Abstract
The metagenomic surveillance of antimicrobial resistance in wastewater has been suggested as a methodological tool to characterize the distribution, status, and trends of antibiotic-resistant bacteria. In this study, a cross-sectional collection of samples of hospital-associated raw and treated wastewater were obtained from February to March 2020. Shotgun metagenomic sequencing and bioinformatic analysis were performed to characterize bacterial abundance and antimicrobial resistance gene analysis. The main bacterial phyla found in all the samples were as follows: Proteobacteria, Bacteroides, Firmicutes, and Actinobacteria. At the species level, ESKAPEE bacteria such as E. coli relative abundance decreased between raw and treated wastewater, but S. aureus, A. baumannii, and P. aeruginosa increased, as did the persistence of K. pneumoniae in both raw and treated wastewater. A total of 172 different ARGs were detected; blaOXA, blaVEB, blaKPC, blaGES, mphE, mef, erm, msrE, AAC(6′), ant(3″), aadS, lnu, PBP-2, dfrA, vanA-G, tet, and sul were found at the highest abundance and persistence. This study demonstrates the ability of ESKAPEE bacteria to survive tertiary treatment processes of hospital wastewater, as well as the persistence of clinically important antimicrobial resistance genes that are spreading in the environment.
1. Introduction
The epidemiological surveillance of antimicrobial resistance (AMR) has been suggested as an essential methodological tool to observe the distribution, status, and trends of antibiotic resistant bacteria [1]. The surveillance of clinically important bacteria in the wastewater environment has gained relevance in hospital settings, community settings, and animal production, mainly because various components such as biomarkers of the microorganisms present, detergents, trace antibiotics, and other substances converge in wastewater systems [2]. Hospital wastewaters are considered the main environment that may contain clinically important antimicrobial-resistant pathogens due to selection pressure for the horizontal gene transfer of AMR [2,3,4].
The detection of bacterial communities, their resistance genes (ARGs), and/or mobile genetic elements (MGEs) in hospital wastewater systems can serve as an early warning tool for the potential spread of AMR to the environment [2,5,6,7].
Among the list of critically important bacteria described by the World Health Organization (WHO) [8] are the ESKAPEE group: Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, Enterobacter cloacae, and Escherichia coli. ESKAPEE bacteria are rapidly acquiring resistance to antibiotics, so their dissemination in the environment through hospital wastewater may pose a serious problem [9,10,11]. Wastewater treatment plants (WWTPs) play a key role in decreasing bacterial abundance because they have been well-described, as well as hotspots that favor AMR through the transfer of genetic material [12,13].
Although WWTPs reduce the abundance of bacteria released into the environment, current wastewater treatment processes do not remove all bacteria, antibiotic-resistant bacteria with their ARGs, and antibiotic residues. Some studies have demonstrated the importance of ESKAPEE group bacteria in hospital, community, or livestock wastewater as important sources of environmental epidemic pathogens [14]. Because hospital wastewater contributes to the burden of multidrug resistance, WWTP may facilitate the presence, persistence, and evolution of ESKAPEE group bacteria [15,16].
The use of metagenomic studies in specific environments such as wastewater has provided insight into the profile of bacterial communities through phylogenetic analyses and their resistome [17,18], which has provided advantages over conventional microbiology based on bacterial cultures with some selection biases. Furthermore, metagenomic studies of AMR bacteria have contributed significantly to existing antimicrobial resistance databases [19,20,21].
The AMR surveillance of ESKAPEE bacteria globally in the hospital wastewater setting is in line with the WHO Global Action Plan on Antimicrobial Resistance with the aim of “strengthening the science and knowledge base through surveillance and research” [22]. Therefore, the aim of this study was to describe the presence and persistence of ESKAPEE group bacteria and antimicrobial resistance genes in raw and treated wastewater from two tertiary-level hospitals in Mexico.
2. Materials and Methods
2.1. Study Design and Sample Collection
This descriptive cross-sectional study was conducted from February to March 2020. Three samples of raw (n = 3) and treated (n = 3) wastewater were collected in the WWTP of the Hospital Regional de Alta Especialidad de Ixtapaluca (HRAEI) with a 19.19116 N latitude and 98.51187 W longitude, and two samples of raw (n = 2) and treated (n = 2) wastewater were obtained from the WWTP of the Instituto Nacional de Cancerología (INCAN) with a 19.17128 N latitude and 99.06314 W longitude, both in central Mexico. One liter of each wastewater sample (raw and treated) was collected using the single grab technique in sterile containers, with an interval of one week between each sample collection. The samples were transported to the laboratory at 4 °C within less than two hours of collection [9,10].
2.2. Wastewater Treatment Plant Characteristics
The layout of both WWTPs begins with a pretreatment process using a grate that eliminates large materials. Then, the water flow is dosed to an aeration process mediated by regulating valves in different chambers. The treatment process continues with a compound sedimentation phase, precipitating the sludge. Tertiary treatment includes calcium hypochlorite tablets, granular activated carbon filters, and ultraviolet light (UVL) in the effluent.
2.3. Sample Processing
Aliquots of 100 mL of raw wastewater and 200 mL of treated wastewater were obtained and centrifuged at 5000× g for 20 min at 4 °C. The supernatant was decanted and one milliliter of EC lysis solution (1M Tris pH 8.0, EDTA, sodium deoxycholate, N-lauryl sarcosyl, RNAase, lysozyme and lysostaphin) was added to the pellet and incubated at 37 °C for four hours. Then, the ESP solution (EDTA + N-lauryl sarcosyl + proteinase K) was added and incubated at 50 °C overnight [23]. The samples were purified with the Wizard® Genomic DNA Purification Kit (PROMEGA Corp., Madison, WI, USA) according to the manufacturer’s instructions. DNA was quantified by fluorometry using the Qubit 4 Fluorometer (Thermo Fisher Scientific, Waltham, MA, USA). The DNA samples were stored at −20 °C until sequencing.
2.4. Sequencing and Bioinformatics Analysis
The ten samples were sequenced by Illumina HiSeq (Illumina, Inc., San Diego, CA, USA), with 2 × 150 configuration (21.4 GB). Libraries were performed using the Nextera DNA protocol. The reads are available from NCBI SRA under BioProject ID PRJNA1010860.
Adapters were removed for raw reads and a Q > 20 was considered with Trimmomatic [24]. Quality control statistics were performed with FastQC [25]. Metagenome assemblies were performed with IDBA-UD (v1.1) [26,27]. Mapping statistics were performed with Bowtie 2 [28] and can be reviewed in Supplementary Material Table S1. For functional annotation, the Trinotate (v3.0.1) pipeline was employed [29,30]. Gene abundance was estimated as coverage by mapping reads to contigs using BWA (v0.7.12-r1039) and the coverBed function of bedtools (v2.25.0) [31,32].
MetaPhlAn (v4.0) was used for taxonomic profiling [33,34]. Differential abundance analysis between conditions at the species level was performed with the MetagenomeSeq R library [35]. Alpha and beta diversity analyses were performed with the vegan (v2.4-6) and phyloseq libraries in [36,37]. The distance matrix for beta diversity was performed with the Bray–Curtis index at the species level. Comparisons between groups were determined with an analysis of similarity (ANOSIM) [37,38]. The base 2 logarithm of the relationship between the two expression values was taken: LFC = log2 (A/B), where A and B represent the relative abundance levels of each genus in different wastewater conditions. Abundance plots and histograms were performed with the ggplot2 and ggpubr library of R [36,39,40].
2.5. Analysis of Antimicrobial Resistance Genes
The ARGs and resistome of the samples were determined using contigs of lengths of >150 bp using ABRicate (v1.0.1) [41], with the Comprehensive Antibiotic Resistance Database (CARD) (v3.2.9) [42] and PlasmidFinder (v2.0) [43]. The identified ARGs were grouped into classes according to the type of antibiotic to which they confer resistance, and quantified by absolute abundance, relative abundance, and normalized abundance expressed as a percentage. The virulence factor database (VFDB) (as of October 2023) and the E. coli O-groups and H-types database (EcOH) (as of October 2023) were used for virulence gene detection [44,45].
3. Results
3.1. Bacterial Composition of the Wastewater
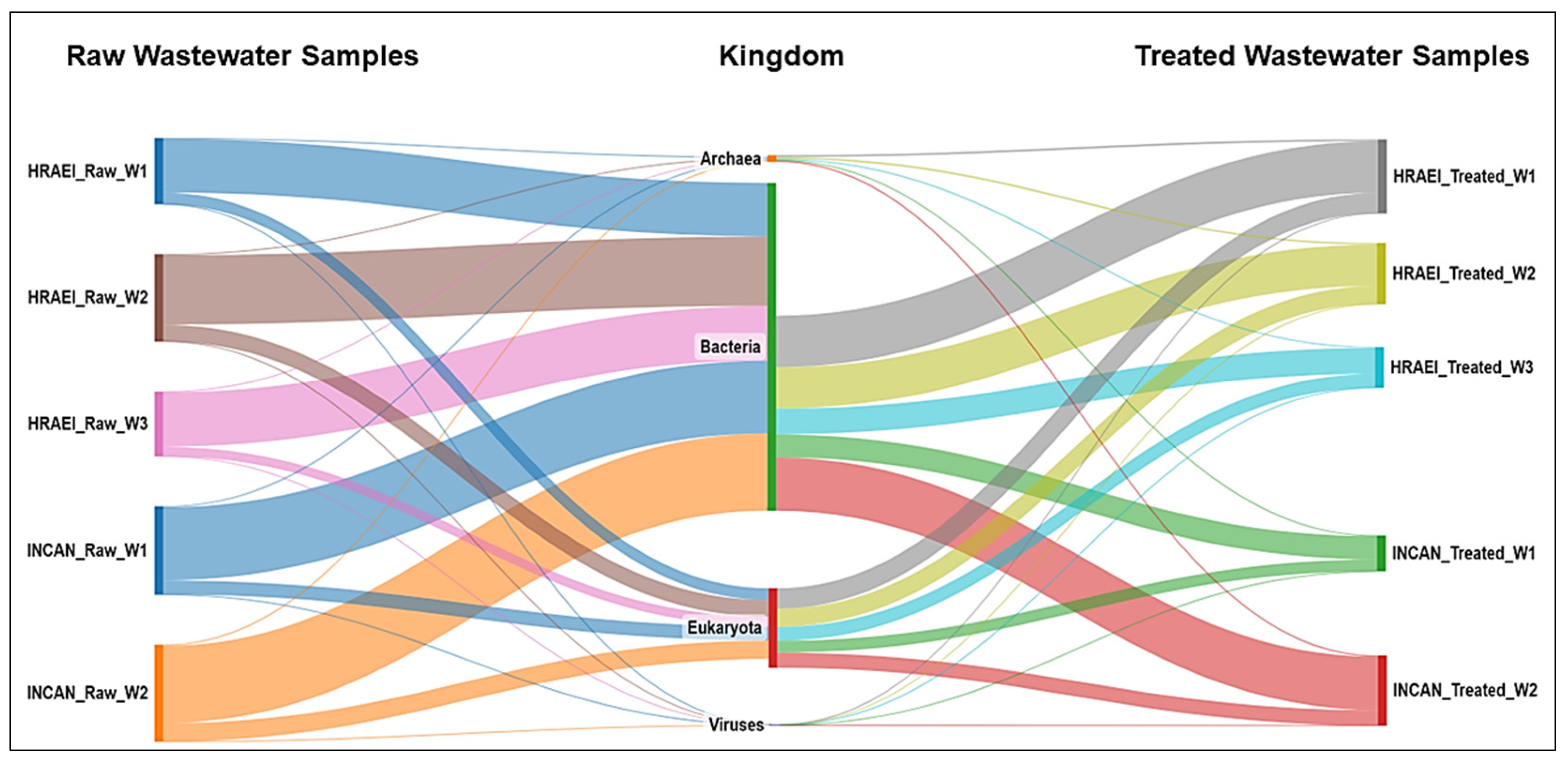
The average abundance relative to the bacterial Kingdom was 81.2 ± 1.9% in the raw wastewater, while in the treated wastewater, it was 67.8 ± 5.1% for both hospitals (Figure 1), which represented an average decrease of 12.8% (14.7% for HRAEI and 11.1% for INCAN).
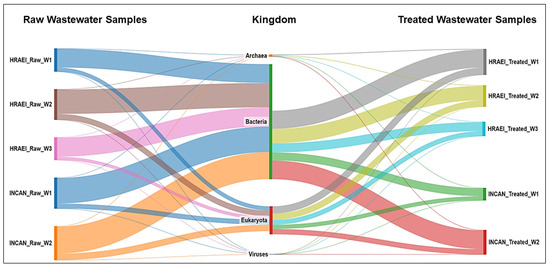
Figure 1.
Bacterial community composition in hospital wastewater samples. Taxonomic annotation at the kingdom level.
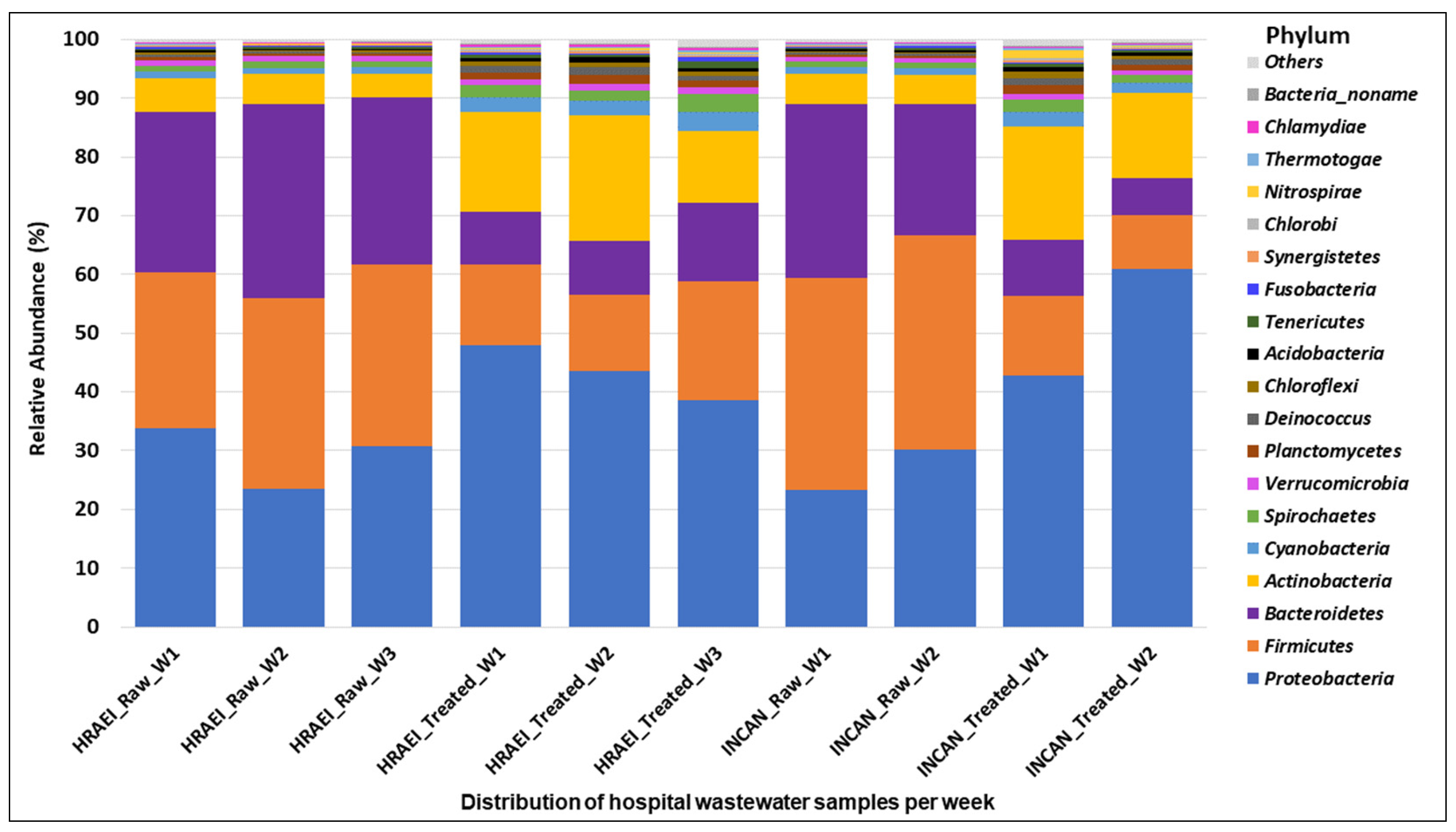
The raw and treated wastewater samples presented a homogeneous pattern, finding 60 classes in 36 phyla of the bacterial kingdom. The main bacterial phyla found in raw wastewater samples above 1% relative abundance were Firmicutes (32 ± 4.1%), Proteobacteria (28 ± 4.7%), Bacteroidetes (28 ± 3.9%), and Actinobacteria (5 ± 0.6%), while in the treated wastewater samples, they were Proteobacteria (46 ± 8.5%), Firmicutes (13 ± 3.9%), Bacteroidetes (9 ± 2.5%), and Actinobacteria (16 ± 3.6%) (Figure 2).
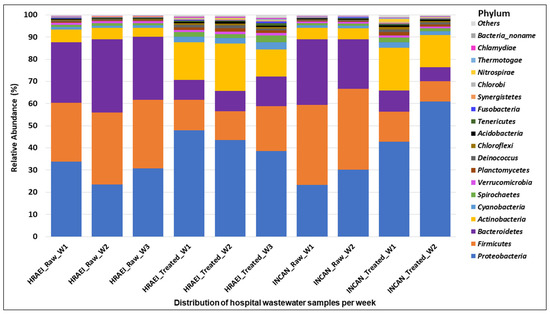
Figure 2.
Bacterial community composition in hospital wastewater samples. Relative abundance at the phylum level.
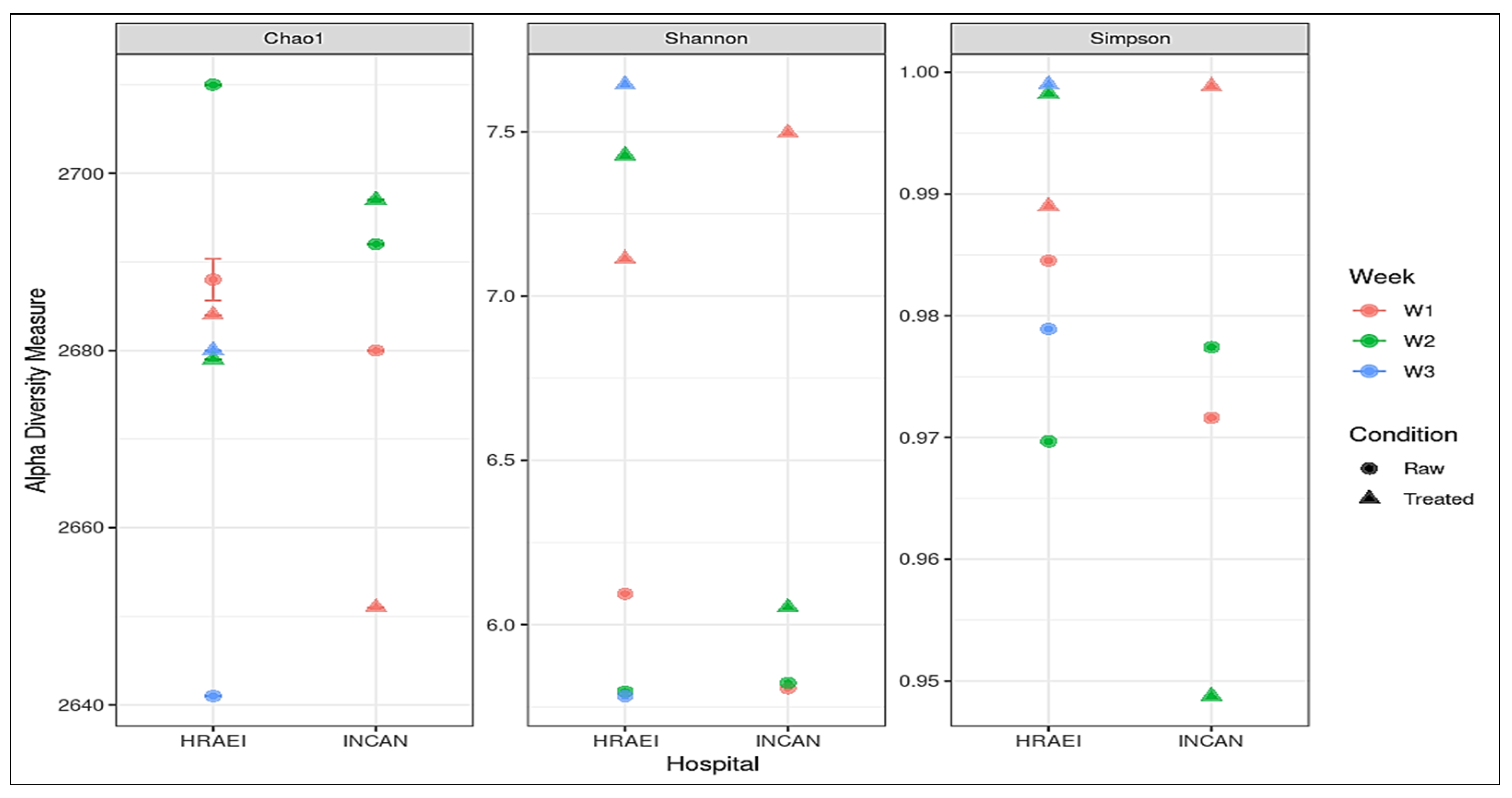
From the taxonomic-level species annotations, alpha diversity was determined with a p-value < 0.05 using Shannon’s index, where a greater richness of bacterial species was observed in the treated wastewater samples. The average observed richness of the wastewater samples was 2680 ± 20 unique species, and the average Chao1 index was 2680 ± 20. The bacterial diversity for all the samples was calculated using the Shannon index value, where we found values within the range of 5.78 to 7.64. The most diverse bacterial communities were those of the raw compared to treated wastewater samples, with species dominance (Simpson’s alpha diversity index), while the Chao1 diversity index showed similar richness between treatments with no significant differences detected (Figure 3).
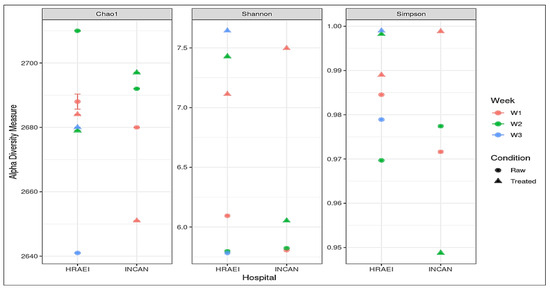
Figure 3.
Alpha diversity indexes of each treatment at the species level, Chao1 diversity index, Shannon’s diversity index, and Simpson diversity index.
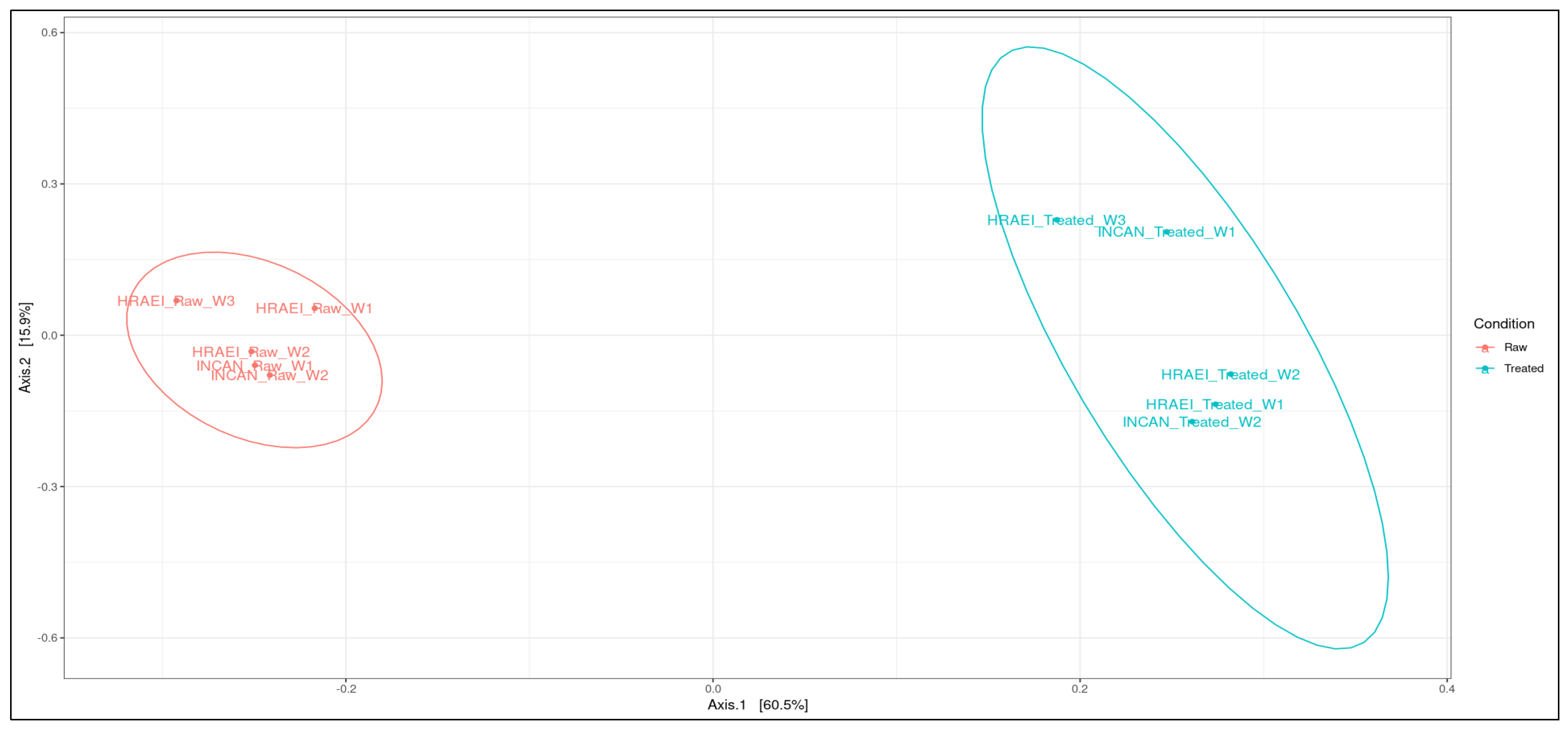
Principal coordinate analysis (PCoA) was performed at the species level to determine the type of wastewater condition (raw and treated), the relationship between samples at the species level, and the time of sampling. In this analysis, it was observed that the wastewater samples from both hospitals were grouped according to wastewater condition (raw and treated), suggesting compositional differences between the two types of waters analyzed. Between sampling time intervals in treated wastewater, the differences in bacterial species composition were observed (p-value < 0.05). In the comparison of the wastewater samples, principal components 1 and 2 explained 76.4% of the variation after the processes in the WWTPs (Figure 4).
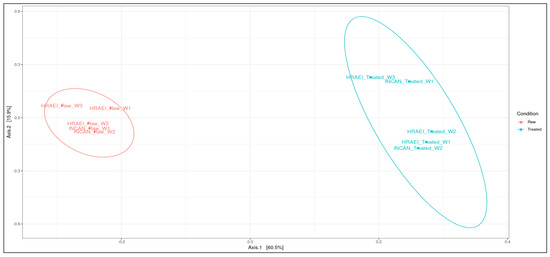
Figure 4.
Analysis of similarity (ANOSIM). Principal coordinate analysis between treatments using the Bray–Curtis distance matrix at the species level.
3.2. Differential Abundance of ESKAPEE Group Genera in Wastewater
The presence of 1004 bacterial genera was detected in the wastewater samples analyzed. The results of the average relative abundances for the bacterial genera of the ESKAPEE group from both hospitals were as follows: Enterococcus spp. 0.4 ± 0.1% in the raw wastewater and 0.5 ± 0.2% in the treated wastewater, Staphylococcus spp. 0.2 ± 0.1% and 0.3 ± 0.2%, respectively, Klebsiella spp. 0.4 ± 0.1% in the raw wastewater and 0.2 + 0.1% in the treated wastewater, Acinetobacter spp. 1.0 ± 0.5% and 1.9 ± 2.9%, respectively, Pseudomonas spp. 1.1 ± 0.2% in the raw wastewater and 7.1 ± 9.3 in the treated wastewater, Enterobacter spp. 0.3 ± 0.1% in the raw wastewater and 0.2 ± 0.1% in the treated wastewater, and Escherichia spp. 1.3 ± 0.0% and 0.2 ± 0.1%, respectively.
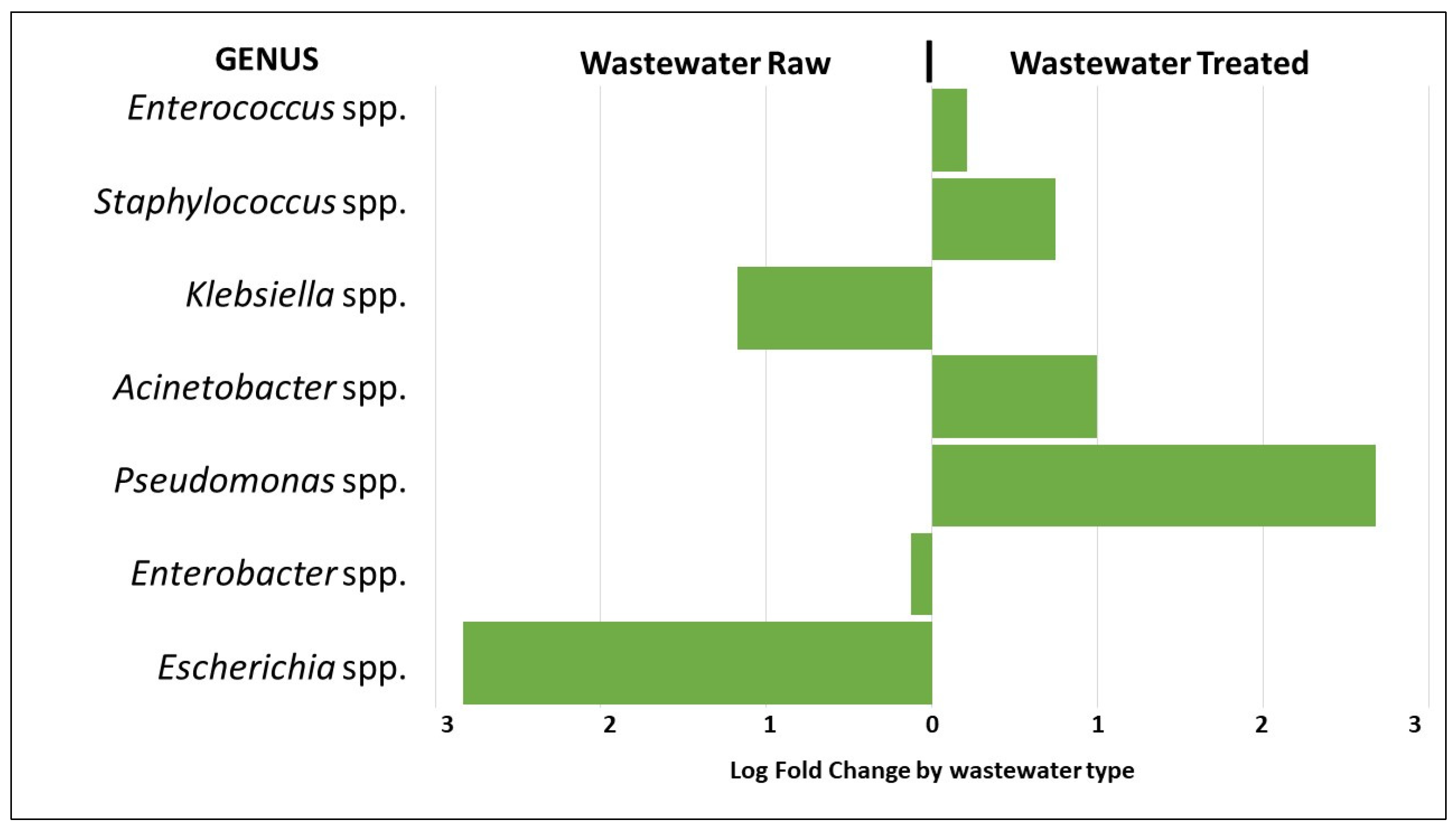
The analysis of differential abundance of bacterial genera belonging to the ESKAPEE group showed approximately a three-fold greater abundance of Pseudomonas spp. in the treated wastewater compared to the raw wastewater. Staphylococcus spp. and Acinetobacter spp. showed approximately a one-fold greater abundance, respectively. On the other hand, the abundance of Escherichia spp. was approximately three-fold greater in the raw wastewater compared to the treated, and for Klebsiella spp., the abundance was approximately one-fold greater, respectively (p < 0.01) (Figure 5).
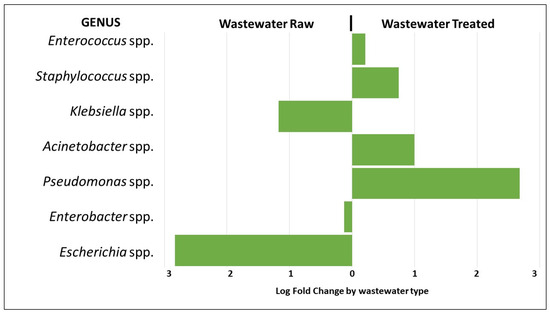
Figure 5.
Analysis of the differential abundance of the genera of the ESKAPEE group by wastewater type.
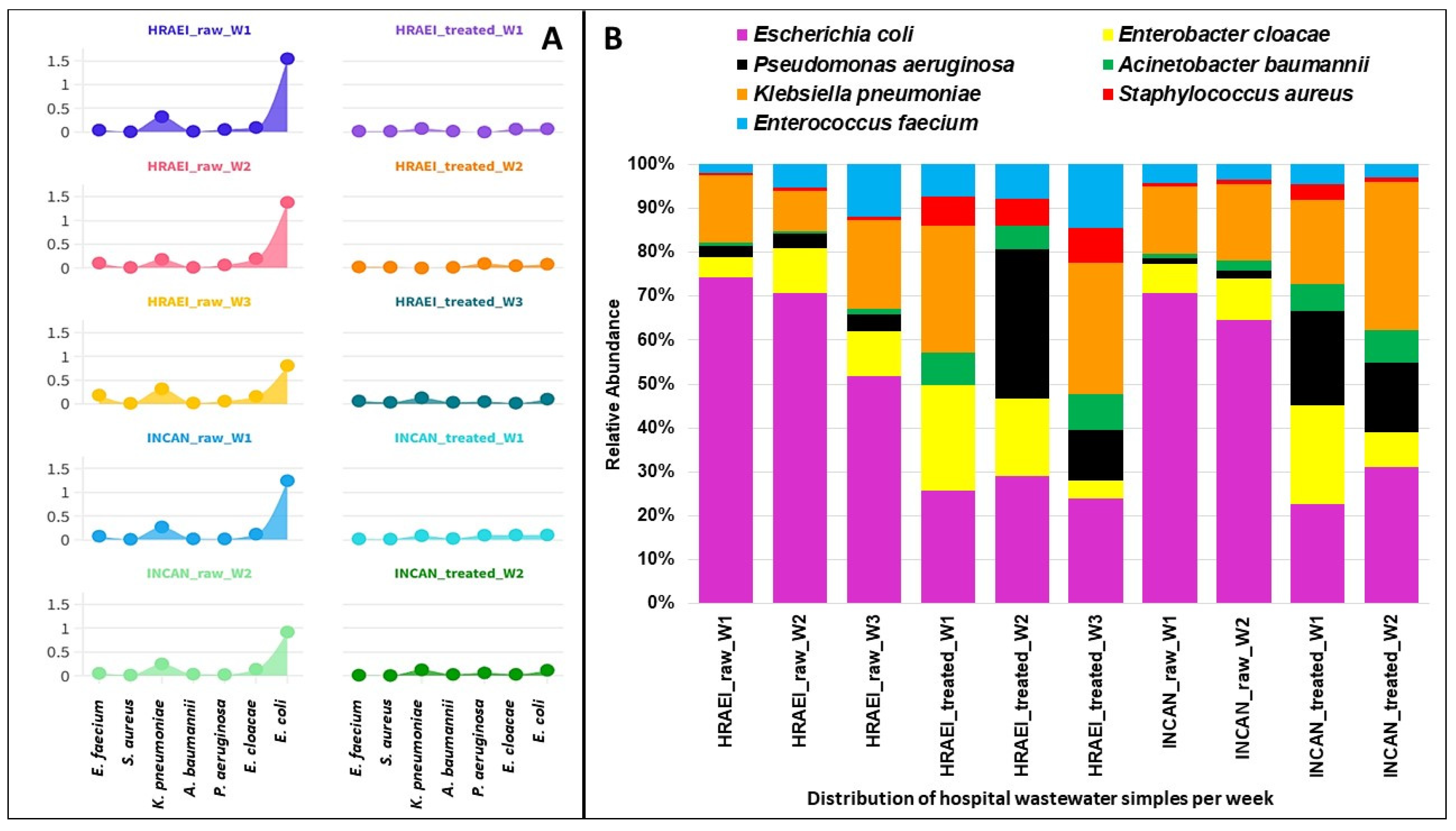
The presence of 2837 bacterial species was detected in the wastewater samples analyzed. The results of the average relative abundances for the bacterial species of the ESKAPEE group from both hospitals were as follows: Enterococcus faecium 0.09 ± 0.05% in the raw wastewater and 0.02 ± 0.01% in the treated wastewater, Staphylococcus aureus 0.01 ± 0.01% and 0.01 ± 0.01%, respectively, Klebsiella pneumoniae 0.26 ± 0.05% in the raw wastewater and 0.08 ± 0.05% in the treated wastewater, Acinetobacter baumannii 0.02 ± 0.01% and 0.02 ± 0.01%, respectively, Pseudomonas aeruginosa 0.04 ± 0.02% in the raw wastewater and 0.05 ± 0.03% in the treated wastewater, Enterobacter cloacae 0.14 ± 0.03% in the raw wastewater and 0.05 ± 0.03% in the treated wastewater, and Escherichia coli 1.17 ± 0.31% and 0.09 ± 0.01%, respectively. A decrease in the relative abundance of E. coli was observed between raw and treated wastewater, while for S. aureus, A. baumannii, and P. aeruginosa, this abundance increased (p < 0.05). For K. pneumoniae, this abundance was similar under both conditions (Figure 6).
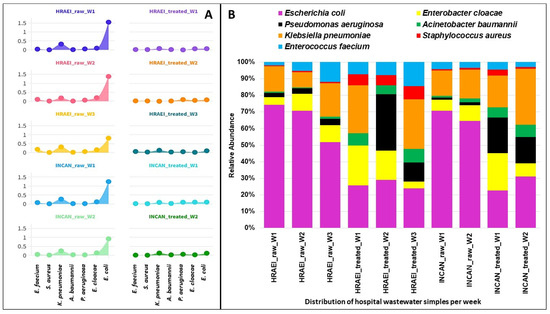
Figure 6.
Relative abundance of ESKAPEE group bacteria at the species level. (A) Relative abundance (log) in each hospital wastewater sample before and after the wastewater treatment plant. (B) Relative abundance of ESKAPEE group bacteria.
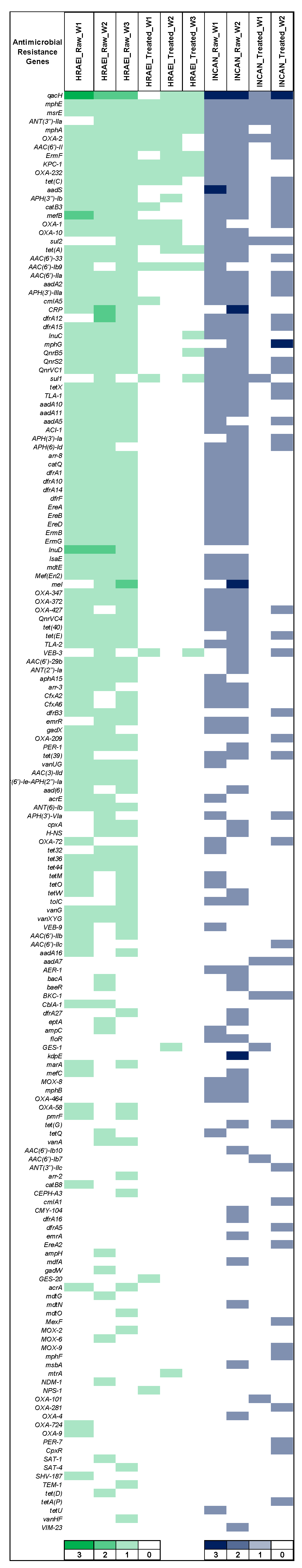
The CARD database was used to determine the presence and identity of ARGs in each sample at various times, for the comparative analysis of the presence, relative abundance, and persistence of ARGs (CARD database). ARGs (172) were detected in both hospitals’ raw and treated wastewater samples. In raw wastewater samples from HRAEI, 102 + 2.8 ARGs and 17.3 + 2.8 ARGs were detected in treated wastewater. On the contrary, in the INCAN raw wastewater samples, 92 + 9.8 ARGs and 32 + 28.2 ARGs were identified in the treated wastewater. The antimicrobial resistance genes blaOXA, blaVEB, blaKPC, blaGES, mphE, mef, erm, msrE, AAC(6′), ant(3″), aadS, lnu, PBP-2, dfrA, vanA-G, tet, and sul had the highest abundance and persistence. These genes encode resistance for β-lactam, macrolides, tetracyclines, sulfonamides, aminoglycosides, lincosamide, and glycopeptides. Other ARGs such as blaNDM, blaTEM, and acrA genes were found at a lower abundance (Figure 7) (Supplementary Material Table S2).
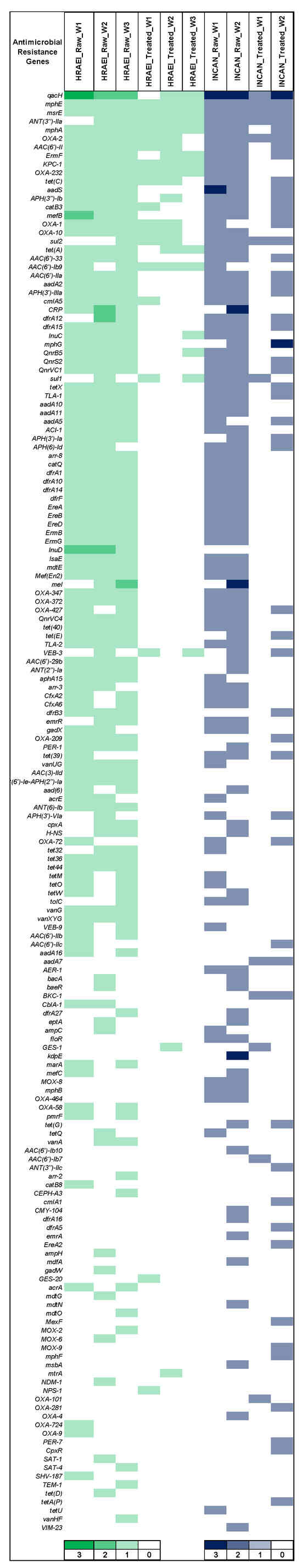
Figure 7.
Relative abundance of antibiotic resistance genes by CARD. Green: HRAEI. Blue: INCAN.
PlasmidFinder analysis of raw and treated wastewater samples detected the persistence of plasmids ColKP3-1 carrying the ARGs blaOXA-131 and blaOXA-232; IncQ2_1 carrying the ARGs sul2, strAB, and tetA; ColRNAI_1 carrying fosA; and Col440I_1 and Col440II_1 carrying the ARGs qnrB, cmlA1 and fosA7, mainly Enterobacterial. The main virulence factor genes persistent in the raw and treated wastewater samples were icmJ associated with endonucleases from Pseudomonas spp., htpB associated with adhesion in different enterobacteria, and fliG associated with adhesion and motility in several enterobacteria (Supplementary Material Table S2).
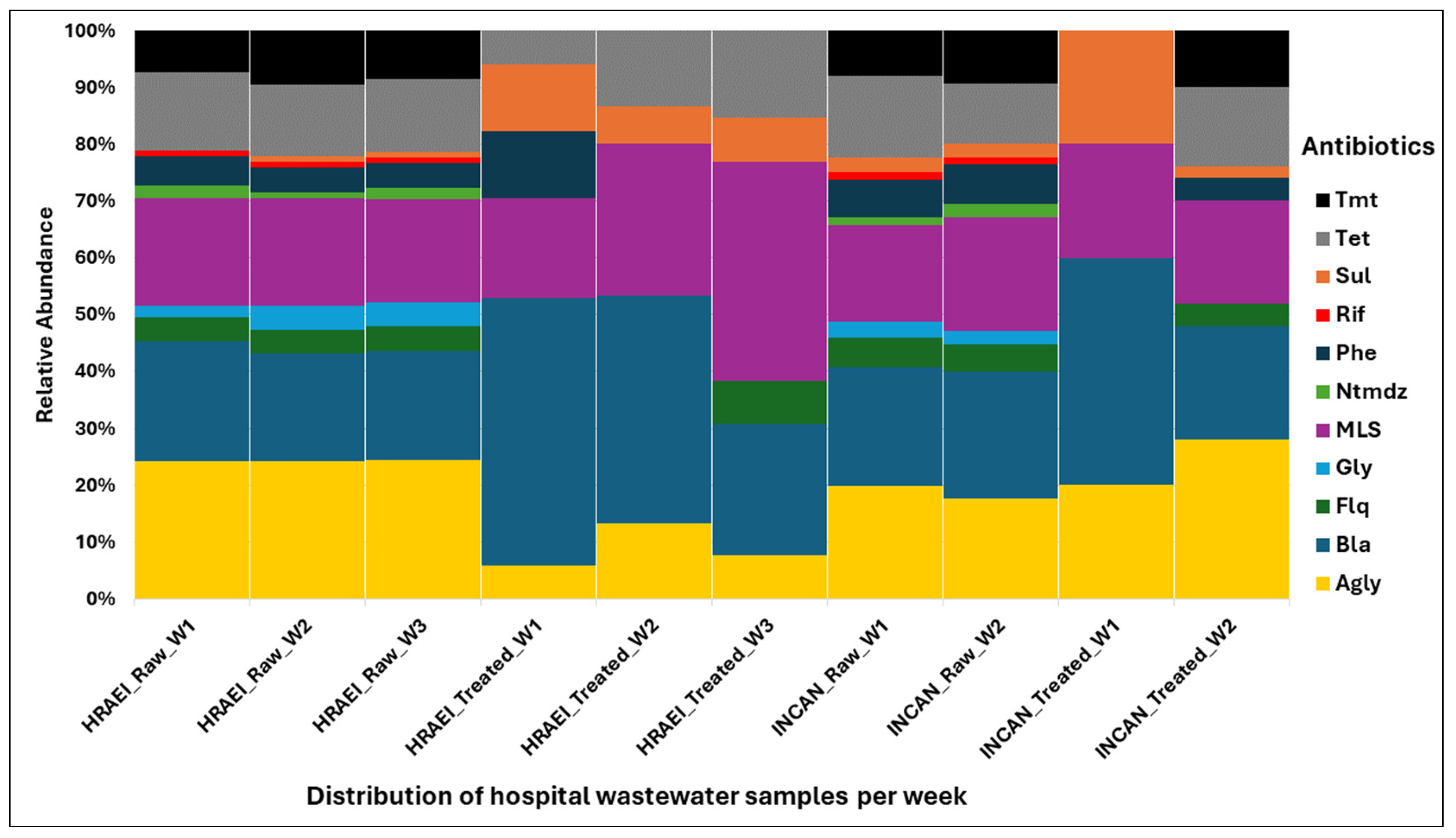
Overall, the ARGs present in each of the raw and treated wastewater samples were grouped in relation to the major antibiotic classes: aminoglycosides (AGly), β-lactams (Bla), fluoroquinolones (Flq), glycopeptides (Gly), macrolides–lincosamides–streptogramines (MLS), nitroimidazole (Ntmdz), phenicol (Phe), rifampicin (Rif), sulfonamides (Sul), tetracycline (Tet), and trimethoprim (Tmt). The highest abundance and persistence of resistance genes were those of aminoglycosides, β-lactams, and MLS compared to the other antibiotic classes detected (Figure 8).
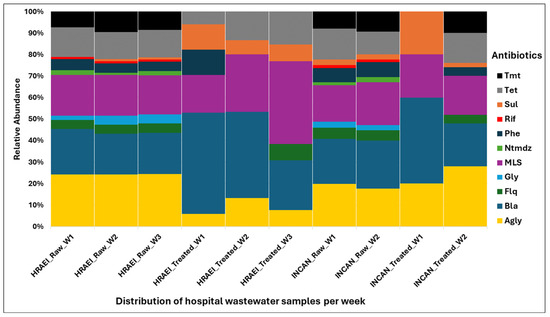
Figure 8.
Relative abundance of resistance genes encoding antibiotic classes in wastewater samples. Drug class: Agly (aminoglycoside), Bla (betalactam), Flq (fluoroquinolana), Gly (glycopeptides), MLS (macrolides–lincosamides–streptogramines), Ntmdz (nitroimidazole), Phe (phenicol), Rif (rifampicin), Sul (sulfonamides), Tet (tetracycline), and Tmt (trimethoprim).
4. Discussion
Wastewater is a vector that transports bacteria and resistance genes from the clinical setting to the environment, so it is essential to adequately treat this water [1]. It is important to evaluate these systems by sequencing techniques to establish the presence and abundance of bacteria and resistance genes in different settings [2,46,47]. This study, conducted in two hospital WWTP environments, showed that these WWTPs reduced the relative abundance of bacteria in treated wastewater by 13% compared to raw wastewater (Figure 1). This result was like that found in other studies conducted in the Netherlands [48], Germany [49], and China [50], in which WWTP processes reduced the relative abundance of pathogenic bacteria.
The microbial composition of the raw wastewater samples analyzed at the phyla level in our study was dominated by Firmicutes (32 ± 4.1%), Proteobacteria (28 ± 4.7%), Bacteroidetes (28 ± 3.9%), and Actinobacteria (5 ± 0.6%), while in the treated wastewater samples, it was Proteobacteria (46 ± 8.5%), Firmicutes (13 ± 3.9%), Bacteroidetes (9 ± 2.5%), and Actinobacteria (16 ± 3.6%). Numberger et al. also reported the predominance of Firmicutes, Proteobacteria, Bacteroidetes, and Actinobacteria, analyzing a hospital WWTP in four times of the year [49]. However, their results on the relative abundance of these phyla were different from those found in our study, where an average relative abundance was observed for Firmicutes (52.2 ± 4.4), Proteobacteria (37.8 ± 4.7%), Bacteroidetes (4.9 ± 1.9%), and Actinobacteria (2.2 ± 0.2%) [48]. The increase in Proteobacteria and Actinobacteria in treated wastewater samples observed in our results could be related to the type of samples associated with human biomes, as previously reported [51].
Particularly, the phyla of Proteobacteria and Firmicutes, which include the genera of the ESKAPEE group, have been referred to as the most abundant [51,52,53,54]. Our results showed that Escherichia spp., Klebsiella spp., and Enterobacter spp. were the predominant genera in the raw wastewater, while Enterococcus spp., Staphylococcus spp., Acinetobacter spp., and Pseudomonas spp. were found in the treated wastewater. In our study, we observed that Escherichia spp. and Klebsiella spp. were significantly reduced (<99% and <50%) in the WWTP. A similar behavior was observed by Verburg I. et al. [48,55], where the reduction in these genera was <90%. In our study, it was also observed that the relative abundance of Acinetobacter spp. and Pseudomonas spp. increased between raw wastewater and treated wastewater. This differs from what was reported by Numberger D. et al., where they found a decrease in the abundance of these two genera (9.5% to 1.3%) between raw wastewater and treated wastewater [48].
The increase in the abundance of Enterococcus spp. and Staphylococcus spp. in the treated wastewater was probably related to lower competition, since the abundance of the other bacterial genera decreased (Figure 6), which has been reported in other studies [56]. Another explanation for this increase could be the ability of these genera (Enterococcus spp., Staphylococcus spp., Acinetobacter spp., and Pseudomonas spp.) to form biofilm [57,58,59,60].
In our study, we observed that the bacterial species of the ESKAPEE group had a relative abundance depending on the bacterial genus detected, where E. coli, K. pneumoniae, and P. aeruginosa were the most abundant species. This is different from what was reported by Hubeny J. et al. [61], who mention that A. baumannii and E. coli were the dominant pathogens. The dominant abundance of Enterobacteriaceae (E. coli, Klebsiella pneumoniae) in raw wastewater samples has been mainly associated with human feces [50,62]. Furthermore, some of these selected bacteria can survive the different WWTP processes and persist due to their own characteristics [18].
It has been suggested that wastewater released into the environment is a major source of antibiotic-resistant bacteria (ARB) and ARGs, with healthcare facilities being a major source of ARB, due to the discharge of antibiotic residues, Karkman et al. mention that human and animal gut microbiota contain a wide range of ARGs, so sewage discharge and fecal contamination have been linked to the increased abundance of ARGs in the aquatic environment [14,46]. In this study, it was observed that raw wastewater had an average ARG abundance of 99.3 ± 9.6% and treated wastewater showed an average ARG abundance of 20.0 ± 17.2%, observing a reduction of 70%. Szczepanowski R et al. [63] reported a 13% reduction in ARG abundance in treated wastewater from a WWTP in Germany. On the other hand, Yang Y. et al. [64] and Gupta S. et al. [65] reported that wastewater treatment eliminated >99% of ARGs in urban WTTPs in Hong Kong and South Korea, respectively.
Our results showed the presence and persistence of blaKPC-1, blaOXA-1, blaOXA-2, blaOXA-10, and blaOXA-232 genes in raw and treated wastewater. The blaKPC and blaOXA genes were previously detected by PCR in the isolation of Klebsiella spp. collected from these same samples, which correlates with the findings obtained by metagenomics [9]. The genes blaTLA-1, blaTLA-2, blaTEM-1, blaMOX-2, blaMOX-6, blaMOX-8, and blaMOX-9 were only present in the raw wastewater, while the genes blaGES-1, blaGES-20, and blaNDM-1 were detected in a lower proportion in treated wastewater samples. The presence of these ARGs has been detected in the bacteria of the ESKAPEE group in hospital wastewater from Romania [66], as well as in urban wastewater in Canada [67] and other countries [15,46,52,68]. Furthermore, it is essential to highlight the presence of environmental bacteria, which play an important role in the abundance of antibiotic-resistant bacteria in aquatic ecosystems, and which, in these water environments, allow for the persistent release of ARGs [69,70,71].
Although the presence of the mcr gene that encodes colistin resistance was not detected in our study, this gene has been significantly detected in urban wastewater in other countries such as Spain [72], France [73], Germany [74], and Tunisia [75].
In this study, the genes mph(A-E), msr(D), mef(B), Inu(b), and ermB, encoding resistance to MLS, were mainly detected in raw wastewater and persisted in treated wastewater, which agrees with the results of Pallares-Vega. R. et al., who detected these same genes in the wastewater of 62 WWTPs studied in the Netherlands [76], with the ermB gene being among the most abundant and persistent. This gene was originally detected in Gram-positive bacteria (Enterococcus spp., Staphylococcus spp., and Streptococcus spp.), but could be transferred to Gram-negative bacteria via a conjugative transposon [18,75].
Furthermore, this study detected the presence of the genes strA, aph(3″), aads, aadA2, and aac3,which encode resistance to aminoglycosides. The presence of these genes was significantly more abundant in the raw wastewater analyzed by Raza S. et al. in South Korea [77] and Pärnänen K. et al. in the European Union [18].
A study published by Berglund F. et al. in 2023 [51] reported that the most abundant ARGs were associated with bacteria from the ESKAPEE group, representing 70% of the total ARGs detected in the samples analyzed; these mainly encode β-lactams, aminoglycosides, and macrolides. These results were similar to those observed in our work, where the highest abundance of ARGs was found to encode these three classes of antibiotic families. The less abundant ARGs were probably not detected in the analyzed samples due to the temporality, number of samples, and low sensitivity of the metagenomic tools.
The pattern of ARGs present in hospital wastewater samples detected by metagenomics could be related to the use of antibiotics prescribed in these hospitals. This is supported by the results of a previous study carried out by the working group, where it was reported that third-generation cephalosporins and carbapenems were the most used antibiotics [78], suggesting that the prescription of antibiotics in the hospital setting influences the microbiome and resistome present in wastewater.
Similar results were observed in a study carried out in 12 hospital WWTPs in the European Union (2022) [18], which found that raw wastewater from hospitals with a high consumption of antibiotics had a significantly higher relative abundance of ARGs compared to hospitals with a low consumption of antibiotics. The study observed that these ARGs decreased after wastewater treatment. If this phenomenon continues (antibiotic prescription–presence of ARGs in wastewater), the analysis of wastewater in other hospitals in Mexico would probably have similar results to those found in this work [79].
The present work has some limitations. The study was based on a cross-sectional design, where two hospitals were sampled. The first hospital (HRAEI) had three replications and the second hospital (INCAN) had only two. Other hospitals were not included due to restrictions due to the COVID-19 pandemic. However, the hospital wastewater analysis allowed us to detect the abundance of bacteria from the ESKAPEE and ARG group as a first approach to the context in our country.
5. Conclusions
Despite the limited number of samples analyzed, our results showed the presence and persistence of ESKAPEE group bacteria in hospital wastewater, as well as the presence of ARGs that code for resistance to antibiotics of clinical importance. This pilot study could serve as a model for the early surveillance of AMR in hospital wastewater in our country. Antimicrobial resistance cannot simply be addressed within healthcare facilities; it must also be addressed beyond the facilities of its waste.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/microorganisms12061231/s1, Table S1: Quality control; Table S2: Resistance genes, plasmids, and virulence factors that persist between raw wastewater and treated wastewater.
Author Contributions
Conceptualization, M.G.-L., M.E.V.-M., A.P.-d.-L., P.S.-H. and C.M.A.-A.; methodology, M.G.-L., M.E.V.-M., E.E.G.-L., A.P.-d.-L., P.S.-H. and C.M.A.-A.; formal analysis, M.G.-L., M.E.V.-M., E.E.G.-L., B.A.C.-Q., P.C.-J., A.S.-G. and C.M.A.-A.; investigation, M.G.-L., M.E.V.-M., E.E.G.-L., B.A.C.-Q., P.C.-J. and C.M.A.-A.; resources, M.G.-L., M.E.V.-M., E.E.G.-L., B.A.C.-Q., P.C.-J., A.S.-G., A.P.-d.-L., P.S.-H. and C.M.A.-A.; data curation, M.G.-L., M.E.V.-M. and E.E.G.-L. writing—original draft preparation, M.G.-L., M.E.V.-M., E.E.G.-L. and C.M.A.-A.; writing—review and editing, M.G.-L., M.E.V.-M., E.E.G.-L., B.A.C.-Q., P.C.-J., A.S.-G., A.P.-d.-L., P.S.-H. and C.M.A.-A.; visualization, M.G.-L., M.E.V.-M., E.E.G.-L., B.A.C.-Q., P.C.-J., A.S.-G., A.P.-d.-L., P.S.-H. and C.M.A.-A.; supervision, A.P.-d.-L. and C.M.A.-A.; project administration, M.E.V.-M. and C.M.A.-A.; funding acquisition, A.S.-G., A.P.-d.-L., P.S.-H. and C.M.A.-A. All authors have read and agreed to the published version of the manuscript.
Funding
This study was financed by FOSISS-CONACYT-2017, N° 290618.
Data Availability Statement
The reads are available from NCBI SRA under BioProject ID PRJNA1010860.
Acknowledgments
We thank the personnel responsible for the wastewater treatment plants and Biolg. Mario Sánchez-Vargas for his collaboration in the water sampling, as well as Alfredo Mendoza and Dora Garnica from the Instituto Nacional de Medicina Genómica (INMEGEN) for the sequencing and processing of the samples. We also thank Ariadna Nava and Lorena Guerrero for their administrative support in this project, and the Department of Bioinformatics in Infectious Diseases of the Center for Research on Infectious Diseases of the Instituto Nacional de Salud Pública. Finally, we thank CONAHCYT for grant no. 712862 awarded to Miguel Galarde-López for the development of his PhD, and the project “Patrones de distribución de genes de resistencia antimicrobiana y virulencia en los linajes de MRSA el microambiente de aguas residuales” with N° 682339 for the development of his postdoctoral studies.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Aarestrup, F.M.; Woolhouse, M.E.J. Using Sewage for Surveillance of Antimicrobial Resistance. Science 2020, 367, 630–632. [Google Scholar] [CrossRef]
- Sinclair, R.G.; Choi, C.Y.; Riley, M.R.; Gerba, C.P. Pathogen Surveillance Through Monitoring of Sewer Systems. In Advances in Applied Microbiology; Academic Press Inc.: Cambridge, MA, USA, 2008; Volume 65, pp. 249–269. ISBN 9780123744296. [Google Scholar]
- Schlüter, A.; Krause, L.; Szczepanowski, R.; Goesmann, A.; Pühler, A. Genetic Diversity and Composition of a Plasmid Metagenome from a Wastewater Treatment Plant. J. Biotechnol. 2008, 136, 65–76. [Google Scholar] [CrossRef]
- Tiedje, J.M.; Wang, F.; Manaia, C.M.; Virta, M.; Sheng, H.; Ma, L.; Zhang, T.; Topp, E. Antibiotic Resistance Genes in the Human-Impacted Environment: A One Health Perspective. Pedosphere 2019, 29, 273–282. [Google Scholar] [CrossRef]
- Daughton, C.G. Wastewater Surveillance for Population-Wide COVID-19: The Present and Future. Sci. Total Environ. 2020, 736, 139631. [Google Scholar] [CrossRef]
- La Rosa, G.; Iaconelli, M.; Mancini, P.; Bonanno Ferraro, G.; Veneri, C.; Bonadonna, L.; Lucentini, L.; Suffredini, E. First Detection of SARS-CoV-2 in Untreated Wastewaters in Italy. Sci. Total Environ. 2020, 736, 139652. [Google Scholar] [CrossRef]
- World Health Organization. Antibiotic Resistance. Available online: https://www.who.int/news-room/fact-sheets/detail/antibiotic-resistance (accessed on 5 May 2020).
- World Health Organization. WHO Publishes List of Bacteria for Which New Antibiotics Are Urgently Needed. Available online: https://www.who.int/es/news-room/detail/27-02-2017-who-publishes-list-of-bacteria-for-which-new-antibiotics-are-urgently-needed (accessed on 26 March 2020).
- Galarde-López, M.; Velazquez-Meza, M.E.; Bobadilla-del-Valle, M.; Carrillo-Quiroz, B.A.; Cornejo-Juárez, P.; Ponce-de-León, A.; Sassoé-González, A.; Alpuche-Aranda, C.M. Surveillance of Antimicrobial Resistance in Hospital Wastewater: Identification of Carbapenemase-Producing Klebsiella spp. Antibiotics 2022, 11, 288. [Google Scholar] [CrossRef]
- Galarde-López, M.; Velazquez-Meza, M.E.; Bobadilla-del-Valle, M.; Cornejo-Juárez, P.; Carrillo-Quiroz, B.A.; Ponce-de-León, A.; Sassoé-González, A.; Saturno-Hernández, P.; Alpuche-Aranda, C.M. Antimicrobial Resistance Patterns and Clonal Distribution of E. coli, Enterobacter spp. and Acinetobacter spp. Strains Isolated from Two Hospital Wastewater Plants. Antibiotics 2022, 11, 601. [Google Scholar] [CrossRef]
- Savin, M.; Bierbaum, G.; Hammerl, J.A.; Heinemann, C.; Parcina, M.; Sib, E.; Voigt, A.; Kreyenschmidt, J. Isolation an Chatracterization of ESKAPE Bacteria and Extended-Spectrum-β-Lactamase-Producing Escherichia coli Isolated from Wastewater and Process Water from German Poultry Slaughterhouses. Appl. Environ. Microbiol. 2020, 86, e02748-19. [Google Scholar] [CrossRef]
- Manaia, C.M.; Rocha, J.; Scaccia, N.; Marano, R.; Radu, E.; Biancullo, F.; Cerqueira, F.; Fortunato, G.; Iakovides, I.C.; Zammit, I.; et al. Antibiotic Resistance in Wastewater Treatment Plants: Tackling the Black Box. Environ. Int. 2018, 115, 312–324. [Google Scholar] [CrossRef]
- Warnes, S.L.; Highmore, C.J.; Keevil, C.W. Horizontal Transfer of Antibiotic Resistance Genes on Abiotic Touch Surfaces: Implications for Public Health. MBio 2012, 3, e00489-12. [Google Scholar] [CrossRef]
- Kraemer, S.A.; Ramachandran, A.; Perron, G.G. Antibiotic Pollution in the Environment: From Microbial Ecology to Public Policy. Microorganisms 2019, 7, 180. [Google Scholar] [CrossRef]
- Marutescu, L.G.; Popa, M.; Gheorghe-Barbu, I.; Barbu, I.C.; Rodríguez-Molina, D.; Berglund, F.; Blaak, H.; Flach, C.F.; Kemper, M.A.; Spießberger, B.; et al. Wastewater Treatment Plants, an “Escape Gate” for ESCAPE Pathogens. Front. Microbiol. 2023, 14, 1193907. [Google Scholar] [CrossRef]
- Bueno, I.; Verdugo, C.; Jimenez-Lopez, O.; Alvarez, P.P.; Gonzalez-Rocha, G.; Lima, C.A.; Travis, D.A.; Wass, B.; Zhang, Q.; Ishii, S.; et al. Role of Wastewater Treatment Plants on Environmental Abundance of Antimicrobial Resistance Genes in Chilean Rivers. Int. J. Hyg. Environ. Health 2020, 223, 56–64. [Google Scholar] [CrossRef]
- Agrawal, K.; Verma, P. Metagenomics: A Possible Solution for Uncovering the “Mystery Box” of Microbial Communities Involved in the Treatment of Wastewater. In Wastewater Treatment: Cutting-Edge Molecular Tools, Techniques and Applied Aspects; Elsevier: Amsterdam, The Netherlands, 2021; pp. 41–53. ISBN 9780128218815. [Google Scholar]
- Pärnänen, K.M.M.; Narciso-da-Rocha, C.; Kneis, D.; Berendonk, T.U.; Cacace, D.; Do, T.T.; Elpers, C.; Fatta-Kassinos, D.; Henriques, I.; Jaeger, T.; et al. Antibiotic Resistance in European Wastewater Treatment Plants Mirrors the Pattern of Clinical Antibiotic Resistance Prevalence. Sci. Adv. 2019, 5, eaau9124. [Google Scholar] [CrossRef]
- Hendriksen, R.S.; Munk, P.; Njage, P.; van Bunnik, B.; McNally, L.; Lukjancenko, O.; Röder, T.; Nieuwenhuijse, D.; Pedersen, S.K.; Kjeldgaard, J.; et al. Global Monitoring of Antimicrobial Resistance Based on Metagenomics Analyses of Urban Sewage. Nat. Commun. 2019, 10, 1124. [Google Scholar] [CrossRef]
- Munk, P.; Brinch, C.; Møller, F.D.; Petersen, T.N.; Hendriksen, R.S.; Seyfarth, A.M.; Kjeldgaard, J.S.; Svendsen, C.A.; van Bunnik, B.; Berglund, F.; et al. Genomic Analysis of Sewage from 101 Countries Reveals Global Landscape of Antimicrobial Resistance. Nat. Commun. 2022, 13, 7251. [Google Scholar] [CrossRef]
- Pham, T.T.H.; Rossi, P.; Dinh, H.D.K.; Pham, N.T.A.; Tran, P.A.; Ho, T.T.K.M.; Dinh, Q.T.; De Alencastro, L.F. Analysis of Antibiotic Multi-Resistant Bacteria and Resistance Genes in the Effluent of an Intensive Shrimp Farm (Long An, Vietnam). J. Environ. Manag. 2018, 214, 149–156. [Google Scholar] [CrossRef]
- World Health Organization. Global Action Plan on Antimicrobial Resistance; World Health Organization: Geneva, Switzerland, 2016. [Google Scholar]
- Chung, M.; De Lencastre, H.; Matthews, P.; Tomasz, A.; Adamsson, I.; Aires de Sousa, M.; Camou, T.; Cocuzza, C.; Corso, A.; Couto, I.; et al. Molecular Typing of Methicillin-Resistant Staphylococcus Aureus by Pulsed-Field Gel Electrophoresis: Comparison of Results Obtained in a Multilaboratory Effort Using Identical Protocols and MRSA Strains. Microb. Drug Resist. 2009, 6, 189–198. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Babraham Institute. Babraham Bioinformatics. FastQC A Quality Control Tool for High Throughput Sequence Data. 2023. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 2 January 2023).
- Peng, Y.; Leung, H.C.M.; Yiu, S.M.; Chin, F.Y.L. IDBA—A Practical Iterative De Bruijn Graph De Novo Assembler. In Proceedings of the 14th Annual International Conference, RECOMB 2010, Lisbon, Portugal, 25–28 April 2010; Volume 6044, pp. 426–440. [Google Scholar]
- Peng, Y.; Leung, H.C.M.; Yiu, S.M.; Chin, F.Y.L. IDBA-UD: A de Novo Assembler for Single-Cell and Metagenomic Sequencing Data with Highly Uneven Depth. Bioinformatics 2012, 28, 1420–1428. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Bryant, D.M.; Johnson, K.; DiTommaso, T.; Tickle, T.; Couger, M.B.; Payzin-Dogru, D.; Lee, T.J.; Leigh, N.D.; Kuo, T.H.; Davis, F.G.; et al. A Tissue-Mapped Axolotl De Novo Transcriptome Enables Identification of Limb Regeneration Factors. Cell Rep. 2017, 18, 762–776. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and Applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A Flexible Suite of Utilities for Comparing Genomic Features. Bioinformatics 2010, 26, 841. [Google Scholar] [CrossRef]
- Truong, D.T.; Tett, A.; Pasolli, E.; Huttenhower, C.; Segata, N. Microbial Strain-Level Population Structure and Genetic Diversity from Metagenomes. Genome Res. 2017, 27, 626–638. [Google Scholar] [CrossRef]
- Blanco-Miguez, A.; Beghini, F.; Cumbo, F.; McIver, L.J.; Thompson, K.N.; Zolfo, M.; Manghi, P.; Dubois, L.; Huang, K.D.; Thomas, A.M.; et al. Extending and Improving Metagenomic Taxonomic Profiling with Uncharacterized Species with MetaPhlAn 4. bioRxiv 2022. [Google Scholar] [CrossRef]
- Paulson, J.N.; Stine, O.C.; Bravo, H.C.; Pop, M. Differential Abundance Analysis for Microbial Marker-Gene Surveys. Nat. Methods 2013, 10, 1200–1202. [Google Scholar] [CrossRef]
- Oksanen, A.J.; Blanchet, F.G.; Kindt, R.; Legen, P.; Minchin, P.R.; Hara, R.B.O.; Simpson, G.L.; Solymos, P.; Stevens, M.H.H. Community Ecology Package. 2022. Available online: http://cran.r-project.org (accessed on 15 March 2023).
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Clarke, K.R.; Gorley, R.N.; Sommerfield, P.J.; Warwick, R.M. Change in Marine Communities—Statistical Analysis, 3rd ed.; Plymouth, P.-E., Ed.; Plymouth Marine Laboratory: Plymouth, UK, 2014. [Google Scholar]
- Valero-Mora, P.M. Ggplot2: Elegant Graphics for Data Analysis. J. Stat. Softw. 2010, 35, 180–185. [Google Scholar] [CrossRef]
- RStudio Team. RStudio: Integrated Development for R; RStudio, Inc.: Boston, MA, USA, 2015; Available online: https://www.rstudio.com/ (accessed on 27 March 2023).
- Seemann, T. Abricate. Github. 2023. Available online: https://github.com/tseemann/abricate (accessed on 13 March 2023).
- Jia, B.; Raphenya, A.R.; Alcock, B.; Waglechner, N.; Guo, P.; Tsang, K.K.; Lago, B.A.; Dave, B.M.; Pereira, S.; Sharma, A.N.; et al. CARD 2017: Expansion and Model-Centric Curation of the Comprehensive Antibiotic Resistance Database. Nucleic Acids Res. 2017, 45, D566–D573. [Google Scholar] [CrossRef]
- Carattoli, A.; Zankari, E.; Garciá-Fernández, A.; Larsen, M.V.; Lund, O.; Villa, L.; Aarestrup, F.M.; Hasman, H. In Silico Detection and Typing of Plasmids Using Plasmidfinder and Plasmid Multilocus Sequence Typing. Antimicrob. Agents Chemother. 2014, 58, 3895–3903. [Google Scholar] [CrossRef]
- Ingle, D.J.; Valcanis, M.; Kuzevski, A.; Tauschek, M.; Inouye, M.; Stinear, T.; Levine, M.M.; Robins-Browne, R.M.; Holt, K.E. In Silico Serotyping of E. coli from Short Read Data Identifies Limited Novel o-Loci but Extensive Diversity of O:H Serotype Combinations within and between Pathogenic Lineages. Microb. Genom. 2016, 2, e000064. [Google Scholar] [CrossRef]
- Chen, L.; Zheng, D.; Liu, B.; Yang, J.; Jin, Q. VFDB 2016: Hierarchical and Refined Dataset for Big Data Analysis—10 Years On. Nucleic Acids Res. 2016, 44, D694–D697. [Google Scholar] [CrossRef]
- Karkman, A.; Pärnänen, K.; Larsson, D.G.J. Fecal Pollution Can Explain Antibiotic Resistance Gene Abundances in Anthropogenically Impacted Environments. Nat. Commun. 2019, 10, 80. [Google Scholar] [CrossRef]
- Lira, F.; Vaz-Moreira, I.; Tamames, J.; Manaia, C.M.; Martínez, J.L. Metagenomic Analysis of an Urban Resistome before and after Wastewater Treatment. Sci. Rep. 2020, 10, 8174. [Google Scholar] [CrossRef]
- Verburg, I.; van Veelen, H.P.J.; Waar, K.; Rossen, J.W.A.; Friedrich, A.W.; Hernández Leal, L.; García-Cobos, S.; Schmitt, H. Effects of Clinical Wastewater on the Bacterial Community Structure from Sewage to the Environment. Microorganisms 2021, 9, 718. [Google Scholar] [CrossRef]
- Numberger, D.; Ganzert, L.; Zoccarato, L.; Mühldorfer, K.; Sauer, S.; Grossart, H.P.; Greenwood, A.D. Characterization of Bacterial Communities in Wastewater with Enhanced Taxonomic Resolution by Full-Length 16S RRNA Sequencing. Sci. Rep. 2019, 9, 9673. [Google Scholar] [CrossRef]
- Ma, X.; Dong, X.; Cai, J.; Fu, C.; Yang, J.; Liu, Y.; Zhang, Y.; Wan, T.; Lin, S.; Lou, Y.; et al. Metagenomic Analysis Reveals Changes in Bacterial Communities and Antibiotic Resistance Genes in an Eye Specialty Hospital and a General Hospital before and after Wastewater Treatment. Front. Microbiol. 2022, 13, 848167. [Google Scholar] [CrossRef]
- Berglund, F.; Ebmeyer, S.; Kristiansson, E.; Larsson, D.G.J. Evidence for Wastewaters as Environments Where Mobile Antibiotic Resistance Genes Emerge. Commun. Biol. 2023, 6, 321. [Google Scholar] [CrossRef]
- Lin, Q.; Xavier, B.B.; Alako, B.T.F.; Mitchell, A.L.; Rajakani, S.G.; Glupczynski, Y.; Finn, R.D.; Cochrane, G.; Malhotra-Kumar, S. Screening of Global Microbiomes Implies Ecological Boundaries Impacting the Distribution and Dissemination of Clinically Relevant Antimicrobial Resistance Genes. Commun. Biol. 2022, 5, 1217. [Google Scholar] [CrossRef]
- Wang, P.; Yu, Z.; Qi, R.; Zhang, H. Detailed Comparison of Bacterial Communities during Seasonal Sludge Bulking in a Municipal Wastewater Treatment Plant. Water Res. 2016, 105, 157–166. [Google Scholar] [CrossRef]
- Osunmakinde, C.O.; Selvarajan, R.; Mamba, B.B.; Msagati, T.A.M. Profiling Bacterial Diversity and Potential Pathogens in Wastewater Treatment Plants Using High-Throughput Sequencing Analysis. Microorganisms 2019, 7, 506. [Google Scholar] [CrossRef]
- Verburg, I.; García-Cobos, S.; Leal, L.H.; Waar, K.; Friedrich, A.W.; Schmitt, H. Abundance and Antimicrobial Resistance of Three Bacterial Species along a Complete Wastewater Pathway. Microorganisms 2019, 7, 312. [Google Scholar] [CrossRef]
- Lakshmanan, V.; Selvaraj, G.; Bais, H.P. Functional Soil Microbiome: Belowground Solutions to an Aboveground Problem. Plant Physiol. 2014, 166, 689–700. [Google Scholar] [CrossRef]
- Lépesová, K.; Kraková, L.; Pangallo, D.; Medveďová, A.; Olejníková, P.; Mackuľak, T.; Tichý, J.; Grabic, R.; Birošová, L. Prevalence of Antibiotic-Resistant Coliform Bacteria, Enterococcus spp. and Staphylococcus spp. in Wastewater Sewerage Biofilm. J. Glob. Antimicrob. Resist. 2018, 14, 145–151. [Google Scholar] [CrossRef]
- Beaudoin, T.; Yau, Y.C.W.; Stapleton, P.J.; Gong, Y.; Wang, P.W.; Guttman, D.S.; Waters, V. Staphylococcus aureus Interaction with Pseudomonas aeruginosa Biofilm Enhances Tobramycin Resistance. NPJ Biofilms Microbiomes 2017, 3, 25. [Google Scholar] [CrossRef]
- Gholami, S.; Tabatabaei, M.; Sohrabi, N. Comparison of Biofilm Formation and Antibiotic Resistance Pattern of Pseudomonas Aeruginosa in Human and Environmental Isolates. Microb. Pathog. 2017, 109, 94–98. [Google Scholar] [CrossRef]
- King, L.B.; Swiatlo, E.; Swiatlo, A.; Mcdaniel, L.S. Serum Resistance and Bio¢lm Formation in Clinical Isolates of Acinetobacter baumannii. FEMS Immunol. Med. Microbiol. 2009, 55, 414–421. [Google Scholar] [CrossRef]
- Hubeny, J.; Korzeniewska, E.; Ciesielski, S.; Płaza, G.; Harnisz, M. The Resistome of ESKAPEE Pathogens in Untreated and Treated Wastewater: A Polish Case Study. Biomolecules 2022, 12, 1160. [Google Scholar] [CrossRef]
- Hubeny, J.; Ciesielski, S.; Harnisz, M.; Korzeniewska, E.; Dulski, T.; Jałowiecki, Ł.; Płaza, G. Impact of Hospital Wastewater on the Occurrence and Diversity of Beta-Lactamase Genes During Wastewater Treatment with an Emphasis on Carbapenemase Genes: A Metagenomic Approach. Front. Environ. Sci. 2021, 9, 738158. [Google Scholar] [CrossRef]
- Szczepanowski, R.; Linke, B.; Krahn, I.; Gartemann, K.H.; Gützkow, T.; Eichler, W.; Pühler, A.; Schlüter, A. Detection of 140 Clinically Relevant Antibiotic-Resistance Genes in the Plasmid Metagenome of Wastewater Treatment Plant Bacteria Showing Reduced Susceptibility to Selected Antibiotics. Microbiology 2009, 155, 2306–2319. [Google Scholar] [CrossRef]
- Yang, Y.; Li, B.; Zou, S.; Fang, H.H.P.; Zhang, T. Fate of Antibiotic Resistance Genes in Sewage Treatment Plant Revealed by Metagenomic Approach. Water Res. 2014, 62, 97–106. [Google Scholar] [CrossRef]
- Gupta, S.K.; Shin, H.; Han, D.; Hur, H.G.; Unno, T. Metagenomic Analysis Reveals the Prevalence and Persistence of Antibiotic- and Heavy Metal-Resistance Genes in Wastewater Treatment Plant. J. Microbiol. 2018, 56, 408–415. [Google Scholar] [CrossRef]
- Gheorghe-Barbu, I.; Barbu, I.C.; Popa, L.I.; Pîrcălăbioru, G.G.; Popa, M.; Măruțescu, L.; Niță-Lazar, M.; Banciu, A.; Stoica, C.; Gheorghe, Ș.; et al. Temporo-Spatial Variations in Resistance Determinants and Clonality of Acinetobacter baumannii and Pseudomonas aeruginosa Strains from Romanian Hospitals and Wastewaters. Antimicrob. Resist. Infect. Control 2022, 11, 115. [Google Scholar] [CrossRef]
- Cooper, A.L.; Carter, C.; McLeod, H.; Wright, M.; Sritharan, P.; Tamber, S.; Wong, A.; Carrillo, C.D.; Blais, B.W. Detection of Carbapenem-Resistance Genes in Bacteria Isolated from Wastewater in Ontario. FACETS 2021, 6, 569–591. [Google Scholar] [CrossRef]
- Kehl, K.; Schallenberg, A.; Szekat, C.; Albert, C.; Sib, E.; Exner, M.; Zacharias, N.; Schreiber, C.; Parčina, M.; Bierbaum, G. Dissemination of Carbapenem Resistant Bacteria from Hospital Wastewater into the Environment. Sci. Total Environ. 2022, 806, 151339. [Google Scholar] [CrossRef]
- Allen, H.K.; Donato, J.; Wang, H.H.; Cloud-Hansen, K.A.; Davies, J.; Handelsman, J. Call of the Wild: Antibiotic Resistance Genes in Natural Environments. Nat. Rev. Microbiol. 2010, 8, 251–259. [Google Scholar] [CrossRef]
- Yitayew, B.; Woldeamanuel, Y.; Asrat, D.; Rahman, A.; Mihret, A.; Aseffa, A.; Olsson, P.-E.; Jass, J. Antimicrobial Resistance Genes in Microbiota Associated with Sediments and Water from the Akaki River in Ethiopia. Environ. Sci. Pollut. Res. 2022, 29, 70040–70055. [Google Scholar] [CrossRef]
- Luo, Y.; Mao, D.; Rysz, M.; Zhou, Q.; Zhang, H.; Xu, L.; Alvarez, P.J.J. Trends in Antibiotic Resistance Genes Occurrence in the Haihe River, China. Environ. Sci. Technol. 2010, 44, 7220–7225. [Google Scholar] [CrossRef]
- Ovejero, C.M.; Delgado-Blas, J.F.; Calero-Caceres, W.; Muniesa, M.; Gonzalez-Zorn, B. Spread of Mcr-1-Carrying Enterobacteriaceae in Sewage Water from Spain. J. Antimicrob. Chemother. 2017, 72, 1050–1053. [Google Scholar] [CrossRef]
- Bréchet, C.; Plantin, J.; Sauget, M.; Thouverez, M.; Talon, D.; Cholley, P.; Guyeux, C.; Hocquet, D.; Bertrand, X. Wastewater Treatment Plants Release Large Amounts of Extended-Spectrum β-Lactamase-Producing Escherichia Coli into the Environment. Clin. Infect. Dis. 2014, 58, 1658–1665. [Google Scholar] [CrossRef]
- Hembach, N.; Schmid, F.; Alexander, J.; Hiller, C.; Rogall, E.T.; Schwartz, T. Occurrence of the Mcr-1 Colistin Resistance Gene and Other Clinically Relevant Antibiotic Resistance Genes in Microbial Populations at Different Municipal Wastewater Treatment Plants in Germany. Front. Microbiol. 2017, 8, 267477. [Google Scholar] [CrossRef]
- Rafraf, I.D.; Lekunberri, I.; Sànchez-Melsió, A.; Aouni, M.; Borrego, C.M.; Balcázar, J.L. Abundance of Antibiotic Resistance Genes in Five Municipal Wastewater Treatment Plants in the Monastir Governorate, Tunisia. Environ. Pollut. 2016, 219, 353–358. [Google Scholar] [CrossRef]
- Pallares-Vega, R.; Blaak, H.; van der Plaats, R.; de Roda Husman, A.M.; Hernandez Leal, L.; van Loosdrecht, M.C.M.; Weissbrodt, D.G.; Schmitt, H. Determinants of Presence and Removal of Antibiotic Resistance Genes during WWTP Treatment: A Cross-Sectional Study. Water Res. 2019, 161, 319–328. [Google Scholar] [CrossRef]
- Raza, S.; Shin, H.; Hur, H.G.; Unno, T. Higher Abundance of Core Antimicrobial Resistant Genes in Effluent from Wastewater Treatment Plants. Water Res. 2022, 208, 117882. [Google Scholar] [CrossRef]
- Zumaya-Estrada, F.A.; Ponce-De-león-Garduño, A.; Ortiz-Brizuela, E.; Tinoco-Favila, J.C.; Cornejo-Juárez, P.; Vilar-Compte, D.; Sassoé-González, A.; Saturno-Hernandez, P.J.; Alpuche-Aranda, C.M. Point Prevalence Survey of Antimicrobial Use in Four Tertiary Care Hospitals in Mexico. Infect. Drug Resist. 2021, 14, 4553–4566. [Google Scholar] [CrossRef]
- Miranda-Novales, M.G.; Flores-Moreno, K.; López-Vidal, Y.; Rodríguez-Álvarez, M.; Solórzano-Santos, F.; Soto-Hernández, J.L.; Ponce de León-Rosales, S. Antimicrobial Resistance and Antibiotic Consumption in Mexican Hospitals. Salud Pública México 2020, 62, 42–49. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).