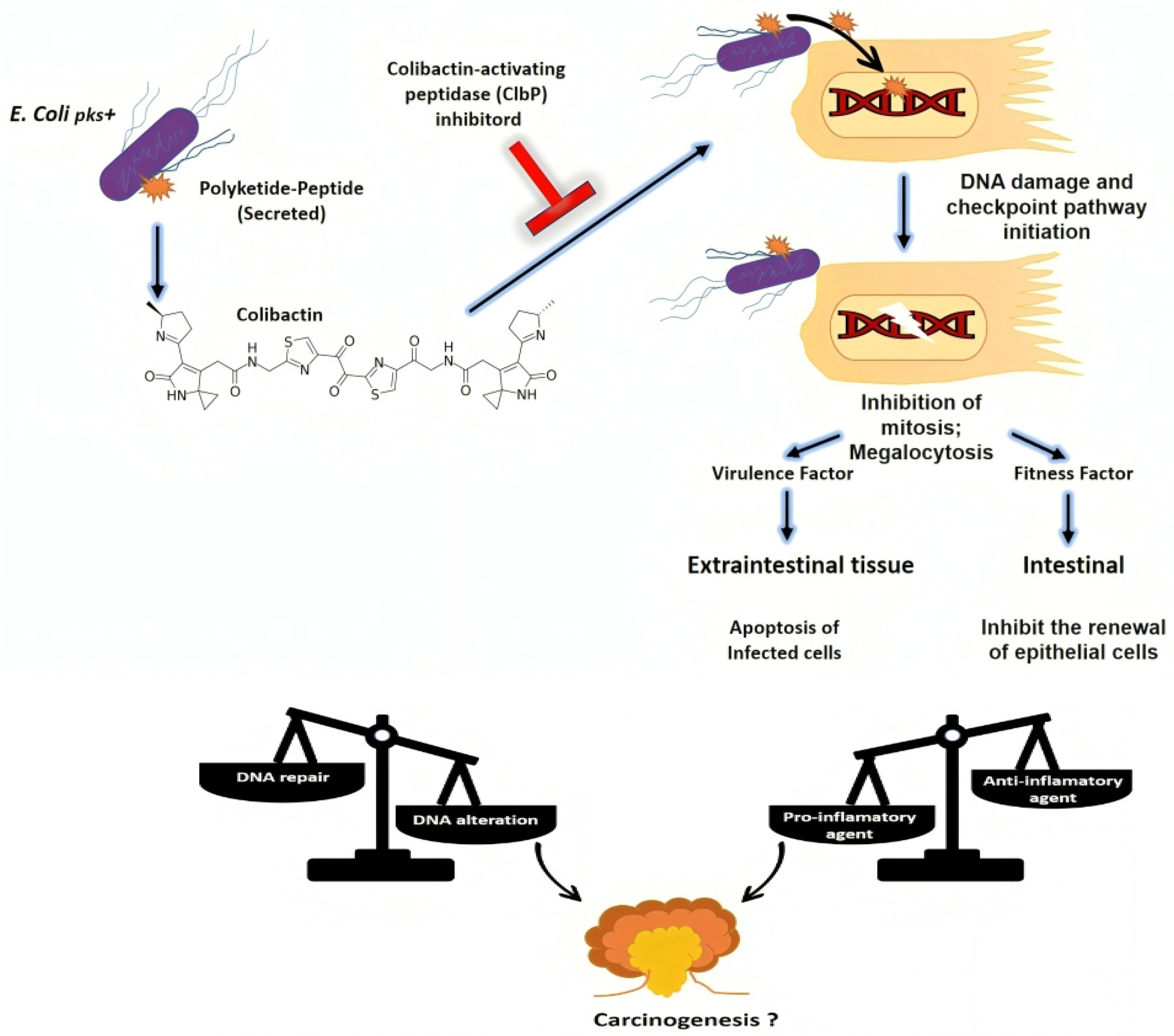
3.6.2. Induction of Genomic Instability
3.6.3. Disruption of Gut Epithelial Barrier and Induction of Inflammation
3.6.4. Modulation of Host Immune Responses
3.6.5. Cell Cycle Effects
3.6.6. β-Glucuronidase Activity
3.6.7. Potential Presence of pks+ in Normal Colon Tissue of Cancer Patients
3.6.8. Heterogeneity of pks+ Bacterial Localization
4. Bacteria Associated with pks+
Colibactin Induces Prophages
5. Prevention and Treatment Strategies
5.1. Inhibiting pks Biosynthesis
5.2. Microbiota Modulation
5.3. Phage Therapy
5.4. Immunomodulator Therapies
6. Screening and Early Detection
6.1. Colorectal Cancer Screening
6.2. Genetic Counseling and Testing
6.3. Evolution of Precision Medicine in CRC
7. Conclusions
Funding
Conflicts of Interest
References
- Rawla, P.; Sunkara, T.; Barsouk, A. Epidemiology of Colorectal Cancer: Incidence, Mortality, Survival, and Risk Factors. Prz. Gastroenterol. 2019, 14, 89–103. [Google Scholar] [CrossRef]
- Sobhani, I.; Rotkopf, H.; Khazaie, K. Bacteria-Related Changes in Host DNA Methylation and the Risk for CRC. Gut Microbes 2020, 12, 1800898. [Google Scholar] [CrossRef] [PubMed]
- Eckburg, P.B.; Bik, E.M.; Bernstein, C.N.; Purdom, E.; Dethlefsen, L.; Sargent, M.; Gill, S.R.; Nelson, K.E.; Relman, D.A. Diversity of the Human Intestinal Microbial Flora. Science 2005, 308, 1635–1638. [Google Scholar] [CrossRef] [PubMed]
- Kawamoto, S.; Maruya, M.; Kato, L.M.; Suda, W.; Atarashi, K.; Doi, Y.; Tsutsui, Y.; Qin, H.; Honda, K.; Okada, T.; et al. Foxp3(+) T Cells Regulate Immunoglobulin a Selection and Facilitate Diversification of Bacterial Species Responsible for Immune Homeostasis. Immunity 2014, 41, 152–165. [Google Scholar] [CrossRef]
- Vallianou, N.G.; Stratigou, T.; Tsagarakis, S. Microbiome and Diabetes: Where Are We Now? Diabetes Res. Clin. Pract. 2018, 146, 111–118. [Google Scholar] [CrossRef]
- Willing, B.; Halfvarson, J.; Dicksved, J.; Rosenquist, M.; Järnerot, G.; Engstrand, L.; Tysk, C.; Jansson, J.K. Twin Studies Reveal Specific Imbalances in the Mucosa-Associated Microbiota of Patients with Ileal Crohn’s Disease. Inflamm. Bowel Dis. 2009, 15, 653–660. [Google Scholar] [CrossRef]
- Jiang, C.; Li, G.; Huang, P.; Liu, Z.; Zhao, B. The Gut Microbiota and Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 58, 1–15. [Google Scholar] [CrossRef]
- Sun, M.-F.; Shen, Y.-Q. Dysbiosis of Gut Microbiota and Microbial Metabolites in Parkinson’s Disease. Ageing Res. Rev. 2018, 45, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Sobhani, I.; Tap, J.; Roudot-Thoraval, F.; Roperch, J.P.; Letulle, S.; Langella, P.; Corthier, G.; Tran Van Nhieu, J.; Furet, J.P. Microbial Dysbiosis in Colorectal Cancer (CRC) Patients. PLoS ONE 2011, 6, e16393. [Google Scholar] [CrossRef]
- Gordon, H.A.; Pesti, L. The Gnotobiotic Animal as a Tool in the Study of Host Microbial Relationships. Bacteriol. Rev. 1971, 35, 390–429. [Google Scholar] [CrossRef]
- Sobhani, I.; Bergsten, E.; Couffin, S.; Amiot, A.; Nebbad, B.; Barau, C.; de’Angelis, N.; Rabot, S.; Canoui-Poitrine, F.; Mestivier, D.; et al. Colorectal Cancer-Associated Microbiota Contributes to Oncogenic Epigenetic Signatures. Proc. Natl. Acad. Sci. USA 2019, 116, 24285–24295. [Google Scholar] [CrossRef] [PubMed]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human Gut Microbiome Viewed across Age and Geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Tlaskalová-Hogenová, H.; Stepánková, R.; Hudcovic, T.; Tucková, L.; Cukrowska, B.; Lodinová-Zádníková, R.; Kozáková, H.; Rossmann, P.; Bártová, J.; Sokol, D.; et al. Commensal Bacteria (Normal Microflora), Mucosal Immunity and Chronic Inflammatory and Autoimmune Diseases. Immunol. Lett. 2004, 93, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Sobhani, I.; Amiot, A.; Le Baleur, Y.; Levy, M.; Auriault, M.-L.; Van Nhieu, J.T.; Delchier, J.C. Microbial Dysbiosis and Colon Carcinogenesis: Could Colon Cancer Be Considered a Bacteria-Related Disease? Ther. Adv. Gastroenterol. 2013, 6, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Sadeghi, M.; Mestivier, D.; Carbonnelle, E.; Benamouzig, R.; Khazaie, K.; Sobhani, I. Loss of Symbiotic and Increase of Virulent Bacteria through Microbial Networks in Lynch Syndrome Colon Carcinogenesis. Front. Oncol. 2024, 13, 1313735. [Google Scholar] [CrossRef] [PubMed]
- Nougayrède, J.-P.; Homburg, S.; Taieb, F.; Boury, M.; Brzuszkiewicz, E.; Gottschalk, G.; Buchrieser, C.; Hacker, J.; Dobrindt, U.; Oswald, E. Escherichia Coli Induces DNA Double-Strand Breaks in Eukaryotic Cells. Science 2006, 313, 848–851. [Google Scholar] [CrossRef] [PubMed]
- Bossuet-Greif, N.; Vignard, J.; Taieb, F.; Mirey, G.; Dubois, D.; Petit, C.; Oswald, E.; Nougayrède, J.-P. The Colibactin Genotoxin Generates DNA Interstrand Cross-Links in Infected Cells. mBio 2018, 9, e02393-17. [Google Scholar] [CrossRef] [PubMed]
- Buc, E.; Dubois, D.; Sauvanet, P.; Raisch, J.; Delmas, J.; Darfeuille-Michaud, A.; Pezet, D.; Bonnet, R. High Prevalence of Mucosa-Associated E. Coli Producing Cyclomodulin and Genotoxin in Colon Cancer. PLoS ONE 2013, 8, e56964. [Google Scholar] [CrossRef] [PubMed]
- Arthur, J.C.; Perez-Chanona, E.; Mühlbauer, M.; Tomkovich, S.; Uronis, J.M.; Fan, T.-J.; Campbell, B.J.; Abujamel, T.; Dogan, B.; Rogers, A.B.; et al. Intestinal Inflammation Targets Cancer-Inducing Activity of the Microbiota. Science 2012, 338, 120–123. [Google Scholar] [CrossRef]
- Kaper, J.B.; Nataro, J.P.; Mobley, H.L. Pathogenic Escherichia Coli. Nat. Rev. Microbiol. 2004, 2, 123–140. [Google Scholar] [CrossRef]
- Penders, J.; Thijs, C.; Vink, C.; Stelma, F.F.; Snijders, B.; Kummeling, I.; van den Brandt, P.A.; Stobberingh, E.E. Factors Influencing the Composition of the Intestinal Microbiota in Early Infancy. Pediatrics 2006, 118, 511–521. [Google Scholar] [CrossRef] [PubMed]
- Massot, M.; Picard, B.; Denamur, E. Diversité des populations d’Escherichia coli et leurs variations au cours du temps au sein du microbiote intestinal. Rev. Francoph. Lab. 2016, 2016, 35–43. [Google Scholar] [CrossRef]
- Darfeuille-Michaud, A. Adherent-Invasive Escherichia Coli: A Putative New E. Coli Pathotype Associated with Crohn’s Disease. Int. J. Med. Microbiol. 2002, 292, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Nataro, J.P.; Kaper, J.B. Diarrheagenic Escherichia coli. Clin. Microbiol. Rev. 1998, 11, 142–201. [Google Scholar] [CrossRef] [PubMed]
- Kenny, B.; Ellis, S.; Leard, A.D.; Warawa, J.; Mellor, H.; Jepson, M.A. Co-Ordinate Regulation of Distinct Host Cell Signalling Pathways by Multifunctional Enteropathogenic Escherichia coli Effector Molecules. Mol. Microbiol. 2002, 44, 1095–1107. [Google Scholar] [CrossRef] [PubMed]
- Kudva, I.T.; Hatfield, P.G.; Hovde, C.J. Characterization of Escherichia coli O157:H7 and Other Shiga Toxin-Producing E. coli Serotypes Isolated from Sheep. J. Clin. Microbiol. 1997, 35, 892–899. [Google Scholar] [CrossRef]
- Sears, C.L.; Kaper, J.B. Enteric Bacterial Toxins: Mechanisms of Action and Linkage to Intestinal Secretion. Microbiol. Rev. 1996, 60, 167–215. [Google Scholar] [CrossRef] [PubMed]
- Nataro, J.P.; Steiner, T.; Guerrant, R.L. Enteroaggregative Escherichia coli. Emerg. Infect. Dis. 1998, 4, 251–261. [Google Scholar] [CrossRef]
- Sweeney, N.J.; Klemm, P.; McCormick, B.A.; Moller-Nielsen, E.; Utley, M.; Schembri, M.A.; Laux, D.C.; Cohen, P.S. The Escherichia coli K-12 gntP Gene Allows E. coli F-18 to Occupy a Distinct Nutritional Niche in the Streptomycin-Treated Mouse Large Intestine. Infect. Immun. 1996, 64, 3497–3503. [Google Scholar] [CrossRef]
- Vial, P.A.; Robins-Browne, R.; Lior, H.; Prado, V.; Kaper, J.B.; Nataro, J.P.; Maneval, D.; Elsayed, A.; Levine, M.M. Characterization of Enteroadherent-Aggregative Escherichia coli, a Putative Agent of Diarrheal Disease. J. Infect. Dis. 1988, 158, 70–79. [Google Scholar] [CrossRef]
- Berben, T.; Sorokin, D.Y.; Ivanova, N.; Pati, A.; Kyrpides, N.; Goodwin, L.A.; Woyke, T.; Muyzer, G. Complete Genome Sequence of Thioalkalivibrio Paradoxus Type Strain ARh 1(T), an Obligately Chemolithoautotrophic Haloalkaliphilic Sulfur-Oxidizing Bacterium Isolated from a Kenyan Soda Lake. Stand. Genom. Sci. 2015, 10, 105. [Google Scholar] [CrossRef] [PubMed]
- Pupo, G.M.; Lan, R.; Reeves, P.R. Multiple Independent Origins of Shigella Clones of Escherichia Coli and Convergent Evolution of Many of Their Characteristics. Proc. Natl. Acad. Sci. USA 2000, 97, 10567–10572. [Google Scholar] [CrossRef] [PubMed]
- Scaletsky, I.C.A.; Fabbricotti, S.H.; Carvalho, R.L.B.; Nunes, C.R.; Maranhão, H.S.; Morais, M.B.; Fagundes-Neto, U. Diffusely Adherent Escherichia Coli as a Cause of Acute Diarrhea in Young Children in Northeast Brazil: A Case-Control Study. J. Clin. Microbiol. 2002, 40, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Bilge, S.S.; Clausen, C.R.; Lau, W.; Moseley, S.L. Molecular Characterization of a Fimbrial Adhesin, F1845, Mediating Diffuse Adherence of Diarrhea-Associated Escherichia coli to HEp-2 Cells. J. Bacteriol. 1989, 171, 4281–4289. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.R.; Stell, A.L. Extended Virulence Genotypes of Escherichia coli Strains from Patients with Urosepsis in Relation to Phylogeny and Host Compromise. J. Infect. Dis. 2000, 181, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Putze, J.; Hennequin, C.; Nougayrède, J.-P.; Zhang, W.; Homburg, S.; Karch, H.; Bringer, M.-A.; Fayolle, C.; Carniel, E.; Rabsch, W.; et al. Genetic Structure and Distribution of the Colibactin Genomic Island among Members of the Family Enterobacteriaceae. Infect. Immun. 2009, 77, 4696–4703. [Google Scholar] [CrossRef] [PubMed]
- Baquero, F.; Martínez, J.-L.; Cantón, R. Antibiotics and Antibiotic Resistance in Water Environments. Curr. Opin. Biotechnol. 2008, 19, 260–265. [Google Scholar] [CrossRef]
- Johnson, C.H.; Dejea, C.M.; Edler, D.; Hoang, L.T.; Santidrian, A.F.; Felding, B.H.; Ivanisevic, J.; Cho, K.; Wick, E.C.; Hechenbleikner, E.M.; et al. Metabolism Links Bacterial Biofilms and Colon Carcinogenesis. Cell Metab. 2015, 21, 891–897. [Google Scholar] [CrossRef] [PubMed]
- Dejea, C.M.; Sears, C.L. Do Biofilms Confer a Pro-Carcinogenic State? Gut Microbes 2016, 7, 54–57. [Google Scholar] [CrossRef] [PubMed]
- Looft, T.; Johnson, T.A.; Allen, H.K.; Bayles, D.O.; Alt, D.P.; Stedtfeld, R.D.; Sul, W.J.; Stedtfeld, T.M.; Chai, B.; Cole, J.R.; et al. In-Feed Antibiotic Effects on the Swine Intestinal Microbiome. Proc. Natl. Acad. Sci. USA 2012, 109, 1691–1696. [Google Scholar] [CrossRef]
- Tenaillon, O.; Skurnik, D.; Picard, B.; Denamur, E. The Population Genetics of Commensal Escherichia coli. Nat. Rev. Microbiol. 2010, 8, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Hudault, S.; Guignot, J.; Servin, A.L. Escherichia Coli Strains Colonising the Gastrointestinal Tract Protect Germfree Mice against Salmonella Typhimurium Infection. Gut 2001, 49, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Wassenaar, T.M.E. E. Coli and Colorectal Cancer: A Complex Relationship That Deserves a Critical Mindset. Crit. Rev. Microbiol. 2018, 44, 619–632. [Google Scholar] [CrossRef] [PubMed]
- Swidsinski, A.; Khilkin, M.; Kerjaschki, D.; Schreiber, S.; Ortner, M.; Weber, J.; Lochs, H. Association between Intraepithelial Escherichia Coli and Colorectal Cancer. Gastroenterology 1998, 115, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Martin, H.M.; Campbell, B.J.; Hart, C.A.; Mpofu, C.; Nayar, M.; Singh, R.; Englyst, H.; Williams, H.F.; Rhodes, J.M. Enhanced Escherichia Coli Adherence and Invasion in Crohn’s Disease and Colon Cancer. Gastroenterology 2004, 127, 80–93. [Google Scholar] [CrossRef]
- Nougayrède, J.-P.; Taieb, F.; De Rycke, J.; Oswald, E. Cyclomodulins: Bacterial Effectors That Modulate the Eukaryotic Cell Cycle. Trends Microbiol. 2005, 13, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, A.; Travaglione, S.; Ballan, G.; Loizzo, S.; Fiorentini, C. The Cytotoxic Necrotizing Factor 1 from E. Coli: A Janus Toxin Playing with Cancer Regulators. Toxins 2013, 5, 1462–1474. [Google Scholar] [CrossRef]
- Marchès, O.; Ledger, T.N.; Boury, M.; Ohara, M.; Tu, X.; Goffaux, F.; Mainil, J.; Rosenshine, I.; Sugai, M.; De Rycke, J.; et al. Enteropathogenic and Enterohaemorrhagic Escherichia coli Deliver a Novel Effector Called Cif, Which Blocks Cell Cycle G2/M Transition. Mol. Microbiol. 2003, 50, 1553–1567. [Google Scholar] [CrossRef] [PubMed]
- Maddocks, O.D.K.; Short, A.J.; Donnenberg, M.S.; Bader, S.; Harrison, D.J. Attaching and Effacing Escherichia coli Downregulate DNA Mismatch Repair Protein in Vitro and Are Associated with Colorectal Adenocarcinomas in Humans. PLoS ONE 2009, 4, e5517. [Google Scholar] [CrossRef]
- Raisch, J. Piste Infectieuse à Escherichia coli Toxinogènes Dans Le Cancer Colorectal. Available online: https://www.semanticscholar.org/paper/Piste-infectieuse-%C3%A0-Escherichia-coli-toxinog%C3%A8nes-le-Raisch/7e546ff34cf670e913a8613ddc4975cdabefb02c (accessed on 16 May 2022).
- Lara-Tejero, M.; Galán, J.E. CdtA, CdtB, and CdtC Form a Tripartite Complex That Is Required for Cytolethal Distending Toxin Activity. Infect. Immun. 2001, 69, 4358–4365. [Google Scholar] [CrossRef]
- Churin, Y.; Al-Ghoul, L.; Kepp, O.; Meyer, T.F.; Birchmeier, W.; Naumann, M. Helicobacter Pylori CagA Protein Targets the C-Met Receptor and Enhances the Motogenic Response. J. Cell Biol. 2003, 161, 249–255. [Google Scholar] [CrossRef]
- Knust, Z.; Schmidt, G. Cytotoxic Necrotizing Factors (CNFs)-A Growing Toxin Family. Toxins 2010, 2, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Falzano, L.; Quaranta, M.G.; Travaglione, S.; Filippini, P.; Fabbri, A.; Viora, M.; Donelli, G.; Fiorentini, C. Cytotoxic Necrotizing Factor 1 Enhances Reactive Oxygen Species-Dependent Transcription and Secretion of Proinflammatory Cytokines in Human Uroepithelial Cells. Infect. Immun. 2003, 71, 4178–4181. [Google Scholar] [CrossRef] [PubMed]
- Flatau, G.; Lemichez, E.; Gauthier, M.; Chardin, P.; Paris, S.; Fiorentini, C.; Boquet, P. Toxin-Induced Activation of the G Protein P21 Rho by Deamidation of Glutamine. Nature 1997, 387, 729–733. [Google Scholar] [CrossRef]
- Schmidt, G.; Selzer, J.; Lerm, M.; Aktories, K. The Rho-Deamidating Cytotoxic Necrotizing Factor 1 from Escherichia Coli Possesses Transglutaminase Activity. Cysteine 866 and Histidine 881 Are Essential for Enzyme Activity. J. Biol. Chem. 1998, 273, 13669–13674. [Google Scholar] [CrossRef]
- Lerm, M.; Selzer, J.; Hoffmeyer, A.; Rapp, U.R.; Aktories, K.; Schmidt, G. Deamidation of Cdc42 and Rac by Escherichia Coli Cytotoxic Necrotizing Factor 1: Activation of c-Jun N-Terminal Kinase in HeLa Cells. Infect. Immun. 1999, 67, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Barbacid, M. RAS Oncogenes: The First 30 Years. Nat. Rev. Cancer 2003, 3, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Thomas, W.; Ascott, Z.K.; Harmey, D.; Slice, L.W.; Rozengurt, E.; Lax, A.J. Cytotoxic Necrotizing Factor from Escherichia Coli Induces RhoA-Dependent Expression of the Cyclooxygenase-2 Gene. Infect. Immun. 2001, 69, 6839–6845. [Google Scholar] [CrossRef] [PubMed]
- Mager, D.L. Bacteria and Cancer: Cause, Coincidence or Cure? A Review. J. Transl. Med. 2006, 4, 14. [Google Scholar] [CrossRef]
- Samaras, V.; Rafailidis, P.I.; Mourtzoukou, E.G.; Peppas, G.; Falagas, M.E. Chronic Bacterial and Parasitic Infections and Cancer: A Review. J. Infect. Dev. Ctries. 2010, 4, 267–281. [Google Scholar] [CrossRef]
- Fabbri, A.; Travaglione, S.; Fiorentini, C. Escherichia Coli Cytotoxic Necrotizing Factor 1 (CNF1): Toxin Biology, In Vivo Applications and Therapeutic Potential. Toxins 2010, 2, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Cao, Y.; Greten, F.R.; Li, Z.-W. NF-kappaB in Cancer: From Innocent Bystander to Major Culprit. Nat. Rev. Cancer 2002, 2, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Benelli, R. Aspirin, COX-2, and the Risk of Colorectal Cancer. N. Engl. J. Med. 2007, 357, 824–825. [Google Scholar] [CrossRef] [PubMed]
- Marchesi, J.R.; Dutilh, B.E.; Hall, N.; Peters, W.H.M.; Roelofs, R.; Boleij, A.; Tjalsma, H. Towards the Human Colorectal Cancer Microbiome. PLoS ONE 2011, 6, e20447. [Google Scholar] [CrossRef] [PubMed]
- Voges, M.; Bachmann, V.; Kammerer, R.; Gophna, U.; Hauck, C.R. CEACAM1 Recognition by Bacterial Pathogens Is Species-Specific. BMC Microbiol. 2010, 10, 117. [Google Scholar] [CrossRef] [PubMed]
- Piteau, M.; Papatheodorou, P.; Schwan, C.; Schlosser, A.; Aktories, K.; Schmidt, G. Correction: Lu/BCAM Adhesion Glycoprotein Is a Receptor for Escherichia Coli Cytotoxic Necrotizing Factor 1 (CNF1). PLOS Pathog. 2014, 10, e1003884. [Google Scholar] [CrossRef] [PubMed]
- Tantillo, E.; Colistra, A.; Vannini, E.; Cerri, C.; Pancrazi, L.; Baroncelli, L.; Costa, M.; Caleo, M. Bacterial Toxins and Targeted Brain Therapy: New Insights from Cytotoxic Necrotizing Factor 1 (CNF1). Int. J. Mol. Sci. 2018, 19, 1632. [Google Scholar] [CrossRef]
- Gordon, D.M. Chapter 1—The Ecology of Escherichia Coli. In Escherichia coli, 2nd ed.; Donnenberg, M.S., Ed.; Academic Press: Boston, MA, USA, 2013; pp. 3–20. ISBN 978-0-12-397048-0. [Google Scholar]
- Jinadasa, R.N.; Bloom, S.E.; Weiss, R.S.; Duhamel, G.E. Cytolethal Distending Toxin: A Conserved Bacterial Genotoxin That Blocks Cell Cycle Progression, Leading to Apoptosis of a Broad Range of Mammalian Cell Lineages. Microbiology 2011, 157, 1851–1875. [Google Scholar] [CrossRef] [PubMed]
- Blazkova, H.; Krejcikova, K.; Moudry, P.; Frisan, T.; Hodny, Z.; Bartek, J. Bacterial Intoxication Evokes Cellular Senescence with Persistent DNA Damage and Cytokine Signalling. J. Cell Mol. Med. 2010, 14, 357–367. [Google Scholar] [CrossRef]
- Cortes-Bratti, X.; Karlsson, C.; Lagergård, T.; Thelestam, M.; Frisan, T. The Haemophilus Ducreyi Cytolethal Distending Toxin Induces Cell Cycle Arrest and Apoptosis via the DNA Damage Checkpoint Pathways. J. Biol. Chem. 2001, 276, 5296–5302. [Google Scholar] [CrossRef]
- Bezine, E.; Vignard, J.; Mirey, G. The Cytolethal Distending Toxin Effects on Mammalian Cells: A DNA Damage Perspective. Cells 2014, 3, 592–615. [Google Scholar] [CrossRef] [PubMed]
- Pérès, S.Y.; Marchès, O.; Daigle, F.; Nougayrède, J.P.; Herault, F.; Tasca, C.; De Rycke, J.; Oswald, E. A New Cytolethal Distending Toxin (CDT) from Escherichia coli Producing CNF2 Blocks HeLa Cell Division in G2/M Phase. Mol. Microbiol. 1997, 24, 1095–1107. [Google Scholar] [CrossRef] [PubMed]
- Comayras, C.; Tasca, C.; Pérès, S.Y.; Ducommun, B.; Oswald, E.; De Rycke, J. Escherichia coli Cytolethal Distending Toxin Blocks the HeLa Cell Cycle at the G2/M Transition by Preventing Cdc2 Protein Kinase Dephosphorylation and Activation. Infect. Immun. 1997, 65, 5088–5095. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Sharipo, A.; Chaves-Olarte, E.; Masucci, M.G.; Levitsky, V.; Thelestam, M.; Frisan, T. The Haemophilus Ducreyi Cytolethal Distending Toxin Activates Sensors of DNA Damage and Repair Complexes in Proliferating and Non-Proliferating Cells. Cell Microbiol. 2002, 4, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Hassane, D.C.; Lee, R.B.; Pickett, C.L. Campylobacter Jejuni Cytolethal Distending Toxin Promotes DNA Repair Responses in Normal Human Cells. Infect. Immun. 2003, 71, 541–545. [Google Scholar] [CrossRef]
- Rodier, F.; Coppé, J.-P.; Patil, C.K.; Hoeijmakers, W.A.M.; Muñoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA Damage Signalling Triggers Senescence-Associated Inflammatory Cytokine Secretion. Nat. Cell Biol. 2009, 11, 973–979. [Google Scholar] [CrossRef] [PubMed]
- Alby, F.; Mazars, R.; de Rycke, J.; Guillou, E.; Baldin, V.; Darbon, J.M.; Ducommun, B. Study of the Cytolethal Distending Toxin (CDT)-Activated Cell Cycle Checkpoint. Involvement of the CHK2 Kinase. FEBS Lett. 2001, 491, 261–265. [Google Scholar] [CrossRef]
- De Rycke, J.; Comtet, E.; Chalareng, C.; Boury, M.; Tasca, C.; Milon, A. Enteropathogenic Escherichia coli O103 from Rabbit Elicits Actin Stress Fibers and Focal Adhesions in HeLa Epithelial Cells, Cytopathic Effects That Are Linked to an Analog of the Locus of Enterocyte Effacement. Infect. Immun. 1997, 65, 2555–2563. [Google Scholar] [CrossRef] [PubMed]
- Jubelin, G.; Chavez, C.V.; Taieb, F.; Banfield, M.J.; Samba-Louaka, A.; Nobe, R.; Nougayrède, J.-P.; Zumbihl, R.; Givaudan, A.; Escoubas, J.-M.; et al. Cycle Inhibiting Factors (CIFs) Are a Growing Family of Functional Cyclomodulins Present in Invertebrate and Mammal Bacterial Pathogens. PLoS ONE 2009, 4, e4855. [Google Scholar] [CrossRef]
- Taieb, F.; Nougayrède, J.-P.; Watrin, C.; Samba-Louaka, A.; Oswald, E. Escherichia Coli Cyclomodulin Cif Induces G2 Arrest of the Host Cell Cycle without Activation of the DNA-Damage Checkpoint-Signalling Pathway. Cell. Microbiol. 2006, 8, 1910–1921. [Google Scholar] [CrossRef]
- Wilson, M.R.; Jiang, Y.; Villalta, P.W.; Stornetta, A.; Boudreau, P.D.; Carrá, A.; Brennan, C.A.; Chun, E.; Ngo, L.; Samson, L.D.; et al. The Human Gut Bacterial Genotoxin Colibactin Alkylates DNA. Science 2019, 363, eaar7785. [Google Scholar] [CrossRef] [PubMed]
- Clay, S.L.; Fonseca-Pereira, D.; Garrett, W.S. Colorectal Cancer: The Facts in the Case of the Microbiota. J. Clin. Investig. 2022, 132, e155101. [Google Scholar] [CrossRef]
- Torres, J.P.; Lin, Z.; Winter, J.M.; Krug, P.J.; Schmidt, E.W. Animal Biosynthesis of Complex Polyketides in a Photosynthetic Partnership. Nat. Commun. 2020, 11, 2882. [Google Scholar] [CrossRef]
- Fischbach, M.A.; Walsh, C.T. Assembly-Line Enzymology for Polyketide and Nonribosomal Peptide Antibiotics: Logic, Machinery, and Mechanisms. Chem. Rev. 2006, 106, 3468–3496. [Google Scholar] [CrossRef] [PubMed]
- Cox, R.J.; Simpson, T.J. Fungal Type I Polyketide Synthases. Methods Enzymol. 2009, 459, 49–78. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.J. Engineering Polyketide Synthases and Nonribosomal Peptide Synthetases. Curr. Opin. Struct. Biol. 2013, 23, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Ogata, H.; Goto, S. Type III Polyketide Synthases: Functional Classification and Phylogenomics. ChemBioChem 2016, 18. [Google Scholar] [CrossRef]
- Pfeifer, B.A.; Wang, C.C.C.; Walsh, C.T.; Khosla, C. Biosynthesis of Yersiniabactin, a Complex Polyketide-Nonribosomal Peptide, Using Escherichia Coli as a Heterologous Host. Appl. Environ. Microbiol. 2003, 69, 6698–6702. [Google Scholar] [CrossRef] [PubMed]
- Crosa, J.H.; Walsh, C.T. Genetics and Assembly Line Enzymology of Siderophore Biosynthesis in Bacteria. Microbiol. Mol. Biol. Rev. 2002, 66, 223–249. [Google Scholar] [CrossRef]
- Addington, E.; Sandalli, S.; Roe, A.J. Current Understandings of Colibactin Regulation. Microbiology 2024, 170, 001427. [Google Scholar] [CrossRef]
- Beld, J.; Sonnenschein, E.C.; Vickery, C.R.; Noel, J.P.; Burkart, M.D. The Phosphopantetheinyl Transferases: Catalysis of a Posttranslational Modification Crucial for Life. Nat. Prod. Rep. 2014, 31, 61–108. [Google Scholar] [CrossRef] [PubMed]
- Chagneau, C.V.; Payros, D.; Tang-Fichaux, M.; Auvray, F.; Nougayrède, J.-P.; Oswald, E. The Pks Island: A Bacterial Swiss Army Knife? Colibactin: Beyond DNA Damage and Cancer. Trends Microbiol. 2022, 30, 1146–1159. [Google Scholar] [CrossRef]
- Brachmann, A.O.; Garcie, C.; Wu, V.; Martin, P.; Ueoka, R.; Oswald, E.; Piel, J. Colibactin Biosynthesis and Biological Activity Depend on the Rare Aminomalonyl Polyketide Precursor. Chem. Commun. 2015, 51, 13138–13141. [Google Scholar] [CrossRef]
- Faïs, T.; Delmas, J.; Barnich, N.; Bonnet, R.; Dalmasso, G. Colibactin: More Than a New Bacterial Toxin. Toxins 2018, 10, E151. [Google Scholar] [CrossRef] [PubMed]
- Cloup, E. Etude de L’effet Mutagène Lors D’une Colonisation Néonatale par Une Souche de Escherichia coli Produisant la Colibactine. Exercise Thesis, Ecole Nationale Vétérinaire de Toulouse, Toulouse, France, 2013. [Google Scholar]
- Brotherton, C.A.; Balskus, E.P. A Prodrug Resistance Mechanism Is Involved in Colibactin Biosynthesis and Cytotoxicity. J. Am. Chem. Soc. 2013, 135, 3359–3362. [Google Scholar] [CrossRef]
- Li, Z.-R.; Li, J.; Gu, J.-P.; Lai, J.Y.H.; Duggan, B.M.; Zhang, W.-P.; Li, Z.-L.; Li, Y.-X.; Tong, R.-B.; Xu, Y.; et al. Divergent Biosynthesis Yields a Cytotoxic Aminomalonate-Containing Precolibactin. Nat. Chem. Biol. 2016, 12, 773–775. [Google Scholar] [CrossRef]
- Mousa, J.J.; Yang, Y.; Tomkovich, S.; Shima, A.; Newsome, R.C.; Tripathi, P.; Oswald, E.; Bruner, S.D.; Jobin, C. MATE Transport of the E. Coli-Derived Genotoxin Colibactin. Nat. Microbiol. 2016, 1, 15009. [Google Scholar] [CrossRef] [PubMed]
- Bian, X.; Plaza, A.; Zhang, Y.; Müller, R. Two More Pieces of the Colibactin Genotoxin Puzzle from Escherichia Coli Show Incorporation of an Unusual 1-Aminocyclopropanecarboxylic Acid Moiety. Chem. Sci. 2015, 6, 3154–3160. [Google Scholar] [CrossRef]
- Bossuet-Greif, N.; Dubois, D.; Petit, C.; Tronnet, S.; Martin, P.; Bonnet, R.; Oswald, E.; Nougayrède, J.-P. Escherichia coli ClbS Is a Colibactin Resistance Protein. Mol. Microbiol. 2016, 99, 897–908. [Google Scholar] [CrossRef]
- Arthur, J.C.; Gharaibeh, R.Z.; Mühlbauer, M.; Perez-Chanona, E.; Uronis, J.M.; McCafferty, J.; Fodor, A.A.; Jobin, C. Microbial Genomic Analysis Reveals the Essential Role of Inflammation in Bacteria-Induced Colorectal Cancer. Nat. Commun. 2014, 5, 4724. [Google Scholar] [CrossRef]
- Yang, Y.; Gharaibeh, R.Z.; Newsome, R.C.; Jobin, C. Amending Microbiota by Targeting Intestinal Inflammation with TNF Blockade Attenuates Development of Colorectal Cancer. Nat. Cancer 2020, 1, 723–734. [Google Scholar] [CrossRef] [PubMed]
- Tronnet, S.; Garcie, C.; Rehm, N.; Dobrindt, U.; Oswald, E.; Martin, P. Iron Homeostasis Regulates the Genotoxicity of Escherichia Coli That Produces Colibactin. Infect. Immun. 2016, 84, 3358–3368. [Google Scholar] [CrossRef] [PubMed]
- Tronnet, S.; Garcie, C.; Brachmann, A.O.; Piel, J.; Oswald, E.; Martin, P. High Iron Supply Inhibits the Synthesis of the Genotoxin Colibactin by Pathogenic Escherichia coli through a Non-Canonical Fur/RyhB-Mediated Pathway. Pathog. Dis. 2017, 75, ftx066. [Google Scholar] [CrossRef] [PubMed]
- Chagneau, C.V.; Garcie, C.; Bossuet-Greif, N.; Tronnet, S.; Brachmann, A.O.; Piel, J.; Nougayrède, J.-P.; Martin, P.; Oswald, E. The Polyamine Spermidine Modulates the Production of the Bacterial Genotoxin Colibactin. mSphere 2019, 4, e00414-19. [Google Scholar] [CrossRef] [PubMed]
- Tang-Fichaux, M.; Chagneau, C.; Bossuet-Greif, N.; Nougayrède, J.-P.; Oswald, E.; Branchu, P. The Polyphosphate Kinase of Escherichia coli Is Required for Full Production of the Genotoxin Colibactin. Msphere 2020, 5, 10–1128. [Google Scholar] [CrossRef]
- Oliero, M.; Calvé, A.; Fragoso, G.; Cuisiniere, T.; Hajjar, R.; Dobrindt, U.; Santos, M.M. Oligosaccharides Increase the Genotoxic Effect of Colibactin Produced by Pks+ Escherichia coli Strains. BMC Cancer 2021, 21, 172. [Google Scholar] [CrossRef] [PubMed]
- Dubinsky, V.; Dotan, I.; Gophna, U. Carriage of Colibactin-Producing Bacteria and Colorectal Cancer Risk. Trends Microbiol. 2020, 28, 874–876. [Google Scholar] [CrossRef] [PubMed]
- Dejea, C.M.; Fathi, P.; Craig, J.M.; Boleij, A.; Taddese, R.; Geis, A.L.; Wu, X.; DeStefano Shields, C.E.; Hechenbleikner, E.M.; Huso, D.L.; et al. Patients with Familial Adenomatous Polyposis Harbor Colonic Biofilms Containing Tumorigenic Bacteria. Science 2018, 359, 592–597. [Google Scholar] [CrossRef] [PubMed]
- Pleguezuelos-Manzano, C.; Puschhof, J.; Rosendahl Huber, A.; van Hoeck, A.; Wood, H.M.; Nomburg, J.; Gurjao, C.; Manders, F.; Dalmasso, G.; Stege, P.B.; et al. Mutational Signature in Colorectal Cancer Caused by Genotoxic Pks+ E. coli. Nature 2020, 580, 269–273. [Google Scholar] [CrossRef]
- Lee-Six, H.; Olafsson, S.; Ellis, P.; Osborne, R.J.; Sanders, M.A.; Moore, L.; Georgakopoulos, N.; Torrente, F.; Noorani, A.; Goddard, M.; et al. The Landscape of Somatic Mutation in Normal Colorectal Epithelial Cells. Nature 2019, 574, 532–537. [Google Scholar] [CrossRef]
- Cougnoux, A.; Dalmasso, G.; Martinez, R.; Buc, E.; Delmas, J.; Gibold, L.; Sauvanet, P.; Darcha, C.; Déchelotte, P.; Bonnet, M.; et al. Bacterial Genotoxin Colibactin Promotes Colon Tumour Growth by Inducing a Senescence-Associated Secretory Phenotype. Gut 2014, 63, 1932–1942. [Google Scholar] [CrossRef] [PubMed]
- Boot, A.; Ng, A.W.T.; Chong, F.T.; Ho, S.-C.; Yu, W.; Tan, D.S.W.; Iyer, N.G.; Rozen, S.G. Characterization of Colibactin-Associated Mutational Signature in an Asian Oral Squamous Cell Carcinoma and in Other Mucosal Tumor Types. Genome Res. 2020, 30, 803–813. [Google Scholar] [CrossRef] [PubMed]
- Cuevas-Ramos, G.; Petit, C.R.; Marcq, I.; Boury, M.; Oswald, E.; Nougayrède, J.-P. Escherichia Coli Induces DNA Damage in Vivo and Triggers Genomic Instability in Mammalian Cells. Proc. Natl. Acad. Sci. USA 2010, 107, 11537–11542. [Google Scholar] [CrossRef] [PubMed]
- Cuevas Ramos, G. Effets Génotoxiques Des Souches de Escherichia Coli Produisant La Colibactine. Ph.D. Thesis, Université de Toulouse, Université Toulouse III—Paul Sabatier, Toulouse, France, 2010. [Google Scholar]
- Volpe, M.R.; Velilla, J.A.; Daniel-Ivad, M.; Yao, J.J.; Stornetta, A.; Villalta, P.W.; Huang, H.-C.; Bachovchin, D.A.; Balbo, S.; Gaudet, R.; et al. A Small Molecule Inhibitor Prevents Gut Bacterial Genotoxin Production. Nat. Chem. Biol. 2023, 19, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Harnack, C.; Berger, H.; Liu, L.; Mollenkopf, H.-J.; Strowig, T.; Sigal, M. Short-Term Mucosal Disruption Enables Colibactin-Producing E. Coli to Cause Long-Term Perturbation of Colonic Homeostasis. Gut Microbes 2023, 15, 2233689. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.G.; Jackson, J.G. SASP: Tumor Suppressor or Promoter? Yes! Trends Cancer 2016, 2, 676–687. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Cai, X.; Zhang, J.; Wang, W.; Sheng, Q.; Hua, H.; Zhou, X. Role of Gut Microbiota in the Development and Treatment of Colorectal Cancer. Digestion 2019, 100, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Feringa, F.M.; Raaijmakers, J.A.; Hadders, M.A.; Vaarting, C.; Macurek, L.; Heitink, L.; Krenning, L.; Medema, R.H. Persistent Repair Intermediates Induce Senescence. Nat. Commun. 2018, 9, 3923. [Google Scholar] [CrossRef] [PubMed]
- Yusof, H.; Zulpa, A.K.; Isa, N.S.M.; Ahmad, F.T.; Kassim, M.N.I. Quality Characteristics, Antioxidant and Anticancer Potential of Stingless Bee Honey and Honeybee Honey from Similar Environmental Conditions. IIUM Med. J. Malays. 2021, 20. [Google Scholar] [CrossRef]
- Kim, D.H.; Jin, Y.H. Intestinal Bacterial Beta-Glucuronidase Activity of Patients with Colon Cancer. Arch. Pharm. Res. 2001, 24, 564–567. [Google Scholar] [CrossRef]
- Chen, B.; Ramazzotti, D.; Heide, T.; Spiteri, I.; Fernandez-Mateos, J.; James, C.; Magnani, L.; Graham, T.A.; Sottoriva, A. Contribution of Pks+ E. Coli Mutations to Colorectal Carcinogenesis. Nat. Commun. 2023, 14, 7827. [Google Scholar] [CrossRef] [PubMed]
- Miyasaka, T.; Yamada, T.; Uehara, K.; Sonoda, H.; Matsuda, A.; Shinji, S.; Ohta, R.; Kuriyama, S.; Yokoyama, Y.; Takahashi, G.; et al. Pks-Positive Escherichia Coli in Tumor Tissue and Surrounding Normal Mucosal Tissue of Colorectal Cancer Patients. Cancer Sci. 2024, 115, 1184–1195. [Google Scholar] [CrossRef] [PubMed]
- Wami, H.; Wallenstein, A.; Sauer, D.; Stoll, M.; von Bünau, R.; Oswald, E.; Müller, R.; Dobrindt, U. Insights into Evolution and Coexistence of the Colibactin- and Yersiniabactin Secondary Metabolite Determinants in Enterobacterial Populations. Microb. Genom. 2021, 7, 000577. [Google Scholar] [CrossRef]
- Silpe, J.E.; Wong, J.W.H.; Owen, S.V.; Baym, M.; Balskus, E.P. The Bacterial Toxin Colibactin Triggers Prophage Induction. Nature 2022, 603, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Bossuet, N.; Guyonnet, C.; Chagneau, C.V.; Tang-Fichaux, M.; Penary, M.; Loubet, D.; Branchu, P.; Oswald, E.; Nougayrede, J.-P. Oxygen Concentration Modulates Colibactin Production. Gut Microbes 2023, 15, 2222437. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Kim, C.S.; Healy, A.R.; Wernke, K.M.; Wang, Z.; Frischling, M.C.; Shine, E.E.; Wang, W.; Herzon, S.B.; Crawford, J.M. Structure Elucidation of Colibactin and Its DNA Cross-Links. Science 2019, 365, eaax2685. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Chen, A.; Chen, J.; Sekhon, A.; Louie, G.V.; Noel, J.P.; La Clair, J.J.; Burkart, M.D. Masked Cerulenin Enables a Dual-Site Selective Protein Crosslink. Chem. Sci. 2023, 14, 10925–10933. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Yuan, Y.; Chen, Q.; Nie, S.; Guo, J.; Ou, Z.; Huang, M.; Deng, Z.; Liu, T.; Ma, T. Metabolic Pathway Assembly Using Docking Domains from Type I Cis-AT Polyketide Synthases. Nat. Commun. 2022, 13, 5541. [Google Scholar] [CrossRef]
- Dupont, H.L.; Jiang, Z.-D.; Dupont, A.W.; Utay, N.S. The intestinal microbiome in human health and disease. Trans. Am. Clin. Climatol. Assoc. 2020, 131, 178–197. [Google Scholar]
- Buffie, C.G.; Pamer, E.G. Microbiota-Mediated Colonization Resistance against Intestinal Pathogens. Nat. Rev. Immunol. 2013, 13, 790–801. [Google Scholar] [CrossRef]
- Gogokhia, L.; Buhrke, K.; Bell, R.; Hoffman, B.; Brown, D.G.; Hanke-Gogokhia, C.; Ajami, N.J.; Wong, M.C.; Ghazaryan, A.; Valentine, J.F.; et al. Expansion of Bacteriophages Is Linked to Aggravated Intestinal Inflammation and Colitis. Cell Host Microbe 2019, 25, 285–299.e8. [Google Scholar] [CrossRef] [PubMed]
- Federici, S.; Kredo-Russo, S.; Valdés-Mas, R.; Kviatcovsky, D.; Weinstock, E.; Matiuhin, Y.; Silberberg, Y.; Atarashi, K.; Furuichi, M.; Oka, A.; et al. Targeted Suppression of Human IBD-Associated Gut Microbiota Commensals by Phage Consortia for Treatment of Intestinal Inflammation. Cell 2022, 185, 2879–2898.e24. [Google Scholar] [CrossRef] [PubMed]
- Stintzing, S. Management of Colorectal Cancer. F1000Prime Rep. 2014, 6, 108. [Google Scholar] [CrossRef]
- Geller, L.T.; Barzily-Rokni, M.; Danino, T.; Jonas, O.H.; Shental, N.; Nejman, D.; Gavert, N.; Zwang, Y.; Cooper, Z.A.; Shee, K.; et al. Potential Role of Intratumor Bacteria in Mediating Tumor Resistance to the Chemotherapeutic Drug Gemcitabine. Science 2017, 357, 1156–1160. [Google Scholar] [CrossRef] [PubMed]
- Lopès, A.; Billard, E.; Casse, A.H.; Villéger, R.; Veziant, J.; Roche, G.; Carrier, G.; Sauvanet, P.; Briat, A.; Pagès, F.; et al. Colibactin-Positive Escherichia Coli Induce a Procarcinogenic Immune Environment Leading to Immunotherapy Resistance in Colorectal Cancer. Int. J. Cancer 2020, 146, 3147–3159. [Google Scholar] [CrossRef]
- Xing, Y.; Clark, J.R.; Chang, J.D.; Zulk, J.J.; Chirman, D.M.; Piedra, F.-A.; Vaughan, E.E.; Hernandez Santos, H.J.; Patras, K.A.; Maresso, A.W. Progress toward a Vaccine for Extraintestinal Pathogenic E. Coli (ExPEC) II: Efficacy of a Toxin-Autotransporter Dual Antigen Approach. Infect. Immun. 2024, 92, e0044023. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, M.W.; Jobin, C. Intestinal Bacteria and Colorectal Cancer: Etiology and Treatment. Gut Microbes 2023, 15, 2185028. [Google Scholar] [CrossRef]
- Valle, L. Genetic Predisposition to Colorectal Cancer: Where We Stand and Future Perspectives. World J. Gastroenterol. 2014, 20, 9828–9849. [Google Scholar] [CrossRef]
- Dienstmann, R.; Vermeulen, L.; Guinney, J.; Kopetz, S.; Tejpar, S.; Tabernero, J. Consensus Molecular Subtypes and the Evolution of Precision Medicine in Colorectal Cancer. Nat. Rev. Cancer 2017, 17, 79–92. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Pathotype | Adhesion Site | Virulence Factors | Mechanism/Effect | Disease Manifestations | References |
|---|---|---|---|---|---|
| Enteropathogenic E. coli (EPEC) | Small bowel enterocytes | Lymphostatin (LifA/Efa1)—EAF plasmid-encoding BFP and per locus—Type III secretion system and effectors (Tir, EspF, EspG, EspH, Map)—Enterotoxin (EspC) | Stimulates Cdc42-dependent filopodia formation, disrupts mitochondrial membrane potential | Diarrhea, intestinal inflammation, increased permeability, loss of absorptive surface area | [20,22,24,25] |
| Enterohaemorrhagic E. coli (EHEC) | Colon | Shiga toxin (Stx), Urease, Cif, —LEE pathogenicity island encoding, Type III secretion system and effectors (Tir, EspF, EspG, EspH, Map)—Enterotoxin (EspC), ToxB, Pet, and StcE, Lymphostatin (LifA/Efa1) | Cell lyse, cleaves ribosomal RNA, disrupting protein synthesis and killing cells, inducing renal damage, apoptosis, and local colonic damage | Bloody diarrhea (hemorrhagic colitis), non-bloody diarrhea, hemolytic uremic syndrome (HUS) | [20,22,24,26] |
| Enterotoxigenic E. coli (ETEC) | Small bowel enterocytes | Heat-stable enterotoxin a (STa), Heat-stable enterotoxin b (STb), Heat-labile enterotoxin (LT) | Increases cyclic AMP/GMP concentration, leading to ion secretion, Increases Ca2+ concentration, Colonization is mediated by colonization factors (CFs), and LT acts similarly to cholera toxin (CT) | Induces watery diarrhea | [20,22,27] |
| Enteroaggregative E. coli (EAEC) | Small and large bowel epithelia in a thick biofilm | Aggregative adherence fimbriae (AAFs), dispersin—Enterotoxins: Pic, ShET1, EAST1, Pet—Virulence factors regulated by AggR | Pathogenesis involves colonization of the intestinal mucosa, secretion of enterotoxins (e.g., EAST1), and induction of mucosal damage. | Causes persistent diarrhea in children and adults | [22,24,28,29,30] |
| Enteroinvasive E. coli (EIEC) | Colonic epithelial cells | Type III secretion system—Plasmid-encoded IcsA (outer-membrane protein), SepA (serine protease)—Virulence plasmid-encoded factors, VirA, IpaA, IpaB, IpaC, IpgD, IpaH | It invades epithelial cells, multiplies intracellularly, and induces apoptosis, Actin depolymerization, activation of Cdc42 and Rac | Causes inflammatory colitis similar to Shigella—Invasion, intracellular multiplication, and dissemination, Dysentery, watery diarrhea | [20,22,31,32] |
| Diffusely adherent E. coli (DAEC) | Epithelial cells diffusely | Adhesin: Dr family (F1845)—Cytotoxic necrotizing factor, autotransporter toxin Pet | Induces cytopathic effects and might impair brush-border enzymes | Implicated in diarrhea, particularly in children >12 months, cytopathic effects, impaired brush-border-associated enzymes | [22,24,33,34] |
| Uropathogenic E. coli (UPEC) | Uroepithelium | Specific adhesins: (P, type 1, F1C, S, M, Dr fimbriae) (Pap), — Toxins: Hemolysin, cytotoxic necrotizing factor (CNF-1, -2), Sat, HIyA | Altered cytoskeleton, necrosis, cell lysis | Uncomplicated cystitis, acute pyelonephritis | [20,22,24,34,35,36] |
| Cyclomodulins Produced by E. coli | Host Gene(s) | Mechanism | Reference |
|---|---|---|---|
| Cytotoxic necrotizing factor | cnf1 | Modifies Rho GTPases, locking them in an active state, inducing cell cycle progression, leading to multinucleation and potentially contributing to carcinogenesis | [47] |
| Cytolethal-distending toxin (CDT) | cdtA, cdtB, and cdtC | Cell cycle arrest, DNA double-strand breaks, and chromosomal aberrations, potentially leading to senescence or apoptosis | [51] |
| Intimin-dependent attachment | eae, Type III secretion system | Effector eae downregulates the DNA mismatch repair system, resulting in DNA strand breaks | [49] |
| Cell cycle-inhibiting factor | cif | Irreversible cell cycle arrest at the G2/M transition through activation of a DNA damage-independent signaling pathway, inhibition of the ubiquitin/proteasome pathway, and accumulation of cyclin-dependent kinase inhibitors. | [48] |
| Colibactin | pks locus | Introduction of DNA strand breaks due to reactive cyclopropane group | [16] |
| Gene Name | Function | References |
|---|---|---|
| ClbA | Phosphopantetheinyl transferase for activation of megasynthases | [93] |
| ClbB | Biosynthesis of NRPS/pks hybrids | [94] |
| ClbC | Biosynthesis of pks | [94] |
| ClbD | Hydroxyl-acyl-CoA dehydrogenase | [95] |
| ClbE | Freestanding acyl carrier protein (ACP) | [95] |
| ClbF | α β dehydrogenase (αβdhg) | [96] |
| ClbG | Acyltransferase (AT) | [96] |
| ClbH | Cyclopropane-formatting synthetase | [92] |
| ClbI | Cyclopropane-formatting synthetase | [92] |
| ClbJ | Biosynthesis of NRPS | [96] |
| ClbK | Biosynthesis of NRPS/pks hybrids | [94] |
| ClbL | Amidase (role not fully known) | [96] |
| ClbM | Efflux pump for exporting pre-colibactin | [94] |
| ClbN | Initiates synthesis using asparagine to generate the prodrug motif N-myristoyl-d-asparagine (NMDA) | [92] |
| ClbO | Final enzymatic module of the assembly line and completes the extension stage of the pre-colibactin skeleton | [92] |
| ClbP | Periplasmic peptidase associated with bacterial cytoplasmic membrane | [96] |
| ClbQ | Thioesterase (TE) | [96] |
| ClbR | LuxR-type transcriptional activator | [92] |
| ClbS | Protects colibactin-producing bacterium from genotoxic effects | [96] |
| Type of Effect | Description | References |
|---|---|---|
| Genotoxic | Colibactin alkylates DNA, leading to interstream crosslinks and DNA double-strand breaks, promoting genomic instability and mutations in critical oncogenes or tumor suppressor genes. | [112,116] |
| Genomic Instability | pks+ bacteria impair DNA repair mechanisms, increase mutagenic events, and promote tumor heterogeneity, facilitating tumor evolution and progression. | [117] |
| Inflammation | pks+ bacteria induce chronic inflammation in the colon, creating a pro-inflammatory microenvironment conducive to tumor development and progression. | [114] |
| Disruption of Gut Epithelial Barrier | pks+ bacteria disrupt the gut epithelial barrier, leading to increased permeability of the intestinal mucosa and leakage of bacterial toxins and antigens, contributing to carcinogenesis. | [120] |
| Modulation of Host Immune Responses | pks+ bacteria modulate host immune responses in the colon, promoting an immunosuppressive microenvironment favorable for tumor growth and progression. | [16] |
| Cell Cycle/Senescence | Colibactin-induced DNA damage triggers cell cycle arrest and senescence, affecting tumor cell proliferation and potentially influencing the anti- or pro-tumor role of senescence. | [115,121] |
| β-glucuronidase Activity | β-glucuronidase activity in E. coli releases carcinogenic methylazoxymethanol in the colon, with inhibitors reducing tumor formation in animal studies. | [125] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sadeghi, M.; Mestivier, D.; Sobhani, I. Contribution of pks+ Escherichia coli (E. coli) to Colon Carcinogenesis. Microorganisms 2024, 12, 1111. https://doi.org/10.3390/microorganisms12061111
Sadeghi M, Mestivier D, Sobhani I. Contribution of pks+ Escherichia coli (E. coli) to Colon Carcinogenesis. Microorganisms. 2024; 12(6):1111. https://doi.org/10.3390/microorganisms12061111
Chicago/Turabian StyleSadeghi, Mohammad, Denis Mestivier, and Iradj Sobhani. 2024. "Contribution of pks+ Escherichia coli (E. coli) to Colon Carcinogenesis" Microorganisms 12, no. 6: 1111. https://doi.org/10.3390/microorganisms12061111
APA StyleSadeghi, M., Mestivier, D., & Sobhani, I. (2024). Contribution of pks+ Escherichia coli (E. coli) to Colon Carcinogenesis. Microorganisms, 12(6), 1111. https://doi.org/10.3390/microorganisms12061111