
Bacterial Diversity and Vertical Distribution Patterns in Sandy Sediments: A Study on the Bacterial Community Structure Based on Environmental Factors in Tributaries of the Yangtze River
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Measurement of Physical and Chemical Indicators
2.3. Bacterial Diversity Measurement
2.4. Fluorescence Quantitative qPCR Assay
2.5. Bacterial 16s rRNA Sequencing
2.6. Data Processing and Analysis
3. Results
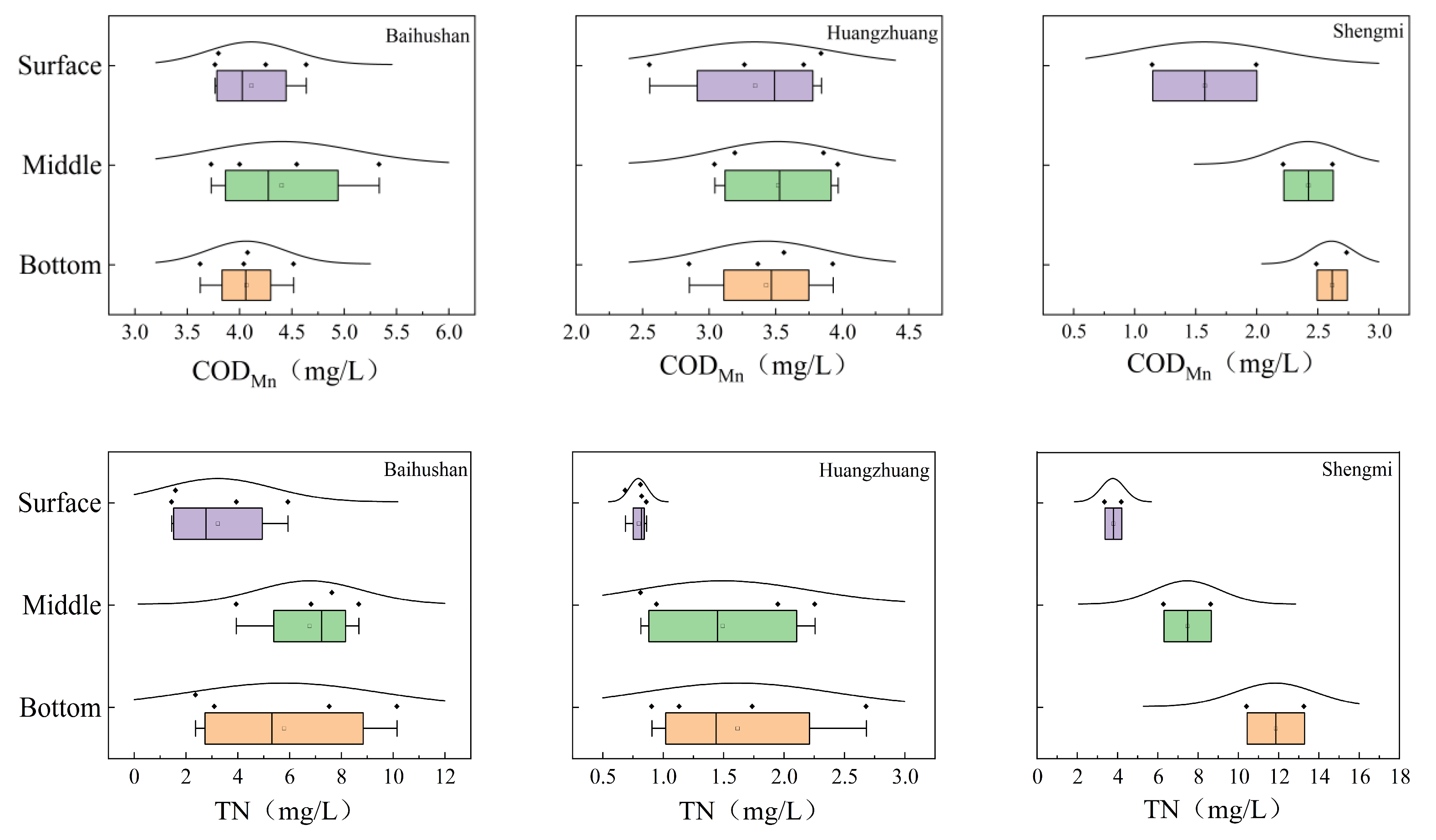
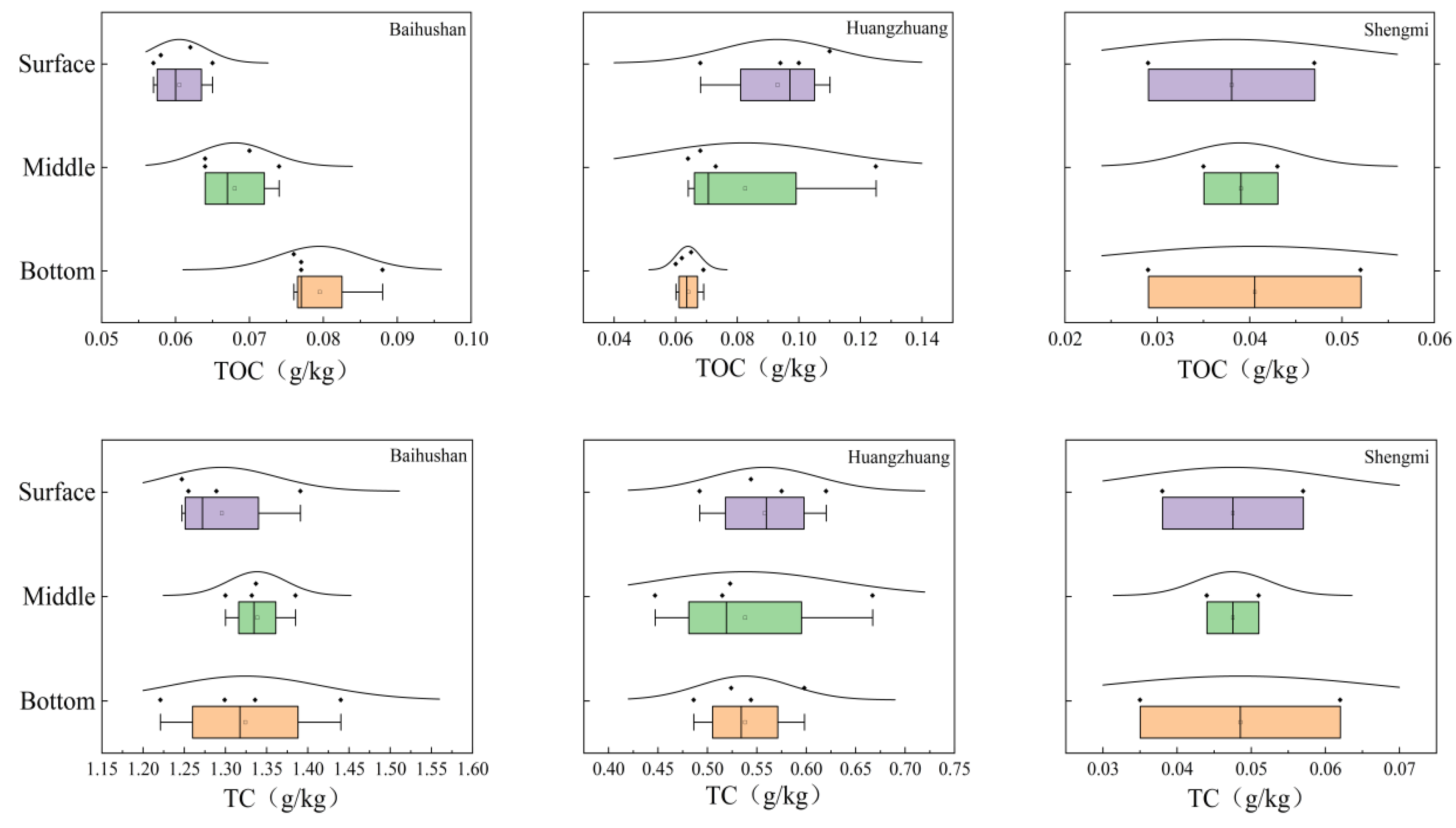
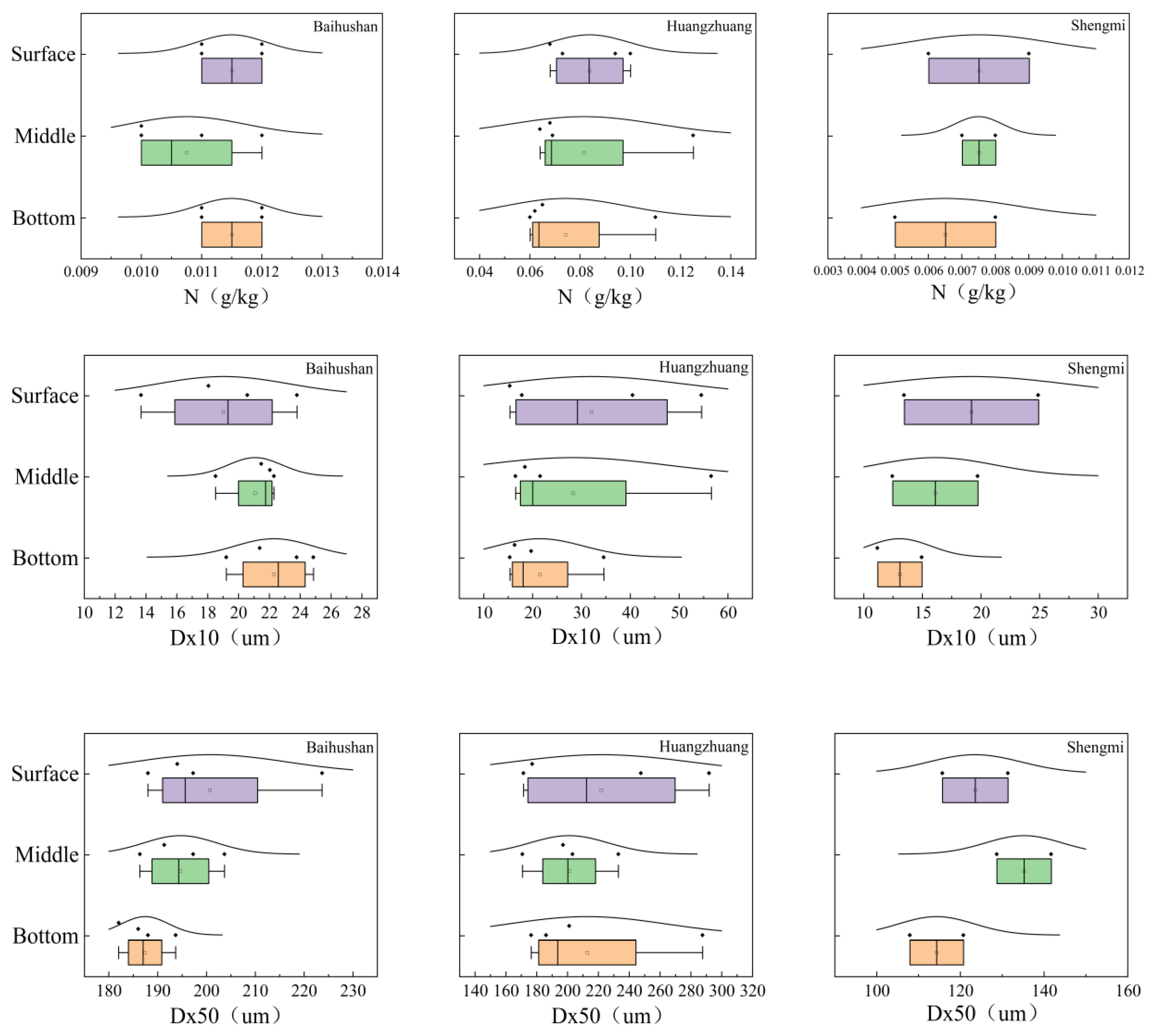
3.1. Changes in Physical and Chemical Indicators at Various Depths
3.2. Variations in Gene Abundance at Different Depths in Bacterial
3.3. Bacterial Community Structure Analysis
3.4. Alpha Diversity Analysis
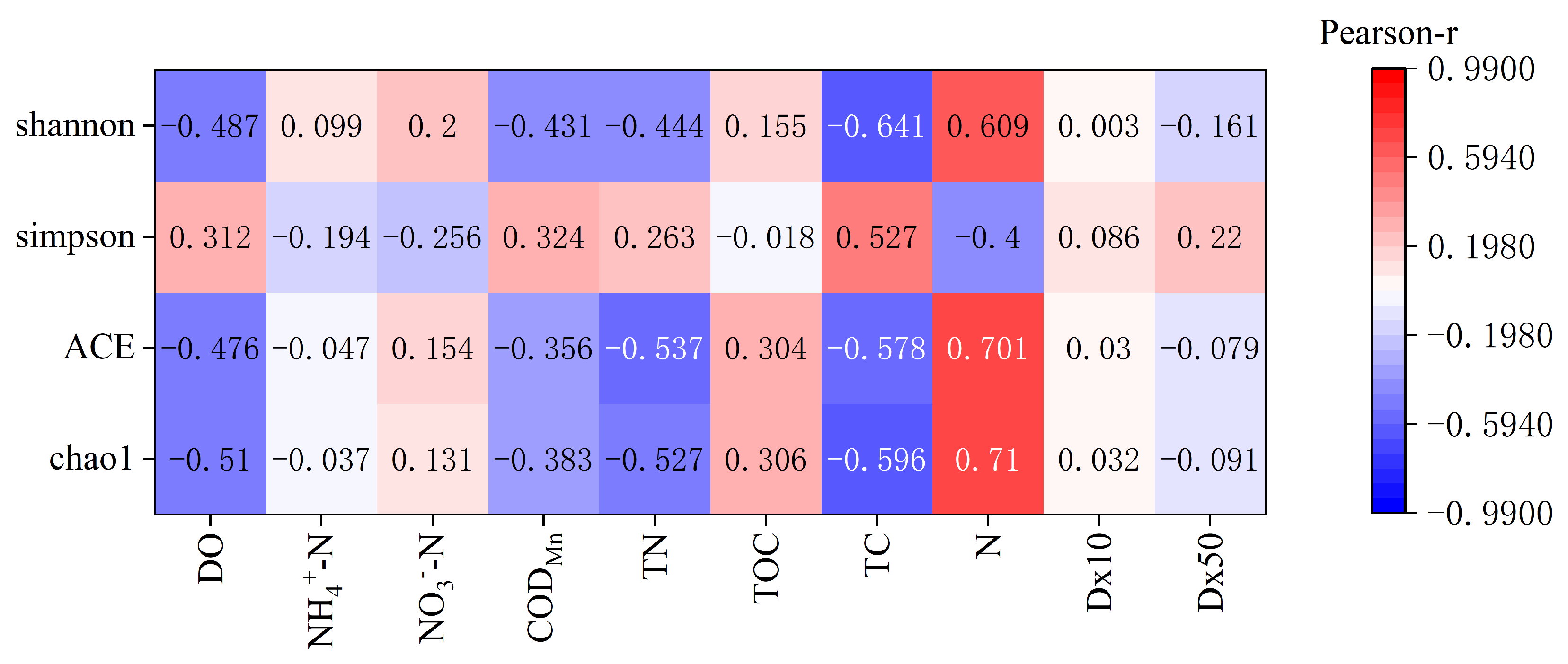
3.5. Effects of Environmental Factors on Bacterial Communities
4. Discussion
4.1. Patterns of Vertical Distribution of Bacterial Diversity
4.2. Bacterial Diversity Impact Factor
5. Conclusions
- (1)
- The abundance and diversity indices of sediment microbes decreased with increasing sediment depth.
- (2)
- The main environmental factors affecting microbial abundance and diversity in the sediments at the sample sites were interstitial water DO, TN concentration and sediment N content.
- (3)
- High oxygen and more nutrient-rich sediment layers provided suitable conditions for Massilia and Flavobacterium to grow and flourish, while Limnobacter preferred low-oxygen or anaerobic conditions.
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Yang, Q.H.; Zhou, H.Y.; Ji, F.W.; Wang, H.; Yang, W.F. Bioturbation in Seabed Sediments and Its Effects on Marine Sedimentary Processes and Records. Adv. Earth Sci. 2008, 9, 932–941. [Google Scholar]
- Kostka, J.E. Controls of the microbial nitrogen cycle in marine sediments and implications for global climate change. Abstr. Pap. Am. Chem. Soc. 2013, 245. [Google Scholar]
- Linn, J.H.; Eike, B.; Philip, W.B.; Keith, A.H. Influence of ocean warming and acidification on trace metal biogeochemistry. Mar. Ecol. Prog. Ser. 2012, 470, 191–205. [Google Scholar]
- Allen, J.G.; Beutel, M.W.; Call, D.R.; Fischer, A.M. Effects of oxygenation on ammonia oxidation potential and microbial diversity in sediment from surface-flow wetland mesocosms. Bioresour. Technol. 2010, 101, 1389–1392. [Google Scholar] [CrossRef] [PubMed]
- Wyness, A.J.; Fortune, I.; Blight, A.J.; Browne, P.; Hartley, M.; Holden, M.; Paterson, D.M. Ecosystem engineers drive differing microbial community composition in intertidal estuarine sediments. PLoS ONE 2021, 16, e0240952. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Li, H.; Wu, H.; Yan, B.; Song, A. Microorganisms in coastal wetland sediments: A review on microbial community structure, functional gene, and environmental potential. Front. Microbiol. 2023, 14, 1163896. [Google Scholar] [CrossRef] [PubMed]
- Hugh, F.; Peter, T.; Gary, A.K. Shifts in composition of microbial communities of subtidal sandy sediments maximise retention of nutrients. FEMS Microbiol. Ecol. 2013, 83, 279–298. [Google Scholar]
- Probandt, D.; Eickhorst, T.; Ellrott, A.; Amann, R.; Knittel, K. Microbial life on a sand grain: From bulk sediment to single grains. ISME J. 2017, 12, 623–633. [Google Scholar] [CrossRef]
- Dighton, J.; White, J.F. The Ecology of Fungi: An Environmental Perspective; CRC Press: Boca Raton, FL, USA, 2017. [Google Scholar]
- Sun, Y.M.; Tang, K.X.; Ma, Y.; Zhu, X.; Li, H.Y.; Zhang, F.; Chen, S.; Huang, H.P. Variations in nutrients and microbes during the occurrence and extinction of algal blooms: A mesocosm experiment with the addition of marine aquaculture sediment. Front. Mar. Sci. 2022, 9, 959161. [Google Scholar] [CrossRef]
- Hong, Y.G.; Wu, J.P.; Wilson, S.; Song, B.K. Vertical Stratification of Sediment Microbial Communities Along Geochemical Gradients of a Subterranean Estuary Located at the Gloucester Beach of Virginia, United States. Front. Microbiol. 2019, 9, 3343. [Google Scholar] [CrossRef]
- Guo, X.P.; Yang, Y.; Niu, Z.S.; Lu, D.P.; Zhu, C.H.; Feng, J.N.; Wu, J.Y. Characteristics of microbial community indicate anthropogenic impact on the sediments along the Yangtze Estuary and its coastal area. Sci. Total Environ. 2019, 648, 306–314. [Google Scholar] [CrossRef]
- Liu, S.; Ren, H.X.; Shen, L.D.; Lou, L.P.; Tian, G.M.; Zheng, P.; Hu, B.L. PH levels drive bacterial community structure in sediments of the Qiantang Riveras determined by 454 pyrosequencing. Front. Microbiol. 2015, 6, 285. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Wang, B.L.; Yang, M.L.; Li, W.Z.; Shi, X.J.; Liu, C.Q. The different responses of planktonic bacteria and archaea to water temperature maintain the stability of their community diversity in dammed rivers. Ecol. Process. 2023, 12, 25. [Google Scholar] [CrossRef]
- Tang, X.Q.; Li, R.; Han, D.; Scholz, M. Response of Eutrophication Development to Variations in Nutrients and Hydrological Regime: A Case Study in the Changjiang River (Yangtze) Basin. Water 2020, 12, 1634. [Google Scholar] [CrossRef]
- State Environmental Protection Administration. Water and Wastewater Monitoring and Analysis Methods; China Environmental Press: Beijing, China, 2002; pp. 258–285. (In Chinese) [Google Scholar]
- Li, S.J.; Xu, C.B.; Qing, S.; Guo, X.; Bai, Y.C.; Guo, F. Molecular characteristics of biochar-derived organic matter sub-fractions extracted by ultrasonication. Sci. Total Environ. 2022, 806, 150190. [Google Scholar] [CrossRef]
- Guan, Y.; Hou, T.; Li, X.; Feng, L.; Wang, Z. Metagenomic insights into comparative study of nitrogen metabolic potential and microbial community between primitive and urban river sediments. Environ. Res. 2022, 212, 113592. [Google Scholar] [CrossRef]
- Li, Y.; Fan, L.H.; Zhang, W.L. How did the bacterial community respond to the level of urbanization along the Yangtze River? Environ. Sci. Process. Impacts 2020, 22, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, J.; Li, H.; Yang, H.; Peng, C.; Peng, Z.; Lu, L. Shift in the microbial community composition of surface water and sediment along an urban river. Sci. Total Environ. 2018, 627, 600–612. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.H.; Yao, P.; Sun, C.; Li, S.Z.; Shi, X.C.; Zhang, X.H.; Liu, J.W. Vertical diversity and association pattern of total, abundant and rare microbial communities in deep-sea sediments. Mol. Ecol. 2021, 30, 2800–2816. [Google Scholar] [CrossRef] [PubMed]
- Wurzbacher, C.; Fuchs, A.; Attermeyer, A.; Frindte, K.; Grossart, H.P.; Hupfer, M.; Casper, P.; Monaghan, M.T. Shifts among Eukaryota, Bacteria, and Archaea define the vertical organization of a lake sediment. Microbiome 2017, 5, 41. [Google Scholar] [CrossRef]
- Shivaji, S.; Kumari, K.; Kishore, K.H.; Pindi, P.K.; Rao, P.S.; Srinivas, T.R.N.; Asthana, R.; Ravindra, R. Vertical distribution of bacteria in a lake sediment from Antarctica by culture-independent and culture-dependent approaches. Res. Microbiol. 2011, 162, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Liu, D.W.; Bai, E. Decreasing soil microbial diversity is associated with decreasing microbial biomass under nitrogen addition. Soil. Biol. Biochem. 2018, 120, 126–133. [Google Scholar] [CrossRef]
- Chernov, T.I.; Tkhakakhova, A.K.; Kutovaya, O.V. Assessment of Diversity Indices for the Characterization of the Soil Prokaryotic Community by Metagenomic Analysis. Eurasian Soil. Sci. 2015, 48, 410–415. [Google Scholar] [CrossRef]
- Louis, B.P.; Maron, P.A.; Menasseri-Aubry, S.; Sarr, A.; Lévêque, A.; Mathieu, O.; Jolivet, C.; Leterme, P.; Viaud, V. Microbial Diversity Indexes Can Explain Soil Carbon Dynamics as a Function of Carbon Source. PLoS ONE 2016, 11, e0161251. [Google Scholar] [CrossRef]
- Wagner, B.D.; Grunwald, G.K.; Zerbe, G.O.; Mikulich-Gilbertson, S.K.; Robertson, C.E.; Zemanick, E.T.; Harris, J.K. On the use of diversity measures in longitudinal sequencing studies of microbial communities. Front. Microbiol. 2018, 9, 01037. [Google Scholar] [CrossRef]
- Ye, Q.; Wu, Y.; Zhu, Z.Y.; Wang, X.N.; Li, Z.Q.; Zhang, J. Bacterial diversity in the surface sediments of the hypoxic zone near the Changjiang Estuary and in the East China Sea. MicrobiologyOpen 2016, 5, 323–339. [Google Scholar] [CrossRef]
- Wilms, R.; Köpke, B.; Sass, H.; Chang, T.S.; Cypionka, H.; Engelen, B.J.E.M. Deep biosphere-related bacteria within the subsurface of tidal flat sediments. Environ. Microbiol. 2006, 8, 709–719. [Google Scholar] [CrossRef] [PubMed]
- Qu, J.H.; Yuan, H.L.; Huang, H.Z.; Wang, E.T. Characteristics of the longitudinal distribution of bacterial communities in the Guanting Reservoir sediment. Sci. China Ser. D-Earth Sci. 2005, 35, 233. (In Chinese) [Google Scholar]
- Liu, T.; Zhang, A.N.; Wang, J.W.; Liu, S.F.; Jiang, X.T.; Dang, C.Y.; Ma, T.; Liu, S.T. Integrated biogeography of planktonic and sedimentary bacterial communities in the Yangtze River. Microbiome 2018, 6, 16. [Google Scholar] [CrossRef]
- Allan, J.D.; Castillo, K.A. Stream Ecology: Structure and Function of Running Waters; Springer Press: Dordrecht, The Netherlands, 2007. [Google Scholar]
- Allan, J.D.; Castillo, M.M.; Capps, K.A. Stream Ecology: Structure and Function of Running Waters; Springer Nature: Berlin/Heidelberg, Germany, 2021. [Google Scholar]
- Liu, W.; Zhang, D.; Wang, S.; Zhao, J.; Yao, H.Y. Risk assessments of emerging contaminants in various waters and changes of microbial diversity in sediments from Yangtze River chemical contiguous zone, Eastern China. Sci. Total Environ. 2022, 803, 149982. [Google Scholar]
- Wang, J.; Shan, S.; Li, D.; Zhang, Z.; Ma, Q. Long-term influence of chloroxylenol on anaerobic microbial community: Performance, microbial interaction, and antibiotic resistance gene behaviors. Sci. Total Environ. 2023, 897, 165330. [Google Scholar] [CrossRef] [PubMed]
- Traving, S.J.; Rowe, O.; Jakobsen, N.M.; Sørensen, H.; Dinasquet, J.; Stedmon, C.A.; Andersson, A.; Riemann, L. The effect of increased loads of dissolved organic matter on estuarine microbial community composition and function. Front. Microbiol. 2017, 8, 351. [Google Scholar] [CrossRef] [PubMed]
- Freixa, A.; Ejarque, E.; Crognale, S.; Amalfitano, S.; Fazi, S.; Butturini, A.; Romanı, A.M. Sediment microbial communities rely on different dissolved organic matter sources along a Mediterranean river continuum. Limnol. Oceanogr. 2016, 61, 1389–1405. [Google Scholar] [CrossRef]
- Wang, K.; Ke, S.Z.; Yuan, H.Z.; Zhu, J.; Li, J.W. Effect of ammonia-nitrogen concentration on bacterial community structure in a MBBR process. Environ. Eng. 2020, 38, 119–125. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Depths | Bacterial |
|---|---|---|
| Baihushan | Surface | Proteobacteria, Bacteroidetes |
| Middle | Proteobacteria, Bacteroidetes | |
| Bottom | Proteobacteria, Bacteroidetes | |
| Huangzhuang | Surface | Proteobacteria, Acidobacteria |
| Middle | Proteobacteria, Acidobacteria | |
| Bottom | Proteobacteria, Acidobacteria | |
| Shengmi | Surface | Proteobacteria, Acidobacteria |
| Middle | Proteobacteria, Acidobacteria | |
| Bottom | Proteobacteria, Acidobacteria |
| Sample | Depths | Bacterial |
|---|---|---|
| Baihushan | Surface | Massilia |
| Middle | Massilia | |
| Bottom | Flavobacterium | |
| Huangzhuang | Surface | Limnobacter |
| Middle | Limnobacter | |
| Bottom | Limnobacter | |
| Shengmi | Surface | Pseudogulbenki |
| Middle | Pseudogulbenki | |
| Bottom | Pseudogulbenki |
| Physicochemical Indicators | Chao1 | ACE | Simpson | Shannon |
|---|---|---|---|---|
| DO | −0.510 ** | −0.476 ** | 0.312 | −0.487 ** |
| NH4+-N | −0.037 | −0.047 | −0.194 | 0.099 |
| NO3−-N | 0.131 | 0.154 | −0.256 | 0.200 |
| CODMn | −0.383 * | −0.356 | 0.324 | −0.431 * |
| TN | −0.527 ** | −0.537 ** | 0.263 | −0.444 * |
| TOC | 0.306 | 0.304 | −0.018 | 0.155 |
| TC | −0.596 ** | −0.578 ** | 0.527 ** | −0.641 ** |
| N | 0.710 ** | 0.701 ** | −0.400 * | 0.609 ** |
| Dx10 | 0.032 | 0.030 | 0.086 | 0.003 |
| Dx50 | −0.091 | −0.079 | 0.220 | −0.161 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, T.; Wang, W.; Leng, Y.; Huang, Y.; Xiong, W.; Chang, F. Bacterial Diversity and Vertical Distribution Patterns in Sandy Sediments: A Study on the Bacterial Community Structure Based on Environmental Factors in Tributaries of the Yangtze River. Microorganisms 2024, 12, 1178. https://doi.org/10.3390/microorganisms12061178
Zhang T, Wang W, Leng Y, Huang Y, Xiong W, Chang F. Bacterial Diversity and Vertical Distribution Patterns in Sandy Sediments: A Study on the Bacterial Community Structure Based on Environmental Factors in Tributaries of the Yangtze River. Microorganisms. 2024; 12(6):1178. https://doi.org/10.3390/microorganisms12061178
Chicago/Turabian StyleZhang, Tian, Weibo Wang, Yifei Leng, Yu Huang, Wen Xiong, and Fengyi Chang. 2024. "Bacterial Diversity and Vertical Distribution Patterns in Sandy Sediments: A Study on the Bacterial Community Structure Based on Environmental Factors in Tributaries of the Yangtze River" Microorganisms 12, no. 6: 1178. https://doi.org/10.3390/microorganisms12061178
APA StyleZhang, T., Wang, W., Leng, Y., Huang, Y., Xiong, W., & Chang, F. (2024). Bacterial Diversity and Vertical Distribution Patterns in Sandy Sediments: A Study on the Bacterial Community Structure Based on Environmental Factors in Tributaries of the Yangtze River. Microorganisms, 12(6), 1178. https://doi.org/10.3390/microorganisms12061178