Shifts in Microbial Community Structure and Co-occurrence Network along a Wide Soil Salinity Gradient


Abstract
:1. Introduction
2. Materials and Methods
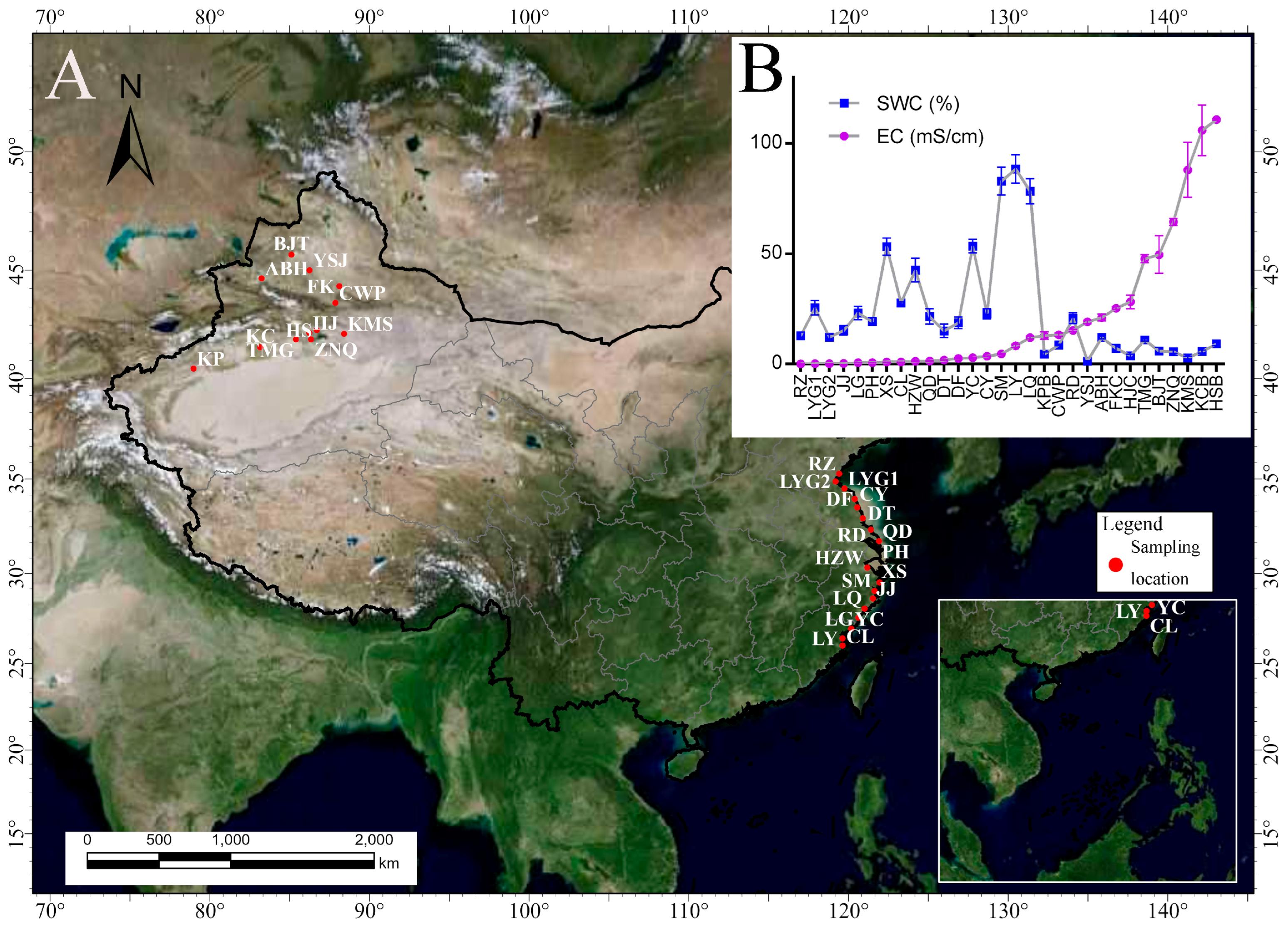
2.1. Studying Area
2.2. Soil Sampling
2.3. Soil Properties Determination
2.4. Metagenomic Sequencing, Assembly, and Annotation
2.5. Statistical Analysis
3. Results
3.1. Changes in Soil Variables in Three Salinity Gradients
3.2. Sequencing Data and Metagenome Assembly
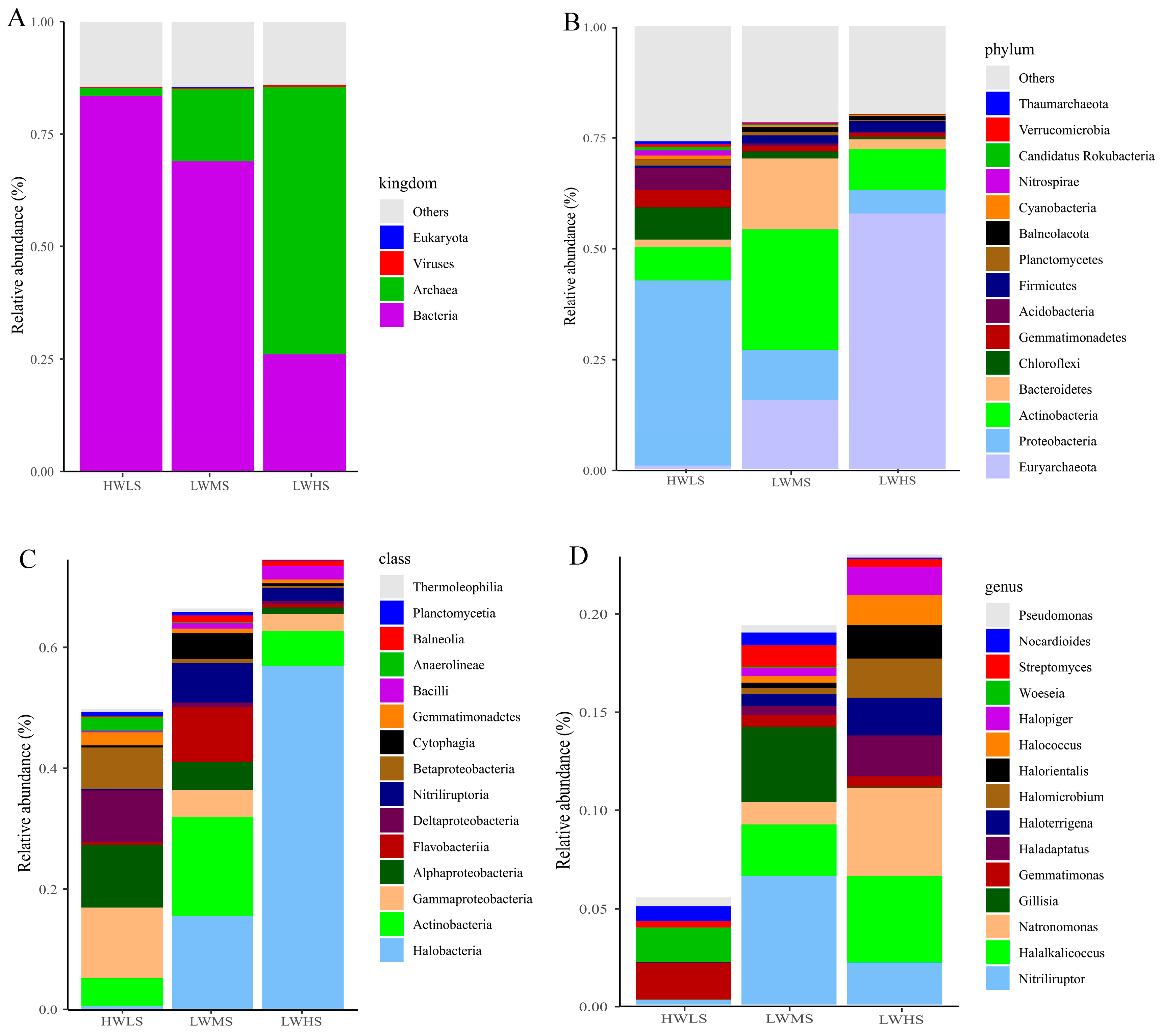
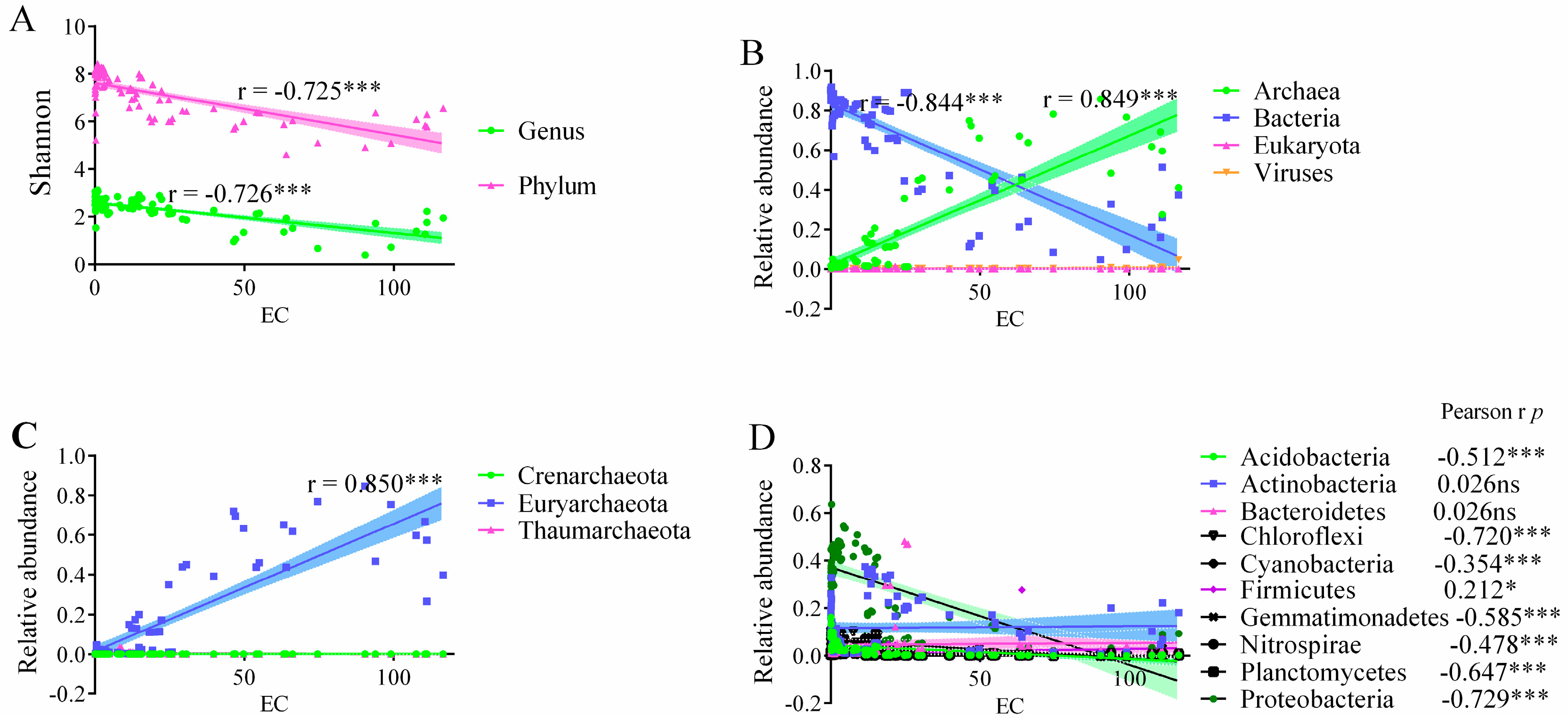
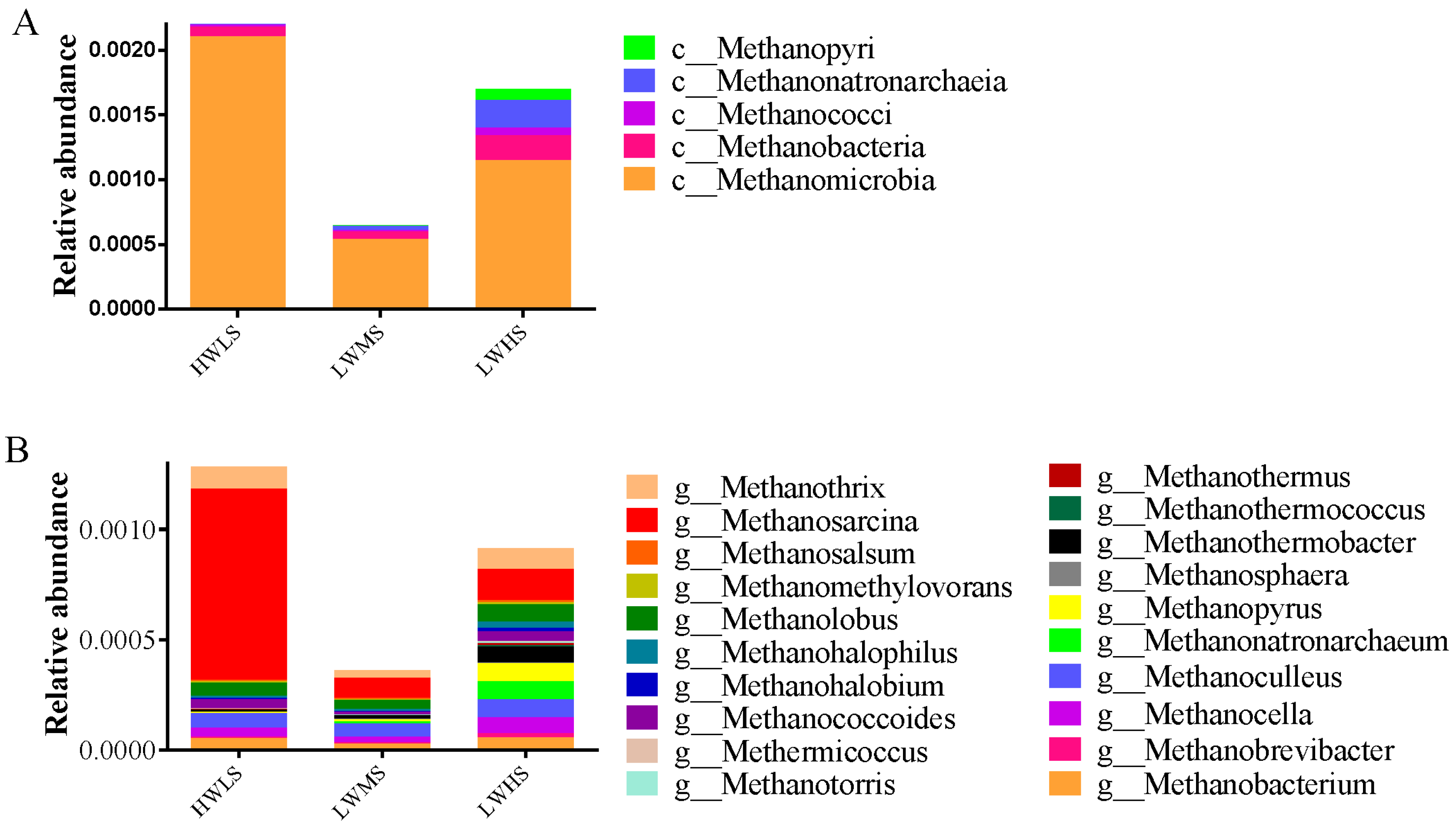
3.3. Microbial Community Composition and Structure
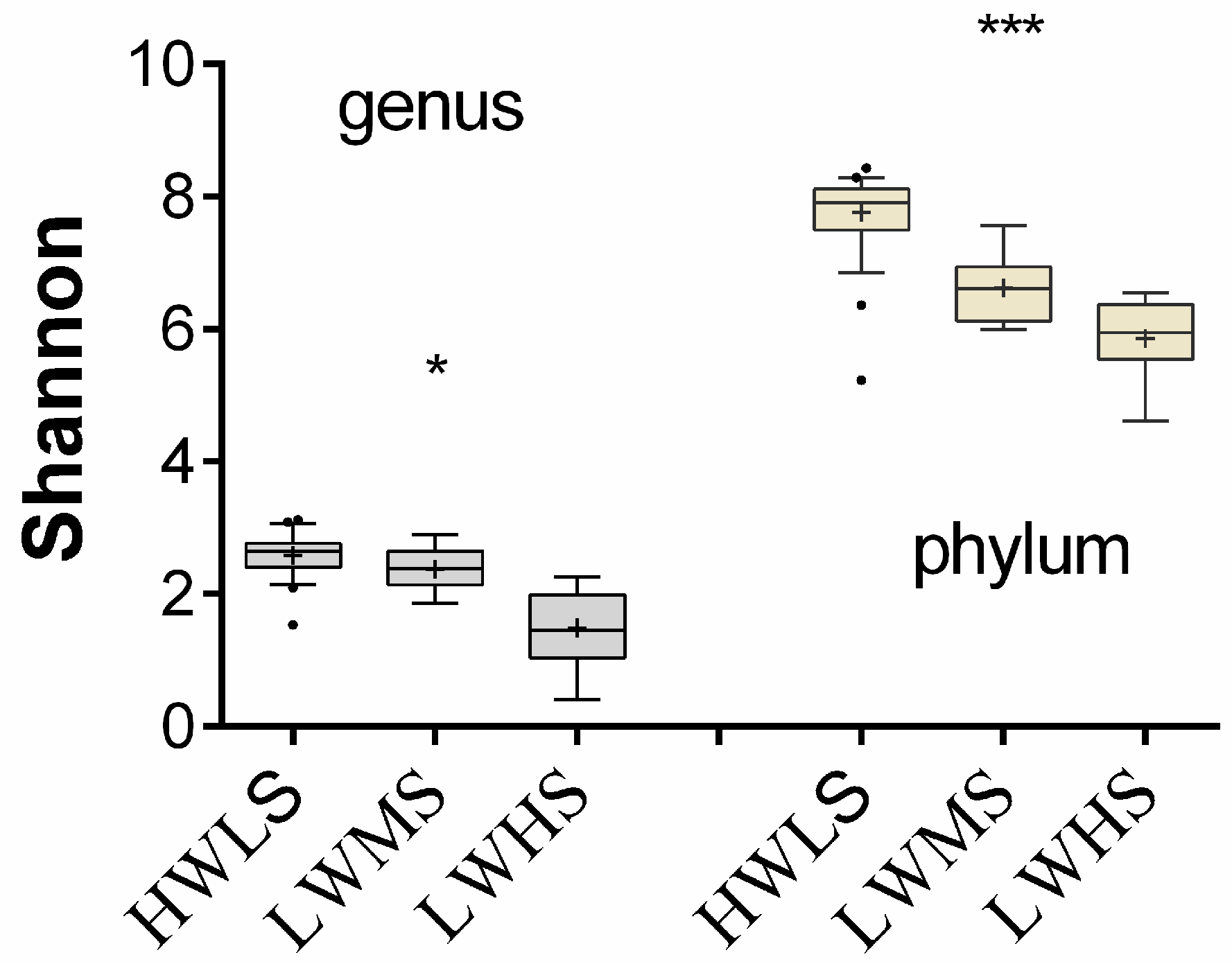
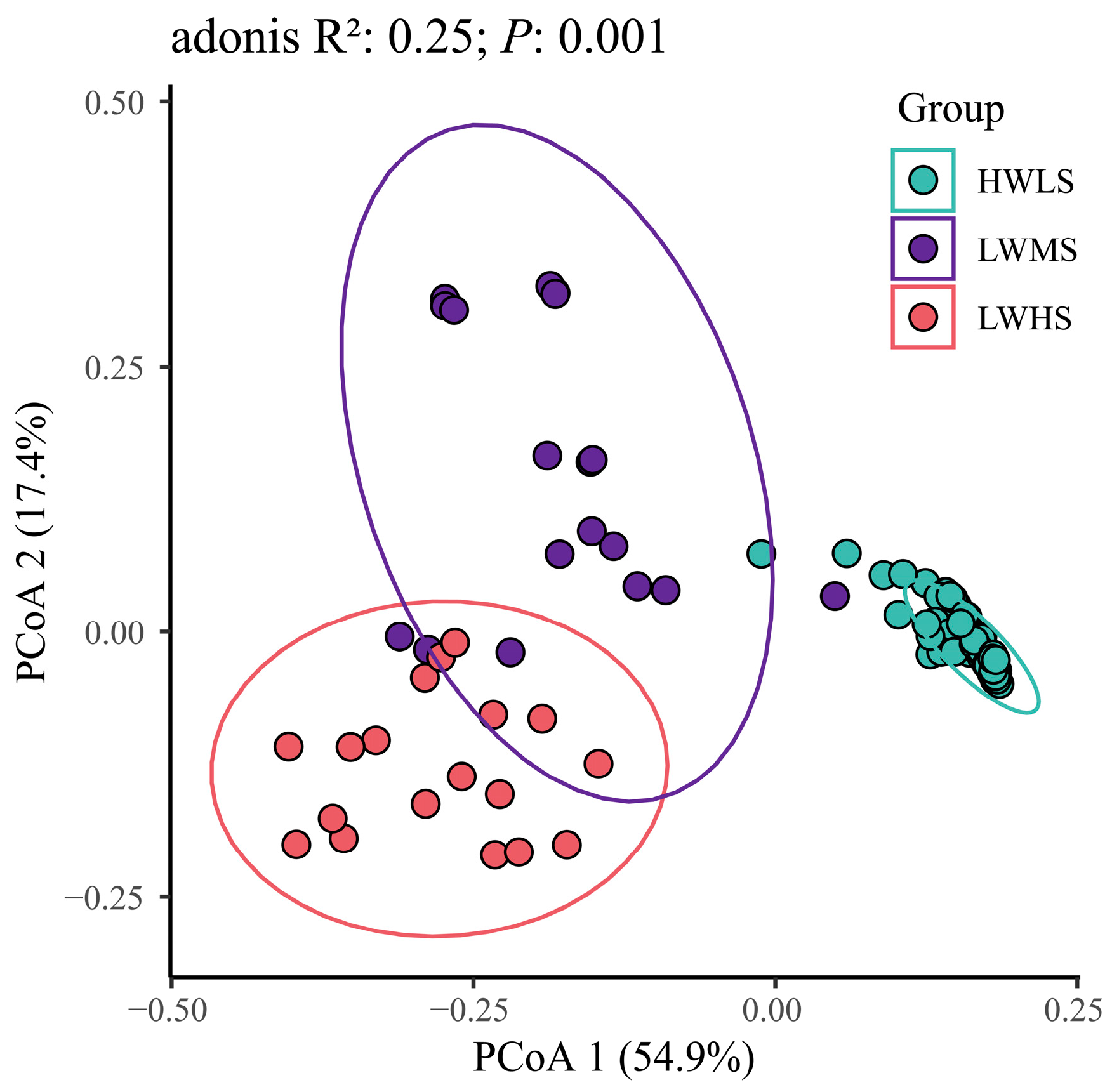
3.4. Microbial Community Structure Difference at Salinity Gradients
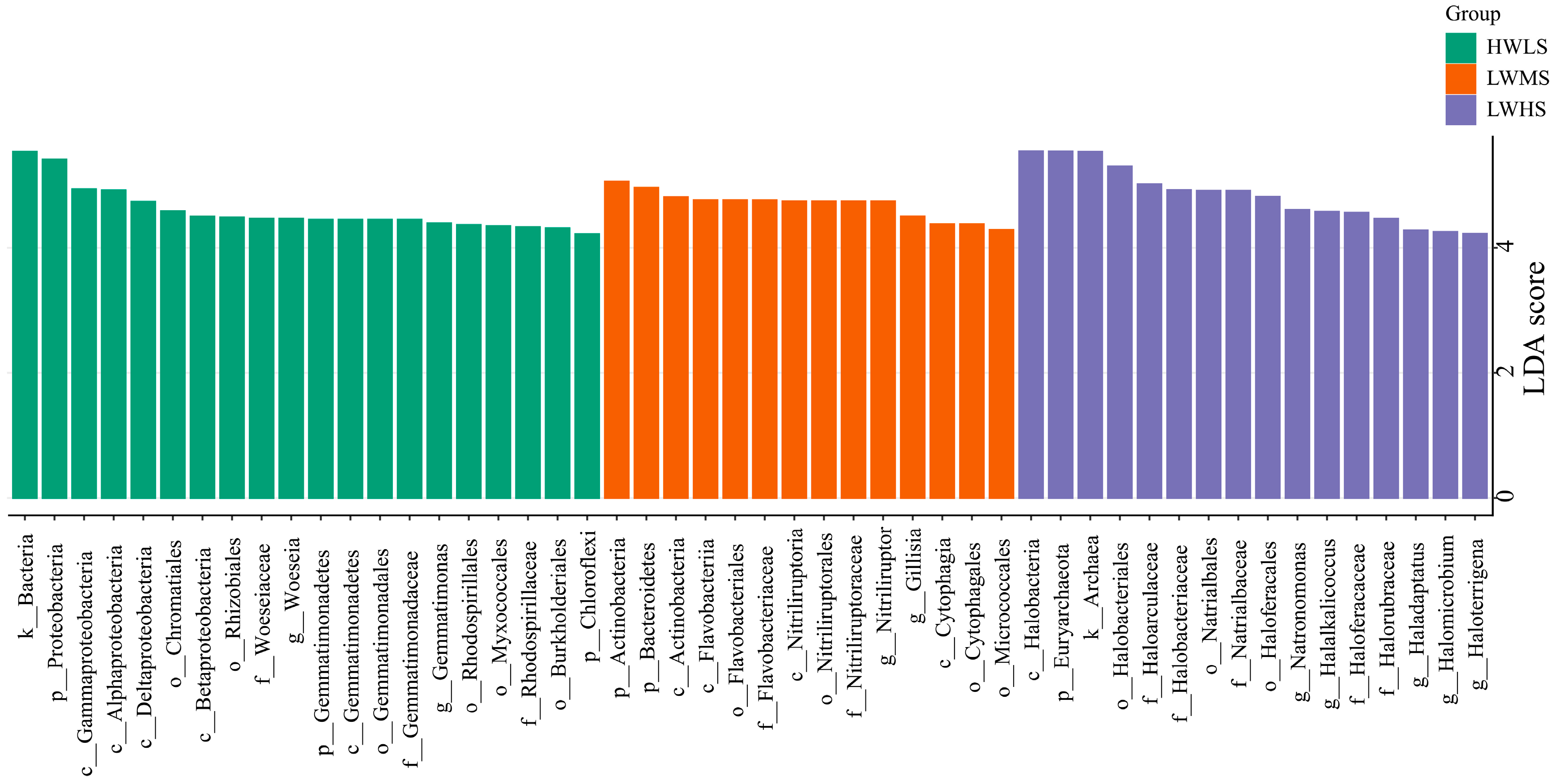
3.5. Biomarkers Identification in Each Salinity Group
3.6. Co-Occurrence Network Relationships of Soil Microbes along Salinity Gradients
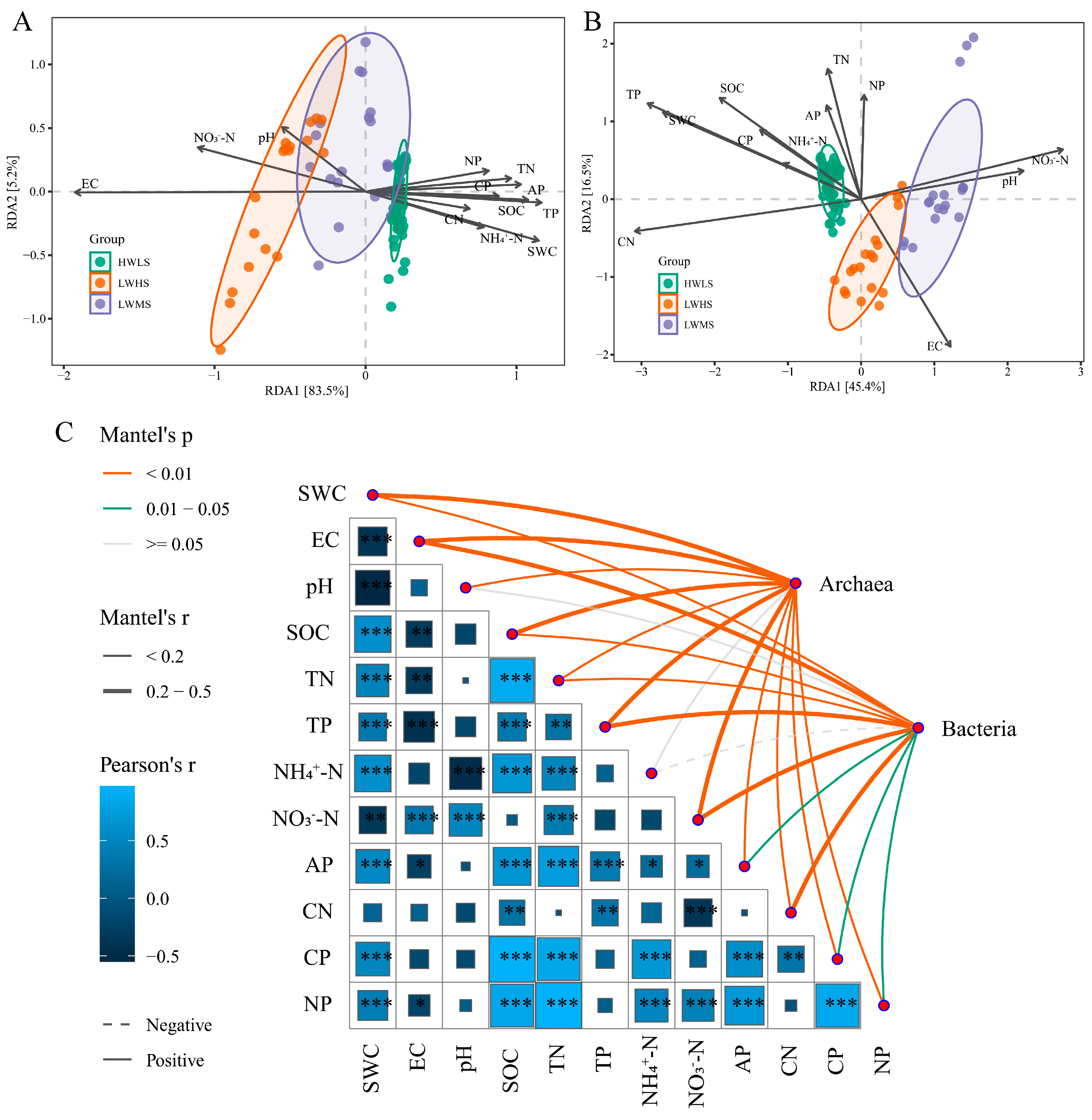
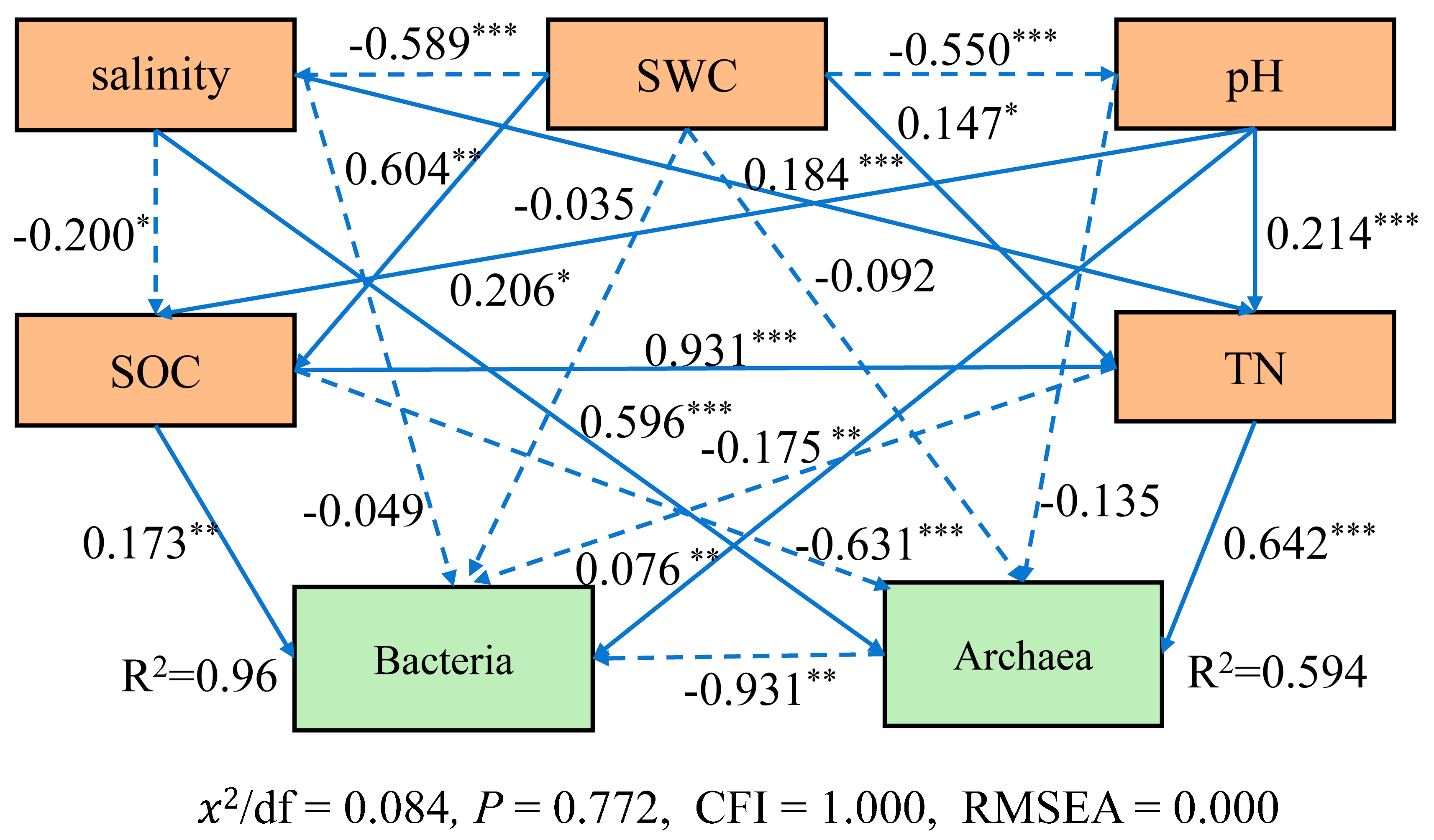
3.7. The Relationships between Soil Variables and Community Structure
4. Discussion
4.1. The Effects of Salinity on Soil Chemical Properties
4.2. The Microbial Community’s Composition at Different Salinity Gradients
4.3. The Effects of Salinity on Microbial Community Network
4.4. The Impacts of Soil Variables on Microbial Community
4.5. Clues for Reclamation of the Degraded Saline Soils
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- An, Y.M.; Yang, X.X.; Zhang, L.S.; Zhang, J.; Du, B.H.; Yao, L.; Li, X.T.; Guo, C.H. Alfalfa MsCBL4 enhances calcium metabolism but not sodium transport in transgenic tobacco under salt and saline-alkali stress. Plant Cell Rep. 2020, 39, 997–1011. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, Q.Y.; Han, Y.C.; Zhang, D.L.; Zhang, C.C.; Hu, C.X. Carbon cycle in the microbial ecosystems of biological soil crusts. Soil Biol. Biochem. 2022, 171, 108729. [Google Scholar] [CrossRef]
- Rengasamy, P. Transient salinity and subsoil constraints to dryland farming in Australian sodic soils: An overview. Aust. J. Exp. Agric. 2002, 42, 351–361. [Google Scholar] [CrossRef]
- Morina, J.C.; Franklin, R.B. Intensity and duration of exposure determine prokaryotic community response to salinization in freshwater wetland soils. Geoderma 2022, 428, 116138. [Google Scholar] [CrossRef]
- Wang, H.C.; Qi, J.F.; Xiao, D.R.; Wang, Y.; Shi, W.Y.; Wang, H. Bacterial community diversity and underlying assembly patterns along vertical soil profiles in wetland and meadow habitats on the Zoige Plateau, China. Soil Biol. Biochem. 2023, 184, 109076. [Google Scholar] [CrossRef]
- Toju, H.; Peay, K.G.; Yamamichi, M.; Narisawa, K.; Hiruma, K.; Naito, K.; Fukuda, S.; Ushio, M.; Nakaoka, S.; Onoda, Y.; et al. Core microbiomes for sustainable agroecosystems. Nat. Plants 2018, 4, 247–257. [Google Scholar] [CrossRef]
- Bardgett, R.D.; van der Putten, W.H. Belowground biodiversity and ecosystem functioning. Nature 2014, 515, 505–511. [Google Scholar] [CrossRef]
- Zheng, W.; Xue, D.M.; Li, X.Z.; Deng, Y.; Rui, J.P.; Feng, K.; Wang, Z.L. The responses and adaptations of microbial communities to salinity in farmland soils: A molecular ecological network analysis. Appl. Soil Ecol. 2017, 120, 239–246. [Google Scholar] [CrossRef]
- Li, Y.Q.; Chai, Y.H.; Wang, X.S.; Huang, L.Y.; Luo, X.M.; Qiu, C.; Liu, Q.H.; Guan, X.Y. Bacterial community in saline farmland soil on the Tibetan plateau: Responding to salinization while resisting extreme environments. BMC Microbiol. 2021, 21, 119. [Google Scholar] [CrossRef]
- Yang, J.; Ma, L.; Jiang, H.C.; Wu, G.; Dong, H.L. Salinity shapes microbial diversity and community structure in surface sediments of the Qinghai-Tibetan Lakes. Sci. Rep. 2016, 6, 25078. [Google Scholar] [CrossRef]
- Zhong, Z.P.; Liu, Y.; Miao, L.L.; Wang, F.; Chu, L.M.; Wang, J.L.; Liu, Z.P. Prokaryotic Community Structure Driven by Salinity and Ionic Concentrations in Plateau Lakes of the Tibetan Plateau. Appl. Environ. Microbiol. 2016, 82, 1846–1858. [Google Scholar] [CrossRef] [PubMed]
- Aerts, J.W.; van Spanning, R.J.M.; Flahaut, J.; Molenaar, D.; Bland, P.A.; Genge, M.J.; Ehrenfreund, P.; Martins, Z. Microbial communities in sediments from four mildly acidic ephemeral salt lakes in the Yilgarn Craton (Australia)—Terrestrial analogs to ancient Mars. Front. Microbiol. 2019, 10, 779. [Google Scholar] [CrossRef] [PubMed]
- Xi, X.F.; Wang, L.; Hu, J.J.; Tang, Y.S.; Hu, Y.; Fu, X.H.; Sun, Y.; Tsang, Y.F.; Zhang, Y.N.; Chen, J.H. Salinity influence on soil microbial respiration rate of wetland in the Yangtze River estuary through changing microbial community. J. Environ. Sci. 2014, 26, 2562–2570. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.L.; Bai, J.H.; Tebbe, C.C.; Zhao, Q.Q.; Jia, J.; Wang, W.; Wang, X.; Yu, L. Salinity controls soil microbial community structure and function in coastal estuarine wetlands. Environ. Microbiol. 2021, 23, 1020–1037. [Google Scholar] [CrossRef] [PubMed]
- Marks, B.M.; Chambers, L.; White, J.R. Effect of fluctuating salinity on potential denitrification in coastal wetland soil and sediments. Soil Sci. Soc. Am. J. 2016, 80, 516–526. [Google Scholar] [CrossRef]
- Yang, J.J.; Li, W.J.; Teng, D.X.; Yang, X.D.; Zhang, Y.J.; Li, Y. Metagenomic Insights into Microbial Community Structure, Function, and Salt Adaptation in Saline Soils of Arid Land, China. Microorganisms 2022, 10, 2183. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.P.; Shi, Y.; Cui, X.Q.; Yue, P.; Li, K.H.; Liu, X.J.; Tripathi, B.M.; Chu, H.Y. Salinity Is a Key Determinant for Soil Microbial Communities in a Desert Ecosystem. Msystems 2019, 4, e00225-18. [Google Scholar] [CrossRef] [PubMed]
- Canfora, L.; Lo Papa, G.; Antisari, L.V.; Bazan, G.; Dazzi, C.; Benedetti, A. Spatial microbial community structure and biodiversity analysis in “extreme” hypersaline soils of a semiarid Mediterranean area. Appl. Soil Ecol. 2015, 93, 120–129. [Google Scholar] [CrossRef]
- Maestre, F.T.; Delgado-Baquerizo, M.; Jeffries, T.C.; Eldridge, D.J.; Ochoa, V.; Gozalo, B.; Quero, J.L.; Garcia-Gomez, M.; Gallardo, A.; Ulrich, W.; et al. Increasing aridity reduces soil microbial diversity and abundance in global drylands. Proc. Natl. Acad. Sci. USA 2015, 112, 15684–15689. [Google Scholar] [CrossRef]
- Liang, X.L.; Wang, X.Y.; Zhang, N.; Li, B.X. Biogeographical Patterns and Assembly of Bacterial Communities in Saline Soils of Northeast China. Microorganisms 2022, 10, 1787. [Google Scholar] [CrossRef]
- Forterre, P.; Brochier, C.; Philippe, H. Evolution of the archaea. Theor. Popul. Biol. 2002, 61, 409–422. [Google Scholar] [CrossRef] [PubMed]
- Spang, A.; Ettema, T.J.G. The methanogenic roots of Archaea. Nat. Microbiol. 2017, 2, 17109. [Google Scholar] [CrossRef]
- Zou, D.Y.; Liu, H.B.; Li, M. Community, Distribution, and ecological roles of estuarine Archaea. Front. Microbiol. 2020, 11, 2060. [Google Scholar] [CrossRef] [PubMed]
- Baker, B.J.; Sheik, C.S.; Taylor, C.A.; Jain, S.; Bhasi, A.; Cavalcoli, J.D.; Dick, G.J. Community transcriptomic assembly reveals microbes that contribute to deep-sea carbon and nitrogen cycling. ISME J. 2013, 7, 1962–1973. [Google Scholar] [CrossRef]
- Ma, B.; Wang, Y.L.; Ye, S.D.; Liu, S.; Stirling, E.; Gilbert, J.A.; Faust, K.; Knight, R.; Jansson, J.K.; Cardona, C.; et al. Earth microbial co-occurrence network reveals interconnection pattern across microbiomes. Microbiome 2020, 8, 82. [Google Scholar] [CrossRef]
- Berry, D.; Widder, S. Deciphering microbial interactions and detecting keystone species with co-occurrence networks. Front. Microbiol. 2014, 5, 219. [Google Scholar] [CrossRef]
- Barberán, A.; Bates, S.T.; Casamayor, E.O.; Fierer, N. Using network analysis to explore co-occurrence patterns in soil microbial communities. ISME J. 2012, 6, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Li, Y.L.; Zhao, X.Y.; Degen, A.A.; Lian, J.; Liu, X.P.; Li, Y.Q.; Duan, Y.L. Fertilizers have a greater impact on the soil bacterial community than on the fungal community in a sandy farmland ecosystem, Inner Mongolia. Ecol. Indic. 2022, 140, 108972. [Google Scholar] [CrossRef]
- Zhao, K.F.; Li, F.Z.; Zhang, F.S. Halophytes in China, 2nd ed.; Science Press: Beijing, China, 2013. [Google Scholar]
- Wang, Z.Q.; Zhu, S.Q.; Yu, R.P. Saline Soil in China; Science Press: Beijing, China, 1993. [Google Scholar]
- Webster, G.; O’Sullivan, L.A.; Meng, Y.; Williams, A.S.; Sass, A.M.; Watkins, A.J.; Parkes, R.J.; Weightman, A.J. Archaeal community diversity and abundance changes along a natural salinity gradient in estuarine sediments. FEMS Microbiol. Ecol. 2015, 91, 1–18. [Google Scholar] [CrossRef]
- Zhao, H.X.; Gu, B.J.; Chen, D.C.; Tang, J.J.; Xu, X.L.; Qiao, Z.; Wang, J.Q. Physicochemical properties and salinization characteristics of soils in coastal land reclamation areas: A case study of China-Singapore Tianjin Eco-City. Heliyon 2022, 8, e12629. [Google Scholar] [CrossRef]
- Mende, D.R.; Waller, A.S.; Sunagawa, S.; Järvelin, A.I.; Chan, M.M.; Arumugam, M.; Raes, J.; Bork, P. Assessment of Metagenomic Assembly Using Simulated Next Generation Sequencing Data. PLoS ONE 2012, 7, e31386. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, H.B.; Almeida, M.; Juncker, A.S.; Rasmussen, S.; Li, J.H.; Sunagawa, S.; Plichta, D.R.; Gautier, L.; Pedersen, A.G.; Le Chatelier, E.; et al. Identification and assembly of genomes and genetic elements in complex metagenomic samples without using reference genomes. Nat. Biotechnol. 2014, 32, 822–828. [Google Scholar] [CrossRef]
- Fu, L.M.; Niu, B.F.; Zhu, Z.W.; Wu, S.T.; Li, W.Z. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Oh, J.; Byrd, A.L.; Deming, C.; Conlan, S.; Kong, H.H.; Segre, J.A.; Progra, N.C.S. Biogeography and individuality shape function in the human skin metagenome. Nature 2014, 514, 59–64. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Mitra, S.; Ruscheweyh, H.J.; Weber, N.; Schuster, S.C. Integrative analysis of environmental sequences using MEGAN4. Genome Res. 2011, 21, 1552–1560. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Hattori, M.; Aoki-Kinoshita, K.F.; Itoh, M.; Kawashima, S.; Katayama, T.; Araki, M.; Hirakawa, M. From genomics to chemical genomics: New developments in KEGG. Nucleic Acids Res. 2006, 34, D354–D357. [Google Scholar] [CrossRef]
- Powell, S.; Forslund, K.; Szklarczyk, D.; Trachana, K.; Roth, A.; Huerta-Cepas, J.; Gabaldón, T.; Rattei, T.; Creevey, C.; Kuhn, M.; et al. eggNOG v4.0: Nested orthology inference across 3686 organisms. Nucleic Acids Res. 2014, 42, D231–D239. [Google Scholar] [CrossRef]
- Paradis, E.; Schliep, K. ape 5.0: An environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 2019, 35, 526–528. [Google Scholar] [CrossRef]
- Peng, G.S.; Wu, J. Optimal network topology for structural robustness based on natural connectivity. Phys. A Stat. Mech. Appl. 2016, 443, 212–220. [Google Scholar] [CrossRef]
- Layeghifard, M.; Hwang, D.M.; Guttman, D.S. Disentangling Interactions in the Microbiome: A network perspective. Trends Microbiol. 2017, 25, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Lefcheck, J.S. piecewiseSEM: Piecewise structural equation modelling in R for ecology, evolution, and systematics. Methods Ecol. Evol. 2016, 7, 573–579. [Google Scholar] [CrossRef]
- Hernández, Z.; Almendros, G. Biogeochemical factors related with organic matter degradation and C storage in agricultural volcanic ash soils. Soil Biol. Biochem. 2012, 44, 130–142. [Google Scholar] [CrossRef]
- Pavloudi, C.; Oulas, A.; Vasileiadou, K.; Sarropoulou, E.; Kotoulas, G.; Arvanitidis, C. Salinity is the major factor influencing the sediment bacterial communities in a Mediterranean lagoonal complex (Amvrakikos Gulf, Ionian Sea). Mar. Genom. 2016, 28, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Malik, A.A.; Puissant, J.; Buckeridge, K.M.; Goodall, T.; Jehmlich, N.; Chowdhury, S.; Gweon, H.S.; Peyton, J.M.; Mason, K.E.; van Agtmaal, M.; et al. Land use driven change in soil pH affects microbial carbon cycling processes. Nat. Commun. 2018, 9, 3591. [Google Scholar] [CrossRef] [PubMed]
- Xiong, C.; Zhu, Y.G.; Wang, J.T.; Singh, B.; Han, L.L.; Shen, J.P.; Li, P.P.; Wang, G.B.; Wu, C.F.; Ge, A.H.; et al. Host selection shapes crop microbiome assembly and network complexity. New Phytol. 2021, 229, 1091–1104. [Google Scholar] [CrossRef] [PubMed]
- Osman, J.R.; Wang, Y.; Jaubert, C.; Nguyen, T.N.; Fernandes, G.R.; DuBow, M.S. The bacterial communities of surface soils from desert sites in the eastern Utah (USA) portion of the Colorado Plateau. Microbiol. Res. 2021, 244, 126664. [Google Scholar] [CrossRef]
- DeBruyn, J.M.; Nixon, L.T.; Fawaz, M.N.; Johnson, A.M.; Radosevich, M. Global Biogeography and Quantitative Seasonal Dynamics of Gemmatimonadetes in Soil. Appl. Environ. Microb. 2011, 77, 6295–6300. [Google Scholar] [CrossRef]
- Araujo, R.; Gupta, V.V.S.R.; Reith, F.; Bissett, A.; Mele, P.; Franco, C.M.M. Biogeography and emerging significance of Actinobacteria in Australia and Northern Antarctica soils. Soil Biol. Biochem. 2020, 146, 107805. [Google Scholar] [CrossRef]
- Xu, J.S.; Gao, W.; Zhao, B.Z.; Chen, M.Q.; Ma, L.; Jia, Z.J.; Zhang, J.B. Bacterial community composition and assembly along a natural sodicity/salinity gradient in surface and subsurface soils. Appl. Soil Ecol. 2021, 157, 103731. [Google Scholar] [CrossRef]
- Oren, A. Microbial life at high salt concentrations: Phylogenetic and metabolic diversity. Saline Syst. 2008, 4, 2. [Google Scholar] [CrossRef] [PubMed]
- GundeCimerman, N.; Oren, A.; Plemenitas, A. Adaptation to Life at High Salt Concentrations in Archaea, Bacteria, and Eukarya. In Adaptation to Life at High Salt Concentrations in Archaea, Bacteria, and Eukarya; GundeCimerman, N., Oren, A., Plemenitas, A., Eds.; Cellular Origin and Life in Extreme Habitats and Astrobiology; Springer: Berlin/Heidelberg, Germany, 2005; Volume 9. [Google Scholar]
- Oren, A. Halophilic Microorganisms and Their Environments; Kluwer Academic Publishers: Norwell, MA, USA, 2002. [Google Scholar]
- Conrad, R. Microbial ecology of methanogens and methanotrophs. In Advances in Agronomy; Donald, L.S., Ed.; Academic Press: Cambridge, MA, USA, 2007; pp. 1–63. [Google Scholar]
- Thauer, R.K.; Kaster, A.K.; Seedorf, H.; Buckel, W.; Hedderich, R. Methanogenic archaea: Ecologically relevant differences in energy conservation. Nat. Rev. Microbiol. 2008, 6, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Kirschke, S.; Bousquet, P.; Ciais, P.; Saunois, M.; Canadell, J.G.; Dlugokencky, E.J.; Bergamaschi, P.; Bergmann, D.; Blake, D.R.; Bruhwiler, L.; et al. Three decades of global methane sources and sinks. Nat. Geosci. 2013, 6, 813–823. [Google Scholar] [CrossRef]
- Liu, Y.Q.; Priscu, J.C.; Xiong, J.B.; Conrad, R.; Vick-Majors, T.; Chu, H.Y.; Hou, J.Z. Salinity drives archaeal distribution patterns in high altitude lake sediments on the Tibetan Plateau. FEMS Microbiol. Ecol. 2016, 92, fiw033. [Google Scholar] [CrossRef] [PubMed]
- Hibbing, M.E.; Fuqua, C.; Parsek, M.R.; Peterson, S.B. Bacterial competition: Surviving and thriving in the microbial jungle. Nat. Rev. Microbiol. 2010, 8, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Ponomarova, O.; Patil, K.R. Metabolic interactions in microbial communities: Untangling the Gordian knot. Curr. Opin. Microbiol. 2015, 27, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Xun, W.B.; Huang, T.; Li, W.; Ren, Y.; Xiong, W.; Ran, W.; Li, D.C.; Shen, Q.R.; Zhang, R.F. Alteration of soil bacterial interaction networks driven by different long-term fertilization management practices in the red soil of South China. Appl. Soil Ecol. 2017, 120, 128–134. [Google Scholar] [CrossRef]
- Szmigiel, I.; Kwiatkowska, D.; Lukaszewicz, M.; Krasowska, A. Xylan Decomposition in Plant Cell Walls as an Inducer of Surfactin Synthesis by Bacillus subtilis. Biomolecules 2021, 11, 239. [Google Scholar] [CrossRef]
- Zlotnikov, A.K.; Shapovalova, Y.N.; Makarov, A.A. Association of Bacillus firmus E3 and Klebsiella terrigena E6 with increased ability for nitrogen fixation. Soil Biol. Biochem. 2001, 33, 1525–1530. [Google Scholar] [CrossRef]
- Fukami, J.; Cerezini, P.; Hungria, M. Azospirillum: Benefits that go far beyond biological nitrogen fixation. AMB Express 2018, 8, 73. [Google Scholar] [CrossRef] [PubMed]
- Han, J.H.; Kim, T.S.; Joung, Y.; Kim, M.N.; Shin, K.S.; Bae, T.; Kim, S.B. Nocardioides endophyticus sp nov and Nocardioides conyzicola sp. nov., isolated from herbaceous plant roots. Int. J. Syst. Evol. Microbiol. 2013, 63, 4730–4734. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Y.; Liu, X.Y.; Liu, S.J. Nocardioides terrae sp. nov., isolated from forest soil. Int. J. Syst. Evol. Microbiol. 2009, 59, 2444–2448. [Google Scholar] [CrossRef]
- Cho, Y.; Jang, G.I.; Cho, B.C. Nocardioides marinquilinus sp. nov., isolated from coastal seawater. Int. J. Syst. Evol. Microbiol. 2013, 63, 2594–2599. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.K.; Pan, Z.; Li, F.N.; Li, X.J.; Liu, S.W.; Tuo, L.; Jiang, M.G.; Sun, C.H. Marmoricola endophyticus sp. nov., an endophytic actinobacterium isolated from Thespesia populnea. Int. J. Syst. Evol. Microbiol. 2017, 67, 4379–4384. [Google Scholar] [CrossRef]
- Gesheva, V.; Vasileva-Tonkova, E. Production of enzymes and antimicrobial compounds by halophilic Antarctic Nocardioides sp grown on different carbon sources. World J. Microbiol. Biotechnol. 2012, 28, 2069–2076. [Google Scholar] [CrossRef] [PubMed]
- Aytas, Z.G.; Tunçer, M.; Kul, Ç.S.; Cilmeli, S.; Aydin, N.; Doruk, T.; Adigüzel, A.O. Partial characterization of ß-glucosidase, ß-xylosidase, and a-larabinofuranosidase from Jiangella alba DSM 45237 and their potential in lignocellulose-based biorefining. Sustain. Chem. Pharm. 2023, 31, 100900. [Google Scholar] [CrossRef]
- Park, D.; Kim, H.; Yoon, S. Nitrous Oxide Reduction by an Obligate Aerobic Bacterium, Gemmatimonas aurantiaca Strain T-27. Appl. Environ. Microb. 2017, 83, e00502-17. [Google Scholar] [CrossRef]
- Gitonga, N.M.; Njeru, E.M.; Cheruiyot, R.; Maingi, J.M. Bradyrhizobium inoculation has a greater effect on soybean growth, production and yield quality in organic than conventional farming systems. Cogent Food Agric. 2021, 7, 1935529. [Google Scholar] [CrossRef]
- Delamuta, J.R.M.; Ribeiro, R.A.; Ormeño-Orrillo, E.; Parma, M.M.; Melo, I.S.; Martínez-Romero, E.; Hungria, M. Bradyrhizobium tropiciagri sp nov and Bradyrhizobium embrapense sp. nov., nitrogen-fixing symbionts of tropical forage legumes. Int. J. Syst. Evol. Microbiol. 2015, 65, 4424–4433. [Google Scholar] [CrossRef]
- Siqueira, A.F.; Minamisawa, K.; Sánchez, C. Anaerobic Reduction of Nitrate to Nitrous Oxide Is Lower in Bradyrhizobium japonicum than in Bradyrhizobium diazoefficiens. Microbes Environ. 2017, 32, 398–401. [Google Scholar] [CrossRef] [PubMed]
- Treude, N.; Rosencrantz, D.; Liesack, W.; Schnell, S. Strain FAc12, a dissimilatory iron-reducing member of the Anaeromyxobacter subgroup of Myxococcales. FEMS Microbiol. Ecol. 2003, 44, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Mori, K.; Suzuki, K.; Yamaguchi, K.; Urabe, T.; Hanada, S. Thiogranum longum gen. nov., sp nov., an obligately chemolithoautotrophic, sulfur-oxidizing bacterium of the family Ectothiorhodospiraceae isolated from a deep-sea hydrothermal field, and an emended description of the genus Thiohalomonas. Int. J. Syst. Evol. Microbiol. 2015, 65, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Minegishi, H.; Echigo, A.; Shimane, Y.; Kamekura, M.; Itoh, T.; Ohkuma, M.; Usami, R. Halococcus agarilyticus sp. nov., an agar-degrading haloarchaeon isolated from commercial salt. Int. J. Syst. Evol. Microbiol. 2015, 65, 1634–1639. [Google Scholar] [CrossRef] [PubMed]
- Moshfegh, M.; Shahverdi, A.R.; Zarrini, G.; Faramarzi, M.A. Biochemical characterization of an extracellular polyextremophilic α-amylase from the halophilic archaeon. Extremophiles 2013, 17, 677–687. [Google Scholar] [CrossRef]
- Gupta, M.; Choudhury, B.; Navani, N.K. Production and characterization of an organic solvent activated protease from haloalkaliphilic bacterium Halobiforma sp. strain BNMIITR. Heliyon 2024, 10, e25084. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.Y.; Zeng, C.L.; Hou, Z.Z.; Wang, Y.; Xu, Q.; Isobe, K.; Senoo, K.; Zhu, L. The complete genome sequence of the archaeal isolate Halomicrobium sp. ZPS1 reveals the nitrogen metabolism characteristics under hypersaline conditions. Ann. Microbiol. 2020, 70, 29. [Google Scholar] [CrossRef]
- Liu, Q.; Chen, C.L.; Chen, Y.H.; Huang, B.C.; Yang, Y.H.; Zhu, H.; Li, Y.H.; Wang, X.P.; Zhang, C.F. Intervention of rhamnolipid improves the rhizosphere microenvironment of cotton in desert saline lands. Environ. Technol. Innov. 2023, 32, 103378. [Google Scholar] [CrossRef]
- Franklin, R.B.; Morrissey, E.M.; Morina, J.C. Changes in abundance and community structure of nitrate-reducing bacteria along a salinity gradient in tidal wetlands. Pedobiologia 2017, 60, 21–26. [Google Scholar] [CrossRef]
- Yan, N.; Marschner, P.; Cao, W.H.; Zuo, C.Q.; Qin, W. Influence of salinity and water content on soil microorganisms. Int. Soil Water Conserv. Res. 2015, 3, 316–323. [Google Scholar] [CrossRef]
- Naylor, D.; Coleman-Derr, D. Drought Stress and Root-Associated Bacterial Communities. Front. Plant Sci. 2018, 8, 303756. [Google Scholar] [CrossRef] [PubMed]
- Van Horn, D.J.; Okie, J.G.; Buelow, H.N.; Gooseff, M.N.; Barrett, J.E.; Takacs-Vesbach, C.D. Soil Microbial Responses to Increased Moisture and Organic Resources along a Salinity Gradient in a Polar Desert. Appl. Environ. Microb. 2014, 80, 3034–3043. [Google Scholar] [CrossRef] [PubMed]
- Lauber, C.L.; Hamady, M.; Knight, R.; Fierer, N. Pyrosequencing-Based Assessment of Soil pH as a Predictor of Soil Bacterial Community Structure at the Continental Scale. Appl. Environ. Microb. 2009, 75, 5111–5120. [Google Scholar] [CrossRef] [PubMed]
- Bhuyan, M.I.; Mia, S.; Supit, I.; Ludwig, F. Spatio-temporal variability in soil and water salinity in the south-central coast of Bangladesh. Catena 2023, 222, 106786. [Google Scholar] [CrossRef]
- Bhuyan, M.I.; Supit, I.; Mia, S.; Mulder, M.; Ludwig, F. Effect of soil and water salinity on dry season boro rice production in the south-central coastal area of Bangladesh. Heliyon 2023, 9, e19180. [Google Scholar] [CrossRef] [PubMed]
- Acik, M.M.; Gryta, A.; Sas-Paszt, L.; Fr, M. New insight into the soil bacterial and fungal microbiome after phosphorus biofertilizer application as an important driver of regenerative agriculture including biodiversity loss reversal and soil health restoration. Appl. Soil Ecol. 2023, 189, 104941. [Google Scholar] [CrossRef]
- Nain, L.; Rana, A.; Joshi, M.; Jadhav, S.D.; Kumar, D.; Shivay, Y.S.; Paul, S.; Prasanna, R. Evaluation of synergistic effects of bacterial and cyanobacterial strains as biofertilizers for wheat. Plant Soil 2010, 331, 217–230. [Google Scholar] [CrossRef]
- Martinez-Hidalgo, P.; Flores-Felix, J.D.; Sanchez-Juanes, F.; Rivas, R.; Mateos, P.F.; Santa Regina, I.; Peix, A.; Martinez-Molina, E.; Igual, J.M.; Velazquez, E. Identification of Canola Roots Endophytic Bacteria and Analysis of Their Potential as Biofertilizers for Canola Crops with Special Emphasis on Sporulating Bacteria. Agronomy 2021, 11, 1796. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Archaea | Bacteria | |||||
|---|---|---|---|---|---|---|
| HWLS | LWMS | LWHS | HWLS | LWMS | LWHS | |
| Nodes | 40 | 40 | 39 | 78 | 90 | 89 |
| Edges | 535 | 593 | 284 | 636 | 574 | 1412 |
| Positive edges percent | 100 | 100 | 100 | 92 | 72 | 98 |
| Average degree | 26.75 | 29.65 | 14.56 | 16.31 | 12.76 | 31.73 |
| Clustering coefficient | 0.76 | 0.95 | 0.82 | 0.79 | 0.55 | 0.81 |
| Average path length | 0.98 | 1.09 | 1.56 | 1.99 | 1.91 | 1.65 |
| Network diameter | 2 | 2 | 4 | 5 | 5 | 5 |
| Density | 0.69 | 0.76 | 0.38 | 0.21 | 0.14 | 0.36 |
| Modularity | 0.12 | 0.08 | 0.36 | 0.33 | 0.41 | 0.20 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Wang, J.; Li, E.; Yang, X.; Yang, J. Shifts in Microbial Community Structure and Co-occurrence Network along a Wide Soil Salinity Gradient. Microorganisms 2024, 12, 1268. https://doi.org/10.3390/microorganisms12071268
Li Y, Wang J, Li E, Yang X, Yang J. Shifts in Microbial Community Structure and Co-occurrence Network along a Wide Soil Salinity Gradient. Microorganisms. 2024; 12(7):1268. https://doi.org/10.3390/microorganisms12071268
Chicago/Turabian StyleLi, Yan, Juan Wang, Eryang Li, Xiaodong Yang, and Jianjun Yang. 2024. "Shifts in Microbial Community Structure and Co-occurrence Network along a Wide Soil Salinity Gradient" Microorganisms 12, no. 7: 1268. https://doi.org/10.3390/microorganisms12071268