
Detection of Klebsiella pneumoniae Carbapenem Resistance Genes by qPCR: Choosing the Right Method for Total DNA Extraction
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. qPCR
2.2. Preparation of DNA Standards
2.3. Cultivation of Klebsiella pneumoniae
2.4. CFU/mL Estimation by OD Measurement
2.5. Isolation of Klebsiella pneumoniae DNA with Different Extraction Methods
2.6. Assessment of Concentration and Quality of DNA Isolated from K. pneumoniae Strains
2.7. Synthetic Stool Matrix as Model for Fecal Contamination
2.8. Processing of qPCR Data
3. Results
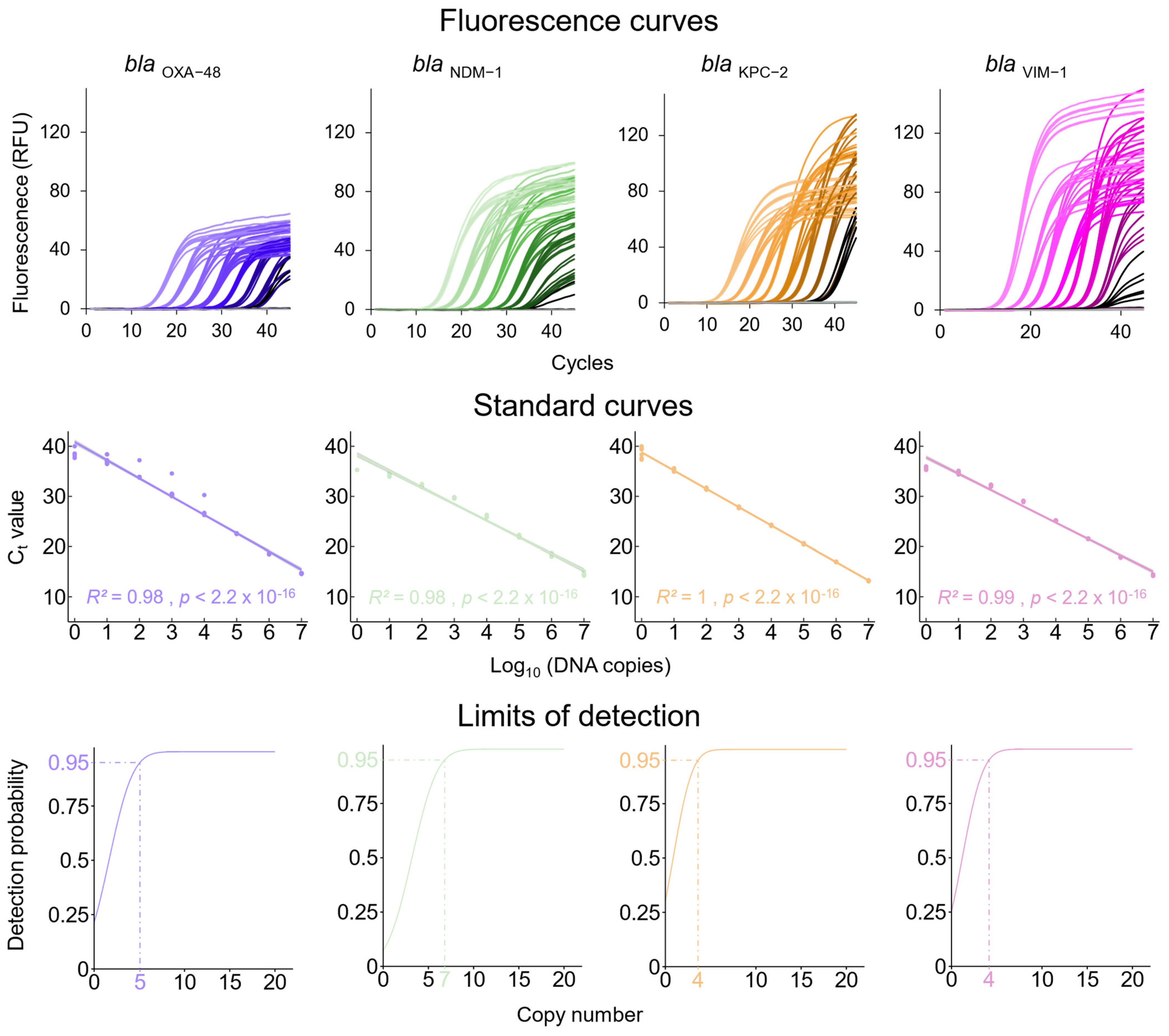
3.1. qPCRs Assays for the Detection of Carbapenem Resistance Genes and the K. pneumoniae Chromosome-Specific khe Gene
3.2. Analytical Sensitivity of the Newly Established qPCR Assays
3.3. Preparation of K. pneumoniae Culture Samples for DNA Extraction
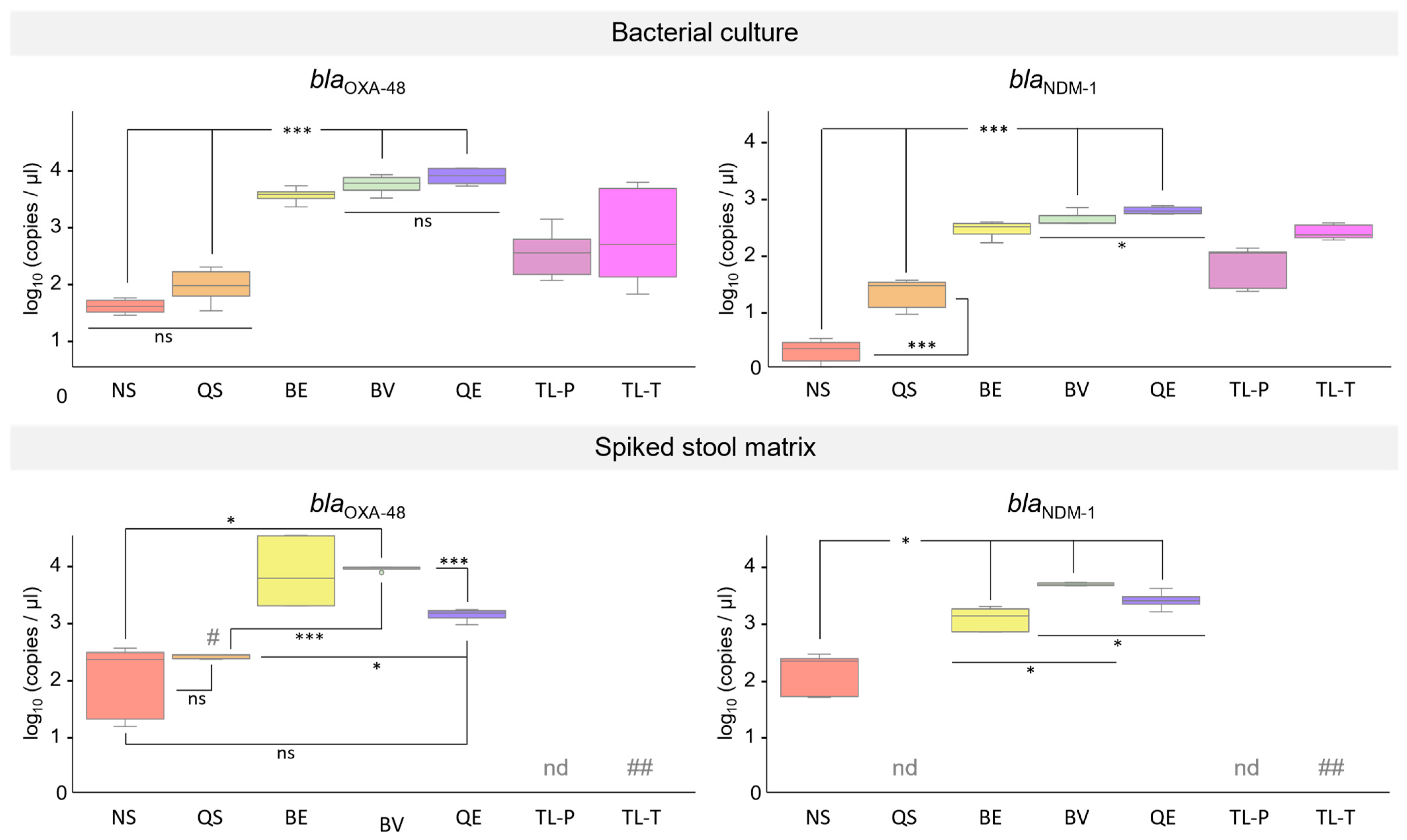
3.4. Estimation of the Quantity and Quality of Total Bacterial DNA Isolated with the Different Extraction Methods
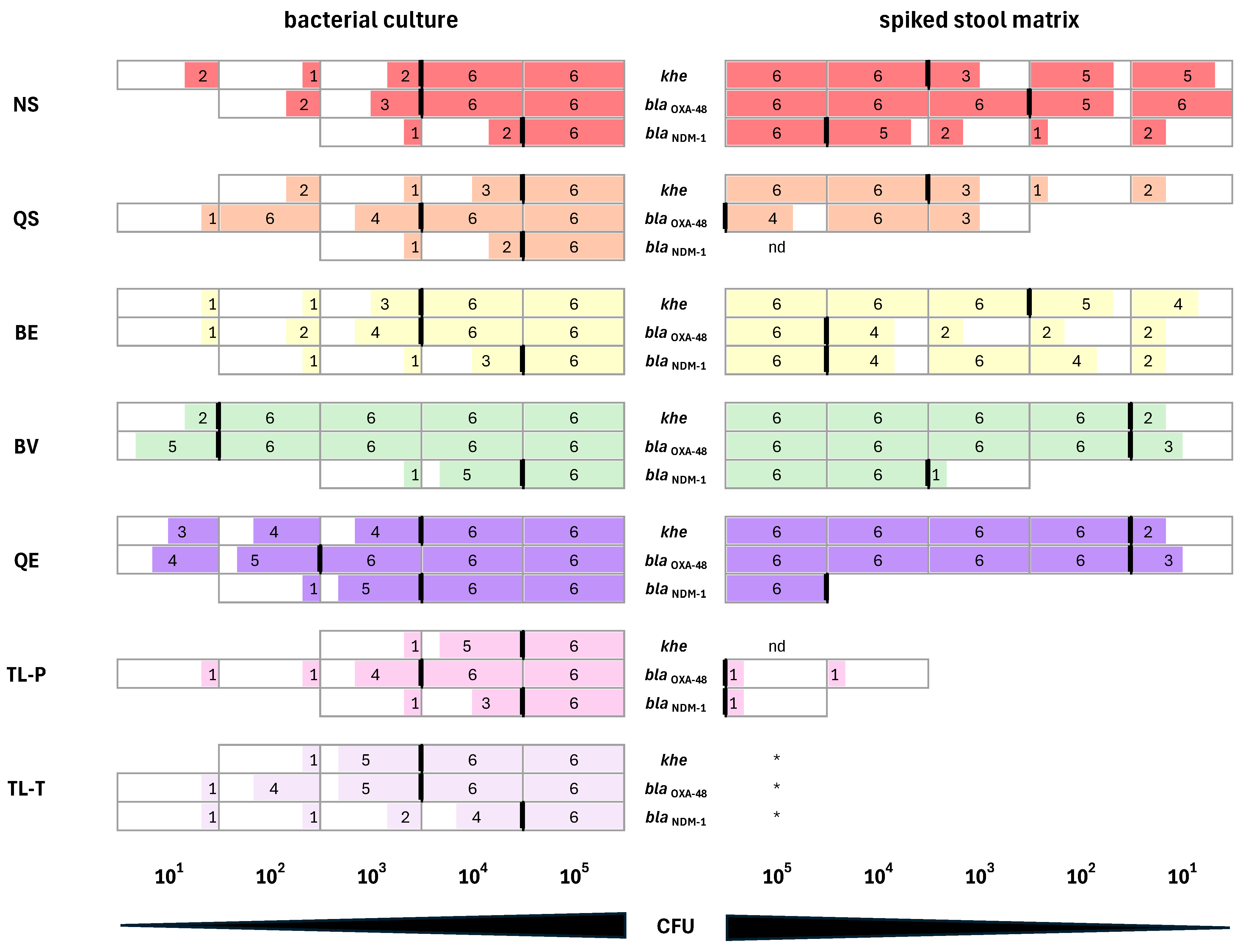
3.5. Performance of DNA Extracted from K. pneumoniae Cultures in qPCR Assays
3.6. Performance of DNA Isolated from Synthetic Stool Matrix Spiked with Cultures of K. pneumoniae Strains Carrying Two Resistance Genes in qPCR Assays
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| A260/A280 ratio | absorbance ratio 260 nm/280 nm |
| BE | EchoLUTION Buccal Swab DNA Kit, BioEcho |
| BV | EchoLUTION Viral RNA/DNA Swab Kit Plus, BioEcho |
| bla | beta-lactamase/β-lactamase |
| CFU | colony-forming unit(s) |
| CIM | carbapenem inactivation method |
| CRE | carbapenem-resistant Enterobacterales |
| Ct | cycle threshold |
| Ctr | raw cycle threshold |
| IMP | imipenem-hydrolyzing metallo-β-lactamase |
| khe | K. pneumoniae hemolysin |
| KPC | K. pneumoniae carbapenemase |
| K. pneumoniae | Klebsiella pneumoniae |
| LAMP | loop-mediated isothermal amplification |
| LOD | limit of detection |
| MRGN | multi-resistant Gram-negative |
| NAAT | Nucleic acid amplification test |
| NDM | New Delhi metallo-β-lactamase |
| NRZ | National Reference Centre for multidrug-resistant Gram-negative bacteria, Germany |
| NS | NucleoSpin DNA Stool kit, Macherey-Nagel |
| NTC | non-template control |
| OD600 | optical density 600 nm |
| OXA | oxacillinase β-lactamase |
| PCR | polymerase chain reaction |
| QE | Quick Extract solution, Lucigen |
| qPCR | quantitative polymerase chain reaction |
| QS | QIAamp Fast DNA Stool Mini kit, Qiagen |
| RFU | relative fluorescence unit(s) |
| RPA | recombinase polymerase amplification |
| rpm | revolutions per minute |
| SD | standard deviation |
| TL-P | thermal lysis PCR, direct PCR |
| TL-T | thermal lysis thermoblock |
| UV/VIS | ultraviolet/visible |
| ve | elution/extraction volume |
| VIM | verona integron-encoded metallo-β-lactamase |
| vs | sample volume |
References
- Daikos, G.L.; Tsaousi, S.; Tzouvelekis, L.S.; Anyfantis, I.; Psichogiou, M.; Argyropoulou, A.; Stefanou, I.; Sypsa, V.; Miriagou, V.; Nepka, M.; et al. Carbapenemase-Producing Klebsiella pneumoniae Bloodstream Infections: Lowering Mortality by Antibiotic Combination Schemes and the Role of Carbapenems. Antimicrob. Agents Chemother. 2014, 58, 2322–2328. [Google Scholar] [CrossRef]
- Tzouvelekis, L.S.; Markogiannakis, A.; Psichogiou, M.; Tassios, P.T.; Daikos, G.L. Carbapenemases in Klebsiella pneumoniae and Other Enterobacteriaceae: An Evolving Crisis of Global Dimensions. Clin. Microbiol. Rev. 2012, 25, 682–707. [Google Scholar] [CrossRef]
- Satlin, M.J.; Jenkins, S.G.; Walsh, T.J. The Global Challenge of Carbapenem-Resistant Enterobacteriaceae in Transplant Recipients and Patients with Hematologic Malignancies. Clin. Infect. Dis. 2014, 58, 1274–1283. [Google Scholar] [CrossRef]
- David, S.; Reuter, S.; Harris, S.R.; Glasner, C.; Feltwell, T.; Argimon, S.; Abudahab, K.; Goater, R.; Giani, T.; Errico, G.; et al. Epidemic of Carbapenem-Resistant Klebsiella pneumoniae in Europe Is Driven by Nosocomial Spread. Nat. Microbiol. 2019, 4, 1919–1929. [Google Scholar] [CrossRef]
- Hansen, G.T. Continuous Evolution: Perspective on the Epidemiology of Carbapenemase Resistance among Enterobacterales and Other Gram-Negative Bacteria. Infect. Dis. Ther. 2021, 10, 75–92. [Google Scholar] [CrossRef]
- Cui, X.; Zhang, H.; Du, H. Carbapenemases in Enterobacteriaceae: Detection and Antimicrobial Therapy. Front. Microbiol. 2019, 10, 1823. [Google Scholar] [CrossRef]
- Lou, T.; Du, X.; Zhang, P.; Shi, Q.; Han, X.; Lan, P.; Yan, R.; Hu, H.; Wang, Y.; Wu, X.; et al. Risk Factors for Infection and Mortality Caused by Carbapenem-Resistant Klebsiella Pneumoniae: A Large Multicentre Case–Control and Cohort Study. J. Infect. 2022, 84, 637–647. [Google Scholar] [CrossRef]
- Ramos-Castañeda, J.A.; Ruano-Ravina, A.; Barbosa-Lorenzo, R.; Paillier-Gonzalez, J.E.; Saldaña-Campos, J.C.; Salinas, D.F.; Lemos-Luengas, E.V. Mortality Due to KPC Carbapenemase-Producing Klebsiella Pneumoniae Infections: Systematic Review and Meta-Analysis: Mortality Due to KPC Klebsiella pneumoniae Infections. J. Infect. 2018, 76, 438–448. [Google Scholar] [CrossRef]
- Banerjee, R.; Humphries, R. Clinical and Laboratory Considerations for the Rapid Detection of Carbapenem-Resistant Enterobacteriaceae. Virulence 2017, 8, 427–439. [Google Scholar] [CrossRef]
- Effah, C.Y.; Sun, T.; Liu, S.; Wu, Y. Klebsiella Pneumoniae: An Increasing Threat to Public Health. Ann. Clin. Microbiol. Antimicrob. 2020, 19, 1. [Google Scholar] [CrossRef]
- Girlich, D.; Poirel, L.; Nordmann, P. Value of the Modified Hodge Test for Detection of Emerging Carbapenemases in Enterobacteriaceae. J. Clin. Microbiol. 2012, 50, 477–479. [Google Scholar] [CrossRef]
- Kaase, M.; Szabados, F.; Wassill, L.; Gatermann, S.G. Detection of Carbapenemases in Enterobacteriaceae by a Commercial Multiplex PCR. J. Clin. Microbiol. 2012, 50, 3115–3118. [Google Scholar] [CrossRef]
- Saliba, R.; Aho-Glélé, L.S.; Karam-Sarkis, D.; Zahar, J.R. Evaluation of Polymerase Chain Reaction Assays for Direct Screening of Carbapenemase-Producing Enterobacteriaceae from Rectal Swabs: A Diagnostic Meta-Analysis. J. Hosp. Infect. 2020, 104, 381–389. [Google Scholar] [CrossRef]
- Nakano, R.; Nakano, A.; Ishii, Y.; Ubagai, T.; Kikuchi-Ueda, T.; Kikuchi, H.; Tansho-Nagakawa, S.; Kamoshida, G.; Mu, X.; Ono, Y. Rapid Detection of the Klebsiella Pneumoniae Carbapenemase (KPC) Gene by Loop-Mediated Isothermal Amplification (LAMP). J. Infect. Chemother. 2015, 21, 202–206. [Google Scholar] [CrossRef]
- Wang, F.; Wang, L.; Chen, H.; Li, N.; Wang, Y.; Li, Y.; Liang, W. Rapid Detection of Bla KPC, Bla NDM, Bla OXA-48-like and Bla IMP Carbapenemases in Enterobacterales Using Recombinase Polymerase Amplification Combined with Lateral Flow Strip. Front. Cell. Infect. Microbiol. 2021, 11, 772966. [Google Scholar] [CrossRef]
- Hrabák, J.; Chudáčková, E.; Papagiannitsis, C.C. Detection of Carbapenemases in Enterobacteriaceae: A Challenge for Diagnostic Microbiological Laboratories. Clin. Microbiol. Infect. 2014, 20, 839–853. [Google Scholar] [CrossRef]
- Sun, D.; Jeannot, K.; Xiao, Y.; Knapp, C.W. Editorial: Horizontal Gene Transfer Mediated Bacterial Antibiotic Resistance. Front. Microbiol. 2019, 10, 478460. [Google Scholar] [CrossRef]
- Nordmann, P.; Naas, T.; Poirel, L. Global Spread of Carbapenemase-Producing Enterobacteriaceae. Emerg. Infect. Dis. 2011, 17, 1791–1798. [Google Scholar] [CrossRef]
- Arpin, C.; Noury, P.; Boraud, D.; Coulange, L.; Manetti, A.; André, C.; M’Zali, F.; Quentin, C. NDM-1-Producing Klebsiella pneumoniae Resistant to Colistin in a French Community Patient without History of Foreign Travel. Antimicrob. Agents Chemother. 2012, 56, 3432–3434. [Google Scholar] [CrossRef]
- David, S.; Cohen, V.; Reuter, S.; Sheppard, A.E.; Giani, T.; Parkhill, J.; Rossolini, G.M.; Feil, E.J.; Grundmann, H.; Aanensen, D.M. Integrated Chromosomal and Plasmid Sequence Analyses Reveal Diverse Modes of Carbapenemase Gene Spread among Klebsiella pneumoniae. Proc. Natl. Acad. Sci. USA 2020, 117, 25043–25054. [Google Scholar] [CrossRef]
- Di Pinto, A.; Forte, V.; Guastadisegni, M.C.; Martino, C.; Schena, F.P.; Tantillo, G. A Comparison of DNA Extraction Methods for Food Analysis. Food Control 2007, 18, 76–80. [Google Scholar] [CrossRef]
- J Shetty, P. The Evolution of DNA Extraction Methods. Am. J. Biomed. Sci. Res. 2020, 8, 39–45. [Google Scholar] [CrossRef]
- Shin, J.H. Nucleic Acid Extraction Techniques. In Advanced Techniques in Diagnostic Microbiology; Springer: Boston, MA, USA, 2012; pp. 209–225. [Google Scholar]
- Qiagen QIAGEN® Plasmid Purification Handbook; QIAGEN: Hilden, Germany, 2023.
- Norman, A.; Riber, L.; Luo, W.; Li, L.L.; Hansen, L.H.; Sørensen, S.J. An Improved Method for Including Upper Size Range Plasmids in Metamobilomes. PLoS ONE 2014, 9, e104405. [Google Scholar] [CrossRef]
- Hwan Shin, J. Nucleic Acid Extraction and Enrichment. In Advanced Techniques in Diagnostic Microbiology: Volume 1: Techniques, 3rd ed.; Springer: Cham, Switzerland, 2018; pp. 273–292. [Google Scholar]
- Qiagen QIAGEN Large-Construct Handbook—April 2012—(EN). Available online: https://www.qiagen.com/us/resources/download.aspx?id=8f67b644-6d21-4ef3-b33e-a60f32623785&lang=en (accessed on 8 June 2024).
- Monteiro, J.; Widen, R.H.; Pignatari, A.C.C.; Kubasek, C.; Silbert, S. Rapid Detection of Carbapenemase Genes by Multiplex Real-Time PCR. J. Antimicrob. Chemother. 2012, 67, 906–909. [Google Scholar] [CrossRef]
- Hindiyeh, M.; Smollen, G.; Grossman, Z.; Ram, D.; Davidson, Y.; Mileguir, F.; Vax, M.; Ben David, D.; Tal, I.; Rahav, G.; et al. Rapid Detection of BlaKPC Carbapenemase Genes by Real-Time PCR. J. Clin. Microbiol. 2008, 46, 2879. [Google Scholar] [CrossRef] [PubMed]
- Bisiklis, A.; Papageorgiou, F.; Frantzidou, F.; Alexiou-Daniel, S. Specific Detection of BlaVIM and BlaIMP Metallo-β-Lactamase Genes in a Single Real-Time PCR. Clin. Microbiol. Infect. 2007, 13, 1201–1203. [Google Scholar] [CrossRef]
- Katevatis, C.; Fan, A.; Klapperich, C.M. Low Concentration DNA Extraction and Recovery Using a Silica Solid Phase. PLoS ONE 2017, 12, e0176848. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.; Hucklenbroich, J. Rapid Purification of High Quality Nucleic Acids from Biological Samples. U.S. Patent Number US 11,578,319 B2, 14 February 2023. [Google Scholar]
- Schurig, S.; Kobialka, R.; Wende, A.; Ashfaq Khan, M.A.; Lübcke, P.; Eger, E.; Schaufler, K.; Daugschies, A.; Truyen, U.; Abd El Wahed, A. Rapid Reverse Purification DNA Extraction Approaches to Identify Microbial Pathogens in Wastewater. Microorganisms 2023, 11, 813. [Google Scholar] [CrossRef]
- Schiwy, M.; Schönfels, C.; Weiter, M.; Torres Benito, L.; Laura Torres Benito, C. Diagnosis of Enteropathogenic Viruses from Clinical Stool Samples Using EchoLUTIONTM Nucleic Acid Extraction Technology. Available online: https://bioecho-prod-media-hp.s3.sbg.perf.cloud.ovh.net/media/21/22/56/1698658588/APN_Entero_viruses_EN_002_online.pdf (accessed on 10 June 2024).
- Srirungruang, S.; Mahajindawong, B.; Nimitpanya, P.; Bunkasem, U.; Ayuyoe, P.; Nuchprayoon, S.; Sanprasert, V. Comparative Study of DNA Extraction Methods for the PCR Detection of Intestinal Parasites in Human Stool Samples. Diagnostics 2022, 12, 2588. [Google Scholar] [CrossRef] [PubMed]
- Hart, M.L.; Meyer, A.; Johnson, P.J.; Ericsson, A.C. Comparative Evaluation of DNA Extraction Methods from Feces of Multiple Host Species for Downstream Next-Generation Sequencing. PLoS ONE 2015, 10, e0143334. [Google Scholar] [CrossRef]
- Roperch, J.P.; Benzekri, K.; Mansour, H.; Incitti, R. Improved Amplification Efficiency on Stool Samples by Addition of Spermidine and Its Use for Non-Invasive Detection of Colorectal Cancer. BMC Biotechnol. 2015, 15, 41. [Google Scholar] [CrossRef]
- Monteiro, L.; Bonnemaison, D.; Vekris, A.; Petry, K.G.; Bonnet, J.; Vidal, R.; Cabrita, J.; Mégraud, F. Complex Polysaccharides as PCR Inhibitors in Feces: Helicobacter Pylori Model. J. Clin. Microbiol. 1997, 35, 995–998. [Google Scholar] [CrossRef] [PubMed]
- Chanderraj, R.; Brown, C.A.; Hinkle, K.; Falkowski, N.; Woods, R.J.; Dickson, R.P. The Bacterial Density of Clinical Rectal Swabs Is Highly Variable, Correlates with Sequencing Contamination, and Predicts Patient Risk of Extraintestinal Infection. Microbiome 2022, 10, 2. [Google Scholar] [CrossRef] [PubMed]
- Warnke, P.; Johanna Pohl, F.P.; Kundt, G.; Podbielski, A. Screening for Gram-Negative Bacteria: Impact of Preanalytical Parameters. Sci. Rep. 2016, 6, 30427. [Google Scholar] [CrossRef] [PubMed]
- Gand, M.; Bloemen, B.; Vanneste, K.; Roosens, N.H.C.; De Keersmaecker, S.C.J. Comparison of 6 DNA Extraction Methods for Isolation of High Yield of High Molecular Weight DNA Suitable for Shotgun Metagenomics Nanopore Sequencing to Detect Bacteria. BMC Genom. 2023, 24, 438. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, I.; Behrmann, O.; Klingenberg-Ernst, M.; Rupnik, M.; Hufert, F.T.; Dame, G.; Weidmann, M. Rapid Isothermal Detection of Pathogenic Clostridioides Difficile Using Recombinase Polymerase Amplification. Anal. Chem. 2024, 96, 3267–3275. [Google Scholar] [CrossRef]
- Hassanain, W.A.; Spoors, J.; Johnson, C.L.; Faulds, K.; Keegan, N.; Graham, D. Rapid Ultra-Sensitive Diagnosis of Clostridium Difficile Infection Using a SERS-Based Lateral Flow Assay. Analyst 2021, 146, 4495. [Google Scholar] [CrossRef] [PubMed]
- Herigstad, B.; Hamilton, M.; Heersink, J. How to Optimize the Drop Plate Method for Enumerating Bacteria. J. Microbiol. Methods 2001, 44, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Smieja, M.; Mahony, J.B.; Goldsmith, C.H.; Chong, S.; Petrich, A.; Chernesky, M. Replicate PCR Testing and Probit Analysis for Detection and Quantitation of Chlamydia Pneumoniae in Clinical Specimens. J. Clin. Microbiol. 2001, 39, 1796–1801. [Google Scholar] [CrossRef]
- Ritz, C.; Spiess, A.N. QpcR: An R Package for Sigmoidal Model Selection in Quantitative Real-Time Polymerase Chain Reaction Analysis. Bioinformatics 2008, 24, 1549–1551. [Google Scholar] [CrossRef]
- Pabinger, S.; Rödiger, S.; Kriegner, A.; Vierlinger, K.; Weinhäusel, A. A Survey of Tools for the Analysis of Quantitative PCR (QPCR) Data. Biomol. Detect. Quantif. 2014, 1, 23–33. [Google Scholar] [CrossRef]
- Behrmann, O.; Hügle, M.; Bronsert, P.; Herde, B.; Heni, J.; Schramm, M.; Hufert, F.T.; Urban, G.A.; Dame, G. A Lab-on-a-Chip for Rapid MiRNA Extraction. PLoS ONE 2019, 14, e0226571. [Google Scholar] [CrossRef]
- Welch, B.L. The Generalisation of “Student’s” Problems When Several Different Population Variances Are Involved. Biometrika 1947, 34, 28–35. [Google Scholar]
- Kuzina, E.S.; Kislichkina, A.A.; Sizova, A.A.; Skryabin, Y.P.; Novikova, T.S.; Ershova, O.N.; Savin, I.A.; Khokhlova, O.E.; Bogun, A.G.; Fursova, N.K. High-Molecular-Weight Plasmids Carrying Carbapenemase Genes BlaNDM-1, BlaKPC-2, and BlaOXA-48 Coexisting in Clinical Klebsiella pneumoniae Strains of ST39. Microorganisms 2023, 11, 459. [Google Scholar] [CrossRef]
- Starkova, P.; Lazareva, I.; Avdeeva, A.; Sulian, O.; Likholetova, D.; Ageevets, V.; Lebedeva, M.; Gostev, V.; Sopova, J.; Sidorenko, S. Emergence of Hybrid Resistance and Virulence Plasmids Harboring New Delhi Metallo-β-Lactamase in Klebsiella Pneumoniae in Russia. Antibiotics 2021, 10, 691. [Google Scholar] [CrossRef]
- Schrader, C.; Schielke, A.; Ellerbroek, L.; Johne, R. PCR Inhibitors—Occurrence, Properties and Removal. J. Appl. Microbiol. 2012, 113, 1014–1026. [Google Scholar] [CrossRef]
- Kreader, C.A. Relief of Amplification Inhibition in PCR with Bovine Serum Albumin or T4 Gene 32 Protein. Appl. Environ. Microbiol. 1996, 62, 1102–1106. [Google Scholar] [CrossRef]
- Becker, L.; Steglich, M.; Fuchs, S.; Werner, G.; Nübel, U. Comparison of Six Commercial Kits to Extract Bacterial Chromosome and Plasmid DNA for MiSeq Sequencing. Sci. Rep. 2016, 6, 10–14. [Google Scholar] [CrossRef]
- Macherey-Nagel NucleoSpin DNA Stool User Manual. Available online: https://www.mn-net.com/media/pdf/e3/88/69/Instruction-NucleoSpin-DNA-Stool.pdf (accessed on 10 May 2024).
- Qiagen QIAamp Fast DNA Stool Mini Handbook—QIAGEN. Available online: https://www.qiagen.com/us/resources/download.aspx?id=2a3f2c0b-2e8a-49fd-b442-829108ae1a4a&lang=en (accessed on 10 May 2024).
- Ma, Z.Y.; Zhang, X.M.; Wang, R.; Wang, M.; Liu, T.; Tan, Z.L. Effects of Chemical and Mechanical Lysis on Microbial DNA Yield, Integrity, and Downstream Amplicon Sequencing of Rumen Bacteria and Protozoa. Front. Microbiol. 2020, 11, 581227. [Google Scholar] [CrossRef] [PubMed]
- Gryp, T.; Glorieux, G.; Joossens, M.; Vaneechoutte, M. Comparison of Five Assays for DNA Extraction from Bacterial Cells in Human Faecal Samples. J. Appl. Microbiol. 2020, 129, 378–388. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Extraction Method (Abbreviation) | Extraction Principle | Intended Use | Elution Volume (µL) | Hands on Time | List Price per Reaction (EUR) | ||
|---|---|---|---|---|---|---|---|
| column-based | NucleoSpin DNA Stool kit, Macherey-Nagel | (NS) | chemical lysis, heating, and mechanical lysis (bead beating); clean-up column; silica spin column (for binding of DNA and subsequent elution) | Isolation of human and bacterial DNA from stool samples | 50 | ≈1 h | 5.52 |
| QIAamp Fast DNA Stool Mini kit, Qiagen | (QS) | chemical lysis, heating; silica spin column (for binding of DNA and subsequent elution) | Isolation of human and bacterial DNA from stool samples | 200 | ≈1 h | 7.22 | |
| EchoLUTION Buccal Swab DNA Kit, BioEcho | (BE) | chemical lysis, heating; reverse elution (DNA in flow-through); spin column format | Isolation of viral DNA from buccal swab samples | 100 | ≈30 min | 3.30 | |
| plate-based | EchoLUTION Viral RNA/DNA Swab Kit Plus, BioEcho | (BV) | chemical and enzymatic lysis, heating; reverse elution (DNA in flow-through); plate format | Isolation of viral DNA/RNA from naso- and oropharyngeal swabs or stool samples | 90 | ≈15 min | 4.62 |
| lysis-only | Quick Extract Solution, Lucigen | (QE) | heating, chemical lysis | Isolation of pro- and eukaryotic DNA from bacterial colonies and eucaryotic tissues | 400 | 9 min | 2.84 |
| Thermal lysis P | (TL-P) | heating (as first step of qPCR) | Isolation of prokaryotic DNA from bacterial colonies | 200 | 6 min | - | |
| Thermal lysis T | (TL-T) | heating (as separate step in thermoblock) | Isolation of prokaryotic DNA from bacterial colonies | 200 | 6 min | - | |
| Oligonucleotide | Sequence (5′ → 3′) | |
|---|---|---|
| blaOXA-48 | forward primer | GATGGACAGACGCGCGATA |
| reverse primer | ACTGAATATTTCATCGCGGTGAT | |
| probe | (6-FAM)-CGCCACTT(BMN-Q535)GGAATCGCGATCATAATC-(BMN-Q535) | |
| blaNDM-1 | forward primer | CGTGCTGGTGGTCGATACC |
| reverse primer | CCTGCTTGATCCAGTTGAGGAT | |
| probe | (6-FAM)-CCTGGACC(BMN-Q535)GATGACCAGACCGC-(BMN-Q535) | |
| blaKPC-2 | forward primer | CGCGGAACCATTCGCTAA |
| reverse primer | CGGTATCCATCGCGTACACA | |
| probe | (6-FAM)-CTCGAACCA(BMN-Q535)GGACTTTGGCGGCTCC-(BMN-Q535) | |
| blaVIM-1 | forward primer | CGCTTCGGTCCAGTAGAGCTCT |
| reverse primer | CCACCGTATAGCACGTTCGCTG | |
| probe | (6-FAM)-TCCTGGTG(BMN-Q535)CTGCGCATTCGACCGACA-(BMN-Q535) | |
| khe | forward primer | GATGAAACGACCTGATTGCATTC |
| reverse primer | CCGGGCTGTCGGGATAAG | |
| probe | (6-FAM)-CGCGAACTGGAAGGGCCCG-(BHQ1) | |
| Extraction Method | Targeted Gene | Ct Value | Copy Number |
|---|---|---|---|
| NS | blaOXA-48 | 35.8 (±0.3) | 10 |
| blaNDM-1 | 36.1 (±0.5) | 3 | |
| blaKPC-2 | 38.4 (±0.6) | 1 | |
| blaVIM-1 | 35.9 (±0.7) | 3 | |
| QS | blaOXA-48 | 33.6 (±1.5) | 70 |
| blaNDM-1 | 33.8 (±0.7) | 20 | |
| blaKPC-2 | 36.9 (±0.6) | 3 | |
| blaVIM-1 | 35.0 (±0.4) | 6 | |
| BE | blaOXA-48 | 31.0 (±1.8) | 380 |
| blaNDM-1 | 30.2 (±0.8) | 210 | |
| blaKPC-2 | 29.2 (±0.3) | 420 | |
| blaVIM-1 | 28.0 (±0.3) | 850 | |
| BV | blaOXA-48 | 28.5 (±1.8) | 2160 |
| blaNDM-1 | 29.0 (±0.8) | 520 | |
| blaKPC-2 | 29.0 (±1.4) | 520 | |
| blaVIM-1 | 28.3 (±0.9) | 820 | |
| QE | blaOXA-48 | 26.7 (±0.7) | 6010 |
| blaNDM-1 | 28.7 (±0.6) | 1340 | |
| blaKPC-2 | 28.1 (±0.2) | 870 | |
| blaVIM-1 | 28.4 (±0.5) | 700 | |
| TL-T | blaOXA-48 | 28.8 (±0.1) | 1560 |
| blaNDM-1 | 30.8 (±0.5) | 140 | |
| blaKPC-2 | 32.1 (±0.7) | 70 | |
| blaVIM-1 | 31.5 (± 0.5) | 80 | |
| TL-P | blaOXA-48 | 28.5 (±0.4) | 1860 |
| blaNDM-1 | 33.1 (±0.3) | 20 | |
| blaKPC-2 | 34.3 (±3.7) | 80 | |
| blaVIM-1 | 33.4 (±0.8) | 20 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heller, C.; Bachmann, I.; Spiegel, M.; Hufert, F.T.; Dame, G. Detection of Klebsiella pneumoniae Carbapenem Resistance Genes by qPCR: Choosing the Right Method for Total DNA Extraction. Microorganisms 2024, 12, 1285. https://doi.org/10.3390/microorganisms12071285
Heller C, Bachmann I, Spiegel M, Hufert FT, Dame G. Detection of Klebsiella pneumoniae Carbapenem Resistance Genes by qPCR: Choosing the Right Method for Total DNA Extraction. Microorganisms. 2024; 12(7):1285. https://doi.org/10.3390/microorganisms12071285
Chicago/Turabian StyleHeller, Cecilia, Iris Bachmann, Martin Spiegel, Frank T. Hufert, and Gregory Dame. 2024. "Detection of Klebsiella pneumoniae Carbapenem Resistance Genes by qPCR: Choosing the Right Method for Total DNA Extraction" Microorganisms 12, no. 7: 1285. https://doi.org/10.3390/microorganisms12071285