
Pangenome Analysis Reveals Novel Contact-Dependent Growth Inhibition System and Phenazine Biosynthesis Operons in Proteus mirabilis BL95 That Are Located in An Integrative and Conjugative Element

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Library Construction, Sequencing, and Assembly
2.2. Annotation and Identification of Genomic Features
2.3. Genomic Comparative Analyses
3. Results
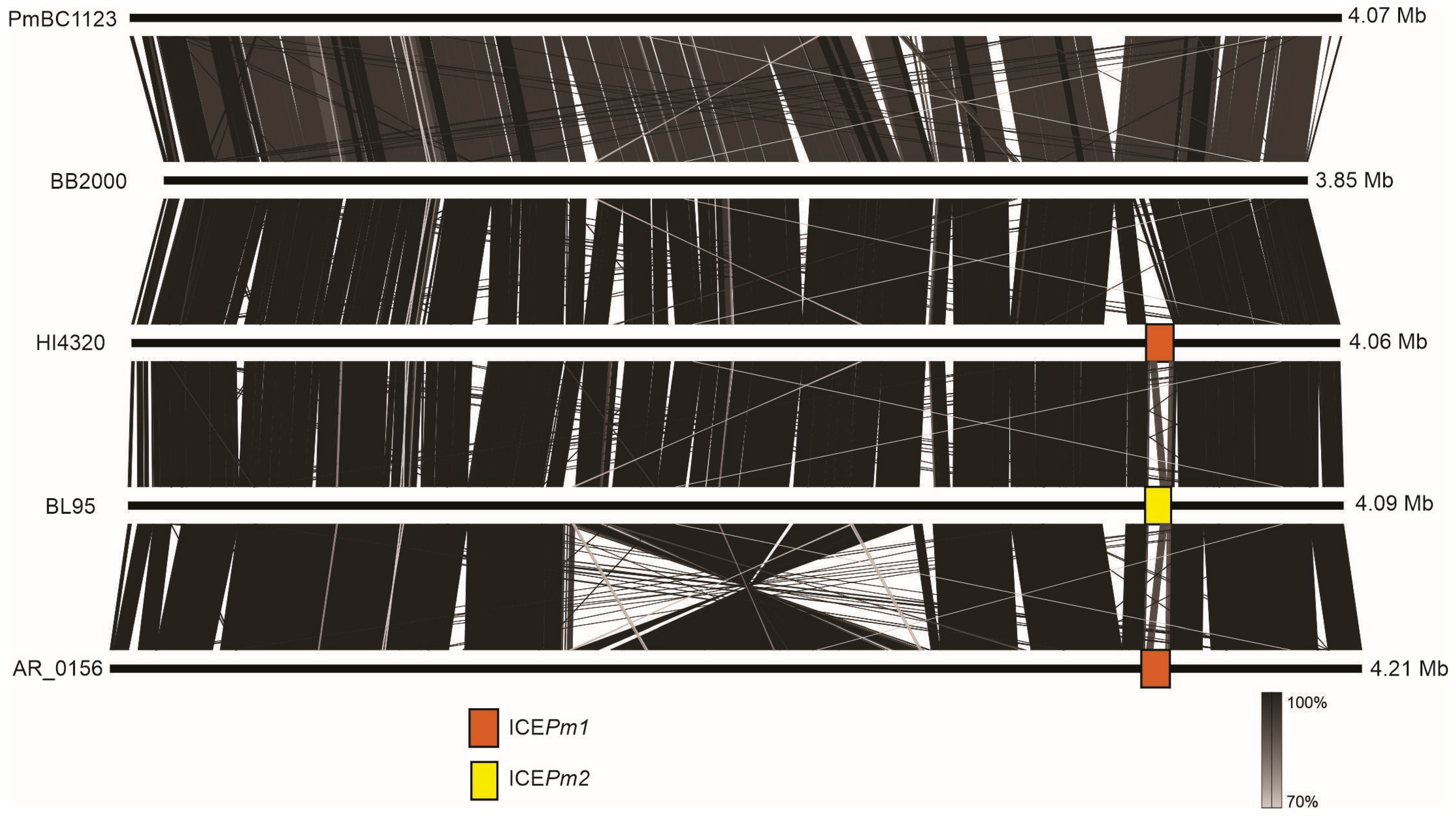
3.1. Phylogenetic Analysis
3.2. Pangenome Analysis and Comparative Genomics
3.3. General Features of BL95 Genome
3.4. Unique Features of BL95 Genome
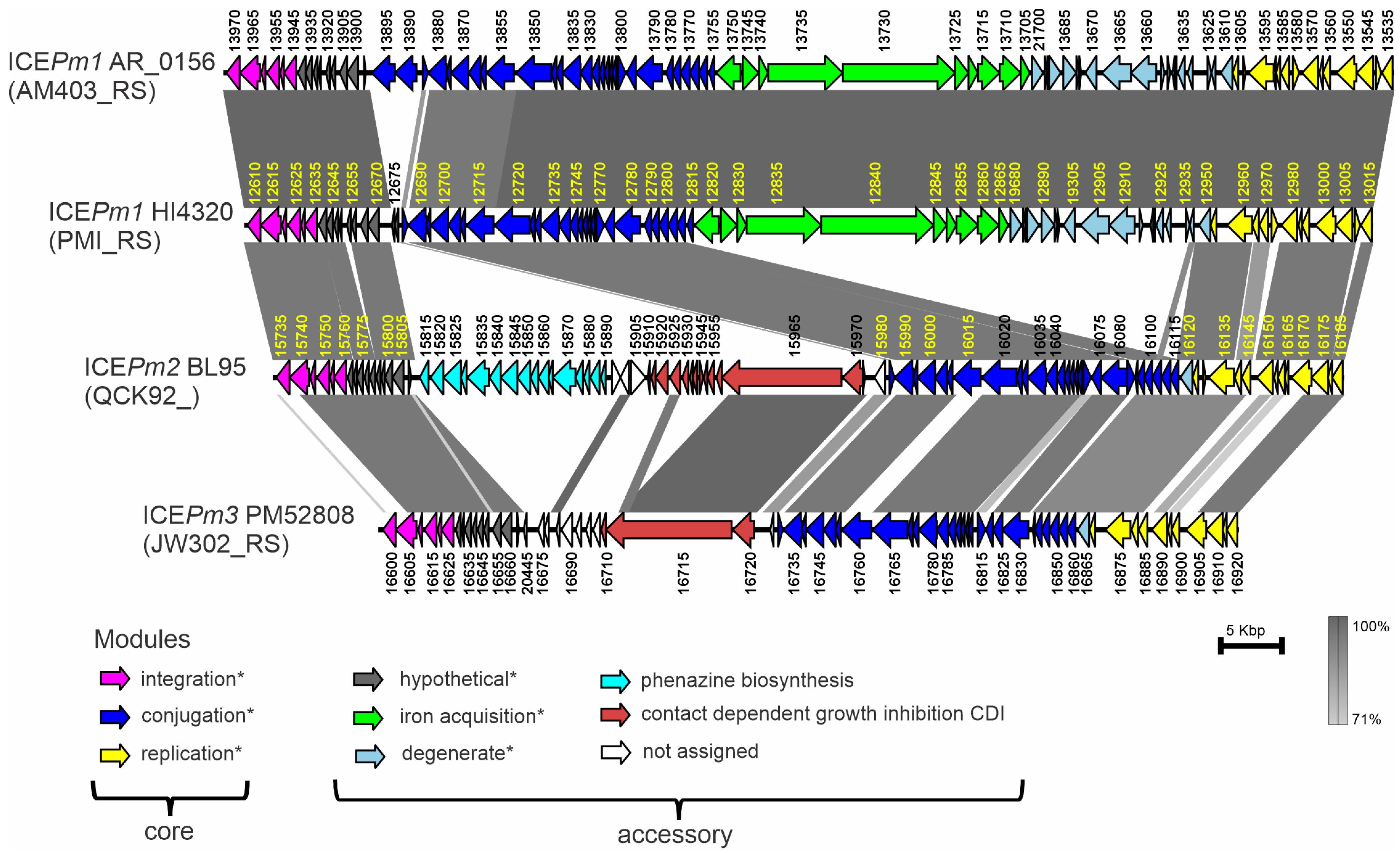
3.5. BL95 Carries a Novel ICE, ICEPm2
3.6. Structure of ICEPm2
3.7. Distribution of ICEPm1, ICEPm2, and ICEPm3 among P. mirabilis Isolates and Variation in Insertion Sites, tRNA-PheV or tRNA-PheU
3.8. Novel Phenazine Biosynthesis and Contact-Dependent Growth Inhibition System Operons in ICEPm2 in P. mirabilis BL95
3.8.1. Phenazine Biosynthesis Gene Operon in ICEPm2
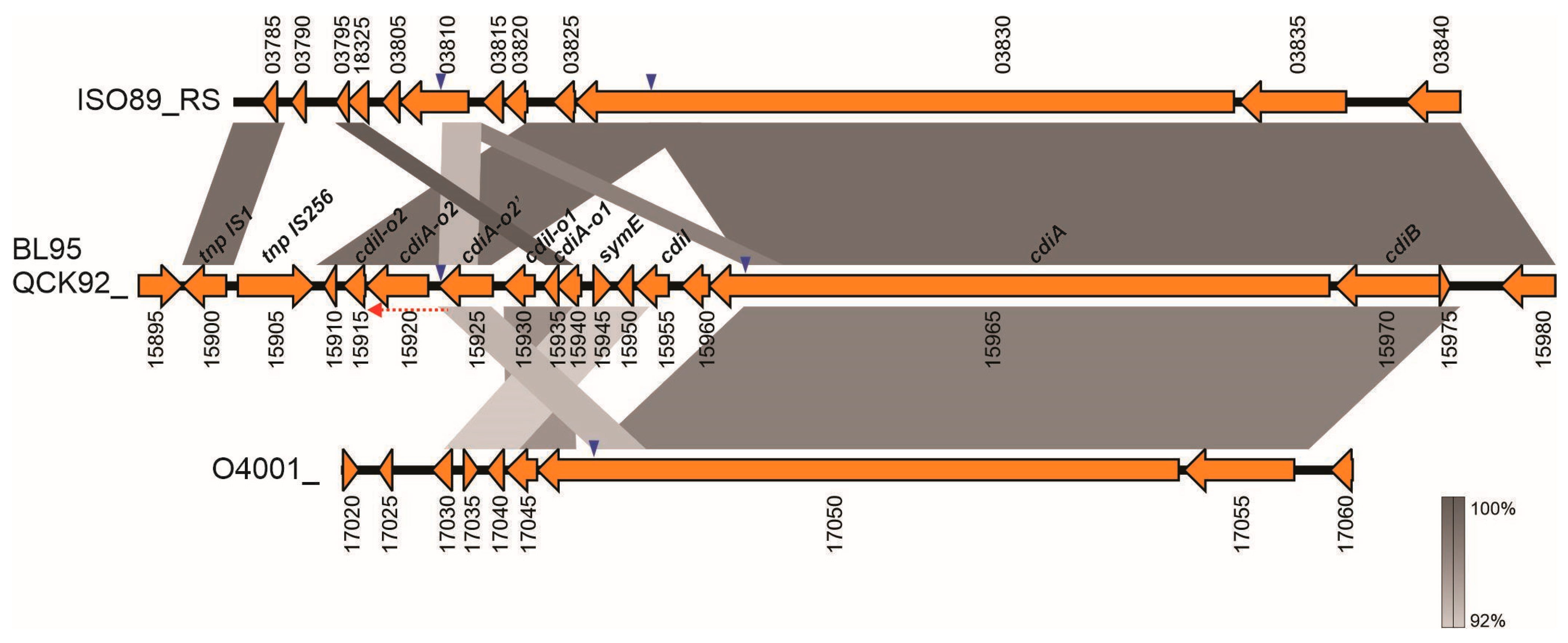
3.8.2. Contact-Dependent Growth Inhibition Gene Cluster in ICEPm2
3.9. Abundance of Secretion Systems in P. mirabilis BL95
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Armbruster, C.E.; Mobley, H.L. Merging mythology and morphology: The multifaceted lifestyle of Proteus mirabilis. Nat. Rev. Microbiol. 2012, 10, 743–754. [Google Scholar] [CrossRef] [PubMed]
- Armbruster, C.E.; Mobley, H.L.T.; Pearson, M.M. Pathogenesis of Proteus mirabilis infection. EcoSal Plus 2018, 8. [Google Scholar] [CrossRef]
- Gibbs, K.A.; Urbanowski, M.L.; Greenberg, E.P. Genetic determinants of self identity and social recognition in bacteria. Science 2008, 321, 256–259. [Google Scholar] [CrossRef] [PubMed]
- Alteri, C.J.; Himpsl, S.D.; Pickens, S.R.; Lindner, J.R.; Zora, J.S.; Miller, J.E.; Arno, P.D.; Straight, S.W.; Mobley, H.L. Multicellular bacteria deploy the type VI secretion system to preemptively strike neighboring cells. PLoS Pathog. 2013, 9, e1003608. [Google Scholar] [CrossRef] [PubMed]
- Mobley, H.L.T. Proteus mirabilis overview. In Proteus mirabilis: Methods and Protocols; Pearson, M.M., Ed.; Humana Press: New York, NY, USA, 2021; pp. 1–4. [Google Scholar] [CrossRef]
- Pearson, M.M.; Sebaihia, M.; Churcher, C.; Quail, M.A.; Seshasayee, A.S.; Luscombe, N.M.; Abdellah, Z.; Arrosmith, C.; Atkin, B.; Chillingworth, T.; et al. Complete genome sequence of uropathogenic Proteus mirabilis, a master of both adherence and motility. J. Bacteriol. 2008, 190, 4027–4037. [Google Scholar] [CrossRef] [PubMed]
- Guerin, J.; Bigot, S.; Schneider, R.; Buchanan, S.K.; Jacob-Dubuisson, F. Two-partner secretion: Combining efficiency and simplicity in the secretion of large proteins for bacteria-host and bacteria-bacteria interactions. Front. Cell. Infect. Microbiol. 2017, 7, 148. [Google Scholar] [CrossRef] [PubMed]
- Hayes, C.S.; Aoki, S.K.; Low, D.A. Bacterial contact-dependent delivery systems. Annu. Rev. Genet. 2010, 44, 71–90. [Google Scholar] [CrossRef]
- Willett, J.L.; Ruhe, Z.C.; Goulding, C.W.; Low, D.A.; Hayes, C.S. Contact-dependent growth inhibition (CDI) and CdiB/CdiA two-partner secretion proteins. J. Mol. Biol. 2015, 427, 3754–3765. [Google Scholar] [CrossRef] [PubMed]
- Juarez, G.E.; Mateyca, C.; Galvan, E.M. Proteus mirabilis outcompetes Klebsiella pneumoniae in artificial urine medium through secretion of ammonia and other volatile compounds. Heliyon 2020, 6, e03361. [Google Scholar] [CrossRef]
- Guimaraes, L.C.; de Jesus, L.B.; Viana, M.V.C.; Silva, A.; Ramos, R.T.J.; Soares, S.D.; Azevedo, V. Inside the pan-genome—Methods and software overview. Curr. Genom. 2015, 16, 245–252. [Google Scholar] [CrossRef]
- Flannery, E.L.; Antczak, S.M.; Mobley, H.L. Self-transmissibility of the integrative and conjugative element ICEPm1 between clinical isolates requires a functional integrase, relaxase, and type IV secretion system. J. Bacteriol 2011, 193, 4104–4112. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.M.; Grossman, A.D. Integrative and Conjugative Elements (ICEs): What they do and how they work. Annu. Rev. Genet. 2015, 49, 577–601. [Google Scholar] [CrossRef]
- Flannery, E.L.; Mody, L.; Mobley, H.L. Identification of a modular pathogenicity island that is widespread among urease-producing uropathogens and shares features with a diverse group of mobile elements. Infect. Immun. 2009, 77, 4887–4894. [Google Scholar] [CrossRef] [PubMed]
- Kiani, D.; Santus, W.; Kiernan, K.A.; Behnsen, J. Proteus mirabilis employs a contact-dependent killing system against competing Enterobacteriaceae. mSphere 2021, 6, e0032121. [Google Scholar] [CrossRef] [PubMed]
- Argueta, F.; Tatarenkov, A.; Mota-Bravo, L. Multiple copies of a qnrB19 gene are carried by tandem repeats of an IS26 composite transposon in an Escherichia coli plasmid. Microbiol. Resour. Announc. 2022, 11, e0066122. [Google Scholar] [CrossRef]
- Maniatis, T.; Fritsch, E.F.; Sambrook, J.K. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1982; pp. 191–195. [Google Scholar]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef]
- Tatusova, T.; DiCuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Olson, R.D.; Assaf, R.; Brettin, T.; Conrad, N.; Cucinell, C.; Davis, J.J.; Dempsey, D.M.; Dickerman, A.; Dietrich, E.M.; Kenyon, R.W.; et al. Introducing the Bacterial and Viral Bioinformatics Resource Center (BV-BRC): A resource combining PATRIC, IRD and ViPR. Nucleic Acids Res. 2023, 51, D678–D689. [Google Scholar] [CrossRef]
- Liu, M.; Li, X.; Xie, Y.; Bi, D.; Sun, J.; Li, J.; Tai, C.; Deng, Z.; Ou, H.Y. ICEberg 2.0: An updated database of bacterial integrative and conjugative elements. Nucleic Acids Res. 2019, 47, D660–D665. [Google Scholar] [CrossRef]
- Abby, S.S.; Cury, J.; Guglielmini, J.; Neron, B.; Touchon, M.; Rocha, E.P. Identification of protein secretion systems in bacterial genomes. Sci. Rep. 2016, 6, 23080. [Google Scholar] [CrossRef] [PubMed]
- Poole, S.J.; Diner, E.J.; Aoki, S.K.; Braaten, B.A.; t’Kint de Roodenbeke, C.; Low, D.A.; Hayes, C.S. Identification of functional toxin/immunity genes linked to contact-dependent growth inhibition (CDI) and rearrangement hotspot (Rhs) systems. PLoS Genet. 2011, 7, e1002217. [Google Scholar] [CrossRef] [PubMed]
- Solovyev, V.; Salamov, A. Automatic annotation of microbial genomes and metagenomic sequences. In Metagenomics and Its Applications in Agriculture, Biomedicine and Environmental Studies; Li, R.W., Ed.; Nova Science Publishers: Hauppauge, NY, USA, 2011; pp. 61–78. [Google Scholar]
- Naville, M.; Ghuillot-Gaudeffroy, A.; Marchais, A.; Gautheret, D. ARNold: A web tool for the prediction of Rho-independent transcription terminators. RNA Biol. 2011, 8, 11–13. [Google Scholar] [CrossRef] [PubMed]
- Stothard, P.; Grant, J.R.; Van Domselaar, G. Visualizing and comparing circular genomes using the CGView family of tools. Brief Bioinform. 2019, 20, 1576–1582. [Google Scholar] [CrossRef]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A genome comparison visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef] [PubMed]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Sullivan, N.L.; Septer, A.N.; Fields, A.T.; Wenren, L.M.; Gibbs, K.A. The complete genome sequence of Proteus mirabilis strain BB2000 reveals differences from the P. mirabilis reference strain. Genome Announc. 2013, 1, e00024-13. [Google Scholar] [CrossRef]
- Di Pilato, V.; Chiarelli, A.; Boinett, C.J.; Riccobono, E.; Harris, S.R.; D’Andrea, M.M.; Thomson, N.R.; Rossolini, G.M.; Giani, T. Complete genome sequence of the first KPC-Type carbapenemase-positive Proteus mirabilis strain from a bloodstream infection. Genome Announc. 2016, 4, e00607-16. [Google Scholar] [CrossRef]
- Bonnin, R.A.; Girlich, D.; Jousset, A.B.; Gauthier, L.; Cuzon, G.; Bogaerts, P.; Haenni, M.; Madec, J.Y.; Couve-Deacon, E.; Barraud, O.; et al. A single Proteus mirabilis lineage from human and animal sources: A hidden reservoir of OXA-23 or OXA-58 carbapenemases in Enterobacterales. Sci. Rep. 2020, 10, 9160. [Google Scholar] [CrossRef]
- Hendry, S.; Steinke, S.; Wittstein, K.; Stadler, M.; Harmrolfs, K.; Adewunmi, Y.; Sahukhal, G.; Elasri, M.; Thomashow, L.; Weller, D.; et al. Functional analysis of phenazine biosynthesis genes in Burkholderia spp. Appl. Environ. Microbiol. 2021, 87, e02348-20. [Google Scholar] [CrossRef] [PubMed]
- Leise, C. The Diversity and Evolution of Phenazine Biosynthesis Pathways in Enterobacterales. Bachelor’s Honor Thesis, University of Southern Mississippi, Hattiesburg, MI, USA, 2021. [Google Scholar]
- Mavrodi, D.V.; Blankenfeldt, W.; Thomashow, L.S. Phenazine compounds in fluorescent Pseudomonas spp. biosynthesis and regulation. Annu. Rev. Phytopathol. 2006, 44, 417–445. [Google Scholar] [CrossRef] [PubMed]
- Price-Whelan, A.; Dietrich, L.E.; Newman, D.K. Rethinking ‘secondary’ metabolism: Physiological roles for phenazine antibiotics. Nat. Chem. Biol. 2006, 2, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Mazzola, M.; Cook, R.J.; Thomashow, L.S.; Weller, D.M.; Pierson, L.S. Contribution of phenazine antibiotic biosynthesis to the ecological competence of fluorescent pseudomonads in soil habitats. Appl. Environ. Microbiol. 1992, 58, 2616–2624. [Google Scholar] [CrossRef] [PubMed]
- Lau, G.W.; Ran, H.; Kong, F.; Hassett, D.J.; Mavrodi, D. Pseudomonas aeruginosa pyocyanin is critical for lung infection in mice. Infect. Immun. 2004, 72, 4275–4278. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, L.E.; Teal, T.K.; Price-Whelan, A.; Newman, D.K. Redox-active antibiotics control gene expression and community behavior in divergent bacteria. Science 2008, 321, 1203–1206. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wilks, J.C.; Danhorn, T.; Ramos, I.; Croal, L.; Newman, D.K. Phenazine-1-carboxylic acid promotes bacterial biofilm development via ferrous iron acquisition. J. Bacteriol. 2011, 193, 3606–3617. [Google Scholar] [CrossRef] [PubMed]
- Aoki, S.K.; Pamma, R.; Hernday, A.D.; Bickham, J.E.; Braaten, B.A.; Low, D.A. Contact-dependent inhibition of growth in Escherichia coli. Science 2005, 309, 1245–1248. [Google Scholar] [CrossRef]
- Cuthbert, B.J.; Hayes, C.S.; Goulding, C.W. Functional and structural diversity of bacterial contact-dependent growth inhibition effectors. Front. Mol. Biosci. 2022, 9, 866854. [Google Scholar] [CrossRef]
- Peters, J.M.; Vangeloff, A.D.; Landick, R. Bacterial transcription terminators: The RNA 3′-end chronicles. J. Mol. Biol. 2011, 412, 793–813. [Google Scholar] [CrossRef]
- Uphoff, T.S.; Welch, R.A. Nucleotide sequencing of the Proteus mirabilis calcium-independent hemolysin genes (hpmA and hpmB) reveals sequence similarity with the Serratia marcescens hemolysin genes (shlA and shlB). J. Bacteriol. 1990, 172, 1206–1216. [Google Scholar] [CrossRef] [PubMed]
- Hibbing, M.E.; Fuqua, C.; Parsek, M.R.; Peterson, S.B. Bacterial competition: Surviving and thriving in the microbial jungle. Nat. Rev. Microbiol. 2010, 8, 15–25. [Google Scholar] [CrossRef]
- Armbruster, C.E.; Forsyth-DeOrnellas, V.; Johnson, A.O.; Smith, S.N.; Zhao, L.; Wu, W.; Mobley, H.L.T. Genome-wide transposon mutagenesis of Proteus mirabilis: Essential genes, fitness factors for catheter-associated urinary tract infection, and the impact of polymicrobial infection on fitness requirements. PLoS Pathog. 2017, 13, e1006434. [Google Scholar] [CrossRef] [PubMed]
- Beaber, J.W.; Burrus, V.; Hochhut, B.; Waldor, M.K. Comparison of SXT and R391, two conjugative integrating elements: Definition of a genetic backbone for the mobilization of resistance determinants. Cell. Mol. Life Sci. 2002, 59, 2065–2070. [Google Scholar] [CrossRef]
- Wozniak, R.A.; Waldor, M.K. Integrative and conjugative elements: Mosaic mobile genetic elements enabling dynamic lateral gene flow. Nat. Rev. Microbiol. 2010, 8, 552–563. [Google Scholar] [CrossRef]
- Bennett, P.M. Plasmid encoded antibiotic resistance: Acquisition and transfer of antibiotic resistance genes in bacteria. Br. J. Pharmacol. 2008, 153 (Suppl. S1), S347–S357. [Google Scholar] [CrossRef]
- Hochhut, B.; Beaber, J.W.; Woodgate, R.; Waldor, M.K. Formation of chromosomal tandem arrays of the SXT element and R391, two conjugative chromosomally integrating elements that share an attachment site. J. Bacteriol. 2001, 183, 1124–1132. [Google Scholar] [CrossRef] [PubMed]
- Pavlovic, G.; Burrus, V.; Gintz, B.; Decaris, B.; Guedon, G. Evolution of genomic islands by deletion and tandem accretion by site-specific recombination: ICESt1-related elements from Streptococcus thermophilus. Microbiology 2004, 150, 759–774. [Google Scholar] [CrossRef]
- Bellanger, X.; Payot, S.; Leblond-Bourget, N.; Guedon, G. Conjugative and mobilizable genomic islands in bacteria: Evolution and diversity. FEMS Microbiol. Rev. 2014, 38, 720–760. [Google Scholar] [CrossRef]
- Nikolakakis, K.; Amber, S.; Wilbur, J.S.; Diner, E.J.; Aoki, S.K.; Poole, S.J.; Tuanyok, A.; Keim, P.S.; Peacock, S.; Hayes, C.S.; et al. The toxin/immunity network of Burkholderia pseudomallei contact-dependent growth inhibition (CDI) systems. Mol. Microbiol. 2012, 84, 516–529. [Google Scholar] [CrossRef]
- Jamet, A.; Nassif, X. New players in the toxin field: Polymorphic toxin systems in bacteria. mBio 2015, 6, e00285-15. [Google Scholar] [CrossRef] [PubMed]
- Robitaille, S.; Trus, E.; Ross, B.D. Bacterial defense against the type VI secretion system. Trends Microbiol. 2021, 29, 187–190. [Google Scholar] [CrossRef] [PubMed]
- Siguier, P.; Perochon, J.; Lestrade, L.; Mahillon, J.; Chandler, M. ISfinder: The reference centre for bacterial insertion sequences. Nucleic Acids Res. 2006, 34, D32–D36. [Google Scholar] [CrossRef] [PubMed]
- Perez-Carrasco, V.; Soriano-Lerma, A.; Soriano, M.; Gutierrez-Fernandez, J.; Garcia-Salcedo, J.A. Urinary microbiome: Yin and yang of the urinary tract. Front. Cell. Infect. Microbiol. 2021, 11, 617002. [Google Scholar] [CrossRef]
- Booth, S.C.; Smith, W.P.J.; Foster, K.R. The evolution of short- and long-range weapons for bacterial competition. Nat. Ecol. Evol. 2023, 7, 2080–2091. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RefSeq Assembly Accession | GenBank Accession | Species | Strain | Insertion Locus of ICEPm1 | Insertion Locus of ICEPm2 | Insertion Locus of ICEPm3 |
|---|---|---|---|---|---|---|
| BL95_P_mirabilis | CP122400 | Proteus mirabilis | BL95 | tRNA-PheV | ||
| GCF_000069965.1 | NC_010554.1 | Proteus mirabilis | HI4320 | tRNA-PheV | ||
| GCF_000444425.1 | NC_022000.1 | Proteus mirabilis | BB2000 | |||
| GCF_000783575.2 | NZ_CP026062.1 | Proteus mirabilis | FDAARGOS_81 | tRNA-PheU | ||
| GCF_000783595.2 | NZ_CP026059.1 | Proteus mirabilis | FDAARGOS_80 | tRNA-PheV | ||
| GCF_000783875.2 | NZ_CP026051.1 | Proteus mirabilis | FDAARGOS_67 | tRNA-PheV | ||
| GCF_000784015.2 | NZ_CP026044.1 | Proteus mirabilis | FDAARGOS_60 | tRNA-PheV | ||
| GCF_001281545.1 | NZ_CP012674.1 | Proteus mirabilis | CYPM1 | |||
| GCF_001281565.1 | NZ_CP012675.1 | Proteus mirabilis | CYPV1 | |||
| GCF_001640985.1 | NZ_CP015347.1 | Proteus mirabilis | AOUC-001 | tRNA-PheU | ||
| GCF_002055685.1 | NZ_CP020052.1 | Proteus mirabilis | AR_0059 | tRNA-PheV | ||
| GCF_002180115.1 | NZ_CP021550.1 | Proteus mirabilis | AR_0159 | tRNA-PheU | ||
| GCF_002180235.1 | NZ_CP021694.1 | Proteus mirabilis | AR_0155 | tRNA-PheV | tRNA-PheU | |
| GCF_002197405.1 | NZ_CP021852.1 | Proteus mirabilis | AR_0156 | tRNA-PheV | ||
| GCF_002310875.1 | NZ_CP017082.1 | Proteus mirabilis | T21 | tRNA-PheV | ||
| GCF_002310895.1 | NZ_CP017085.1 | Proteus mirabilis | T18 | tRNA-PheV | ||
| GCF_002944495.1 | NZ_CP026571.1 | Proteus mirabilis | BC11-24 | |||
| GCF_002945235.1 | NZ_CP026581.1 | Proteus mirabilis | GN2 | |||
| GCF_003073935.1 | NZ_CP029133.1 | Proteus mirabilis | AR379 | tRNA-PheU | tRNA-PheU | |
| GCF_003204115.1 | NZ_CP029725.1 | Proteus mirabilis | AR_0029 | tRNA-PheU | ||
| GCF_003855615.1 | NZ_CP034091.1 | Proteus mirabilis | PmBC1123 | |||
| GCF_003855635.1 | NZ_CP034090.1 | Proteus mirabilis | PmSC1111 | |||
| GCF_008041895.1 | NZ_CP042907.1 | Proteus mirabilis | VAC | tRNA-PheV | ||
| GCF_008195605.1 | NZ_CP043332.1 | Proteus mirabilis | CRPM10 | tRNA-PheU | ||
| GCF_008705195.1 | NZ_CP044028.1 | Proteus mirabilis | K817 | |||
| GCF_009429045.2 | NZ_CP045538.2 | Proteus mirabilis | CRE14IB | |||
| GCF_009684665.1 | NZ_CP044136.1 | Proteus mirabilis | ENT1157 | tRNA-PheV | tRNA-PheU | |
| GCF_009806715.1 | NZ_CP047112.1 | Proteus mirabilis | SCBX1.1 | tRNA-PheV | tRNA-PheU | |
| GCF_010442675.1 | NZ_CP048404.1 | Proteus mirabilis | N18-00201 | |||
| GCF_010692865.1 | NZ_CP048787.1 | Proteus mirabilis | CC15031 | |||
| GCF_011045575.1 | NZ_CP042857.1 | Proteus mirabilis | 1701092 | |||
| GCF_011045855.1 | NZ_CP047352.1 | Proteus mirabilis | ZA25 | |||
| GCF_011149675.1 | NZ_CP049753.1 | Proteus mirabilis | PmBR607 | tRNA-PheU | ||
| GCF_011383025.1 | NZ_CP049941.1 | Proteus mirabilis | XH1568 | tRNA-PheV | ||
| GCF_011383045.1 | NZ_CP049942.1 | Proteus mirabilis | XH1569 | tRNA-PheV | ||
| GCF_012516515.1 | NZ_CP051260.1 | Proteus mirabilis | STP3 | |||
| GCF_013255765.1 | NZ_CP053894.1 | Proteus mirabilis | JPM24 | |||
| GCF_013256075.1 | NZ_CP053898.1 | Proteus mirabilis | YPM35 | |||
| GCF_013343255.1 | NZ_CP045257.1 | Proteus mirabilis | L90-1 | tRNA-PheV | ||
| GCF_013357405.1 | NZ_CP053615.1 | Proteus mirabilis | MPE0734 | |||
| GCF_013357425.1 | NZ_CP053616.1 | Proteus mirabilis | MPE0767 | |||
| GCF_013357445.1 | NZ_CP053614.1 | Proteus mirabilis | S74-1(++)-2 | |||
| GCF_013357465.1 | NZ_CP053681.1 | Proteus mirabilis | M3-1-17 | |||
| GCF_013357485.1 | NZ_CP053682.1 | Proteus mirabilis | MPE0156 | |||
| GCF_013357505.1 | NZ_CP053683.1 | Proteus mirabilis | MPE0027 | |||
| GCF_013357525.1 | NZ_CP053718.1 | Proteus mirabilis | MPE4069 | |||
| GCF_013357545.1 | NZ_CP053684.1 | Proteus mirabilis | MPE5139 | |||
| GCF_013357565.1 | NZ_CP053685.1 | Proteus mirabilis | MPE5203 | |||
| GCF_013357585.1 | NZ_CP053719.1 | Proteus mirabilis | MPE0346 | tRNA-PheU | ||
| GCF_013358795.1 | NZ_CP046048.1 | Proteus mirabilis | HN2p | |||
| GCF_014843115.1 | NZ_CP062146.1 | Proteus mirabilis | S012 | |||
| GCF_014931585.1 | NZ_CP047929.1 | Proteus mirabilis | ChSC1905 | |||
| GCF_015169015.1 | NZ_CP063440.1 | Proteus mirabilis | Yak_2019 | |||
| GCF_015693865.1 | NZ_CP065147.1 | Proteus mirabilis | PmBJ015-2 | |||
| GCF_015693965.1 | NZ_CP065148.1 | Proteus mirabilis | PmBJ012-2 | |||
| GCF_015693985.1 | NZ_CP065144.1 | Proteus mirabilis | PmBJ024-1 | |||
| GCF_015694005.1 | NZ_CP065145.1 | Proteus mirabilis | PmBJ023-2 | |||
| GCF_015694025.1 | NZ_CP065146.1 | Proteus mirabilis | PmBJ020-1 | |||
| GCF_016725905.1 | NZ_CP068152.1 | Proteus mirabilis | FDAARGOS_1079 | tRNA-PheU | ||
| GCF_016772335.1 | NZ_CP044436.1 | Proteus mirabilis | C55 | |||
| GCF_016772355.1 | NZ_CP044437.1 | Proteus mirabilis | C74 | |||
| GCF_016939715.1 | NZ_CP070569.1 | Proteus mirabilis | PM52260 | tRNA-PheU | tRNA-PheV | |
| GCF_016939735.1 | NZ_CP070572.1 | Proteus mirabilis | PM52808 | tRNA-PheU | tRNA-PheV | |
| GCF_017161055.1 | NZ_CP046049.1 | Proteus mirabilis | DY.F1.2 | |||
| GCF_017808555.1 | NZ_CP066833.1 | Proteus mirabilis | RGF134-1 | |||
| GCF_017901195.1 | NZ_CP072779.1 | Proteus mirabilis | 1035 | |||
| GCF_018138945.1 | NZ_CP073248.1 | Proteus mirabilis | N292 | |||
| GCF_018138985.1 | NZ_CP073246.1 | Proteus mirabilis | N639-2X | |||
| GCF_018139005.1 | NZ_CP073247.1 | Proteus mirabilis | S62-3-2-2 | |||
| GCF_018139105.1 | NZ_CP073245.1 | Proteus mirabilis | S74-3-2 | |||
| GCF_018336495.1 | NZ_CP047589.1 | Proteus mirabilis | SNYG35 | |||
| GCF_018972025.2 | NZ_CP065039.2 | Proteus mirabilis | XH1653 | |||
| GCF_019192645.1 | NZ_CP077963.1 | Proteus mirabilis | 6Pmi283 | |||
| GCF_019443785.1 | NZ_CP048692.1 | Proteus mirabilis | HNS2p | |||
| GCF_021228935.1 | NZ_CP089317.1 | Proteus mirabilis | PM1162 | tRNA-PheV | ||
| GCF_022353845.1 | NZ_CP055009.1 | Proteus mirabilis | STIN_74 | |||
| GCF_022354605.1 | NZ_CP055095.1 | Proteus mirabilis | SWHIN_109 | tRNA-PheU | ||
| GCF_022453625.1 | NZ_CP092652.1 | Proteus mirabilis | PM8762 | |||
| GCF_023093855.1 | NZ_CP095765.1 | Proteus mirabilis | T1010 | |||
| GCF_023242175.1 | NZ_CP096775.1 | Proteus mirabilis | HURS-181823 | tRNA-PheV | tRNA-PheU | |
| GCF_023242195.1 | NZ_CP096776.1 | Proteus mirabilis | HURS-186083 | tRNA-PheV | tRNA-PheU | |
| GCF_023702555.1 | NZ_CP098446.1 | Proteus mirabilis | FZP2826 | |||
| GCF_023702575.1 | NZ_CP098447.1 | Proteus mirabilis | FZP2936 | tRNA-PheU | ||
| GCF_023702595.1 | NZ_CP098450.1 | Proteus mirabilis | FZP3115 | tRNA-PheU | ||
| GCF_024138795.1 | NZ_CP071773.1 | Proteus mirabilis | swupm1 | |||
| GCF_024138815.1 | NZ_CP071777.1 | Proteus mirabilis | swupm2 | |||
| GCF_024138835.1 | NZ_CP071780.1 | Proteus mirabilis | swupm3 | |||
| GCF_025264285.1 | NZ_CP031846.1 | Proteus mirabilis | XH983 | tRNA-PheV | ||
| GCF_025398955.1 | NZ_CP104698.1 | Proteus mirabilis | NG-ABK-32 | |||
| GCF_025490355.1 | NZ_CP104986.1 | Proteus mirabilis | W47 | |||
| GCF_025998255.1 | NZ_AP026827.1 | Proteus mirabilis | NUITM-VP1 | |||
| GCF_026016045.1 | NZ_CP110371.1 | Proteus mirabilis | CZP17 | |||
| GCF_026016065.1 | NZ_CP110372.1 | Proteus mirabilis | CZP44 | |||
| GCF_026016085.1 | NZ_CP110373.1 | Proteus mirabilis | CZP26 | |||
| GCF_026016105.1 | NZ_CP110376.1 | Proteus mirabilis | NYP69 | |||
| GCF_026016125.1 | NZ_CP110377.1 | Proteus mirabilis | NYP73 | |||
| GCF_026016145.1 | NZ_CP110375.1 | Proteus mirabilis | NYP6 | |||
| GCF_026167565.1 | NZ_CP110673.1 | Proteus mirabilis | DP2019 | |||
| GCF_900635965.1 | NZ_LR134205.1 | Proteus mirabilis | NCTC4199 | tRNA-PheV | ||
| GCF_000754995.1 | NZ_KN150745.1 | Proteus vulgaris | ATCC_49132 | |||
| GCF_004116975.1 | NZ_CP026364.1 | Proteus hauseri | 15H5D-4a | |||
| GCF_022369495.1 | NZ_CP059690.1 | Proteus penneri | S178-2 |
| Locus_Tag | Genome Nucleotide Positions | Length | Direction | Product (GenBank Annotation) | Product (Prokka Annotation) | Genes * |
|---|---|---|---|---|---|---|
| QCK92_00170 | 29,331–29,576 | 246 | reverse | AlpA family phage regulatory protein | hypothetical protein | |
| QCK92_00175 | 29,665–30,540 | 876 | reverse | hypothetical protein | hypothetical protein | |
| QCK92_00180 | 30,633–31,898 | 1266 | reverse | tyrosine-type recombinase/integrase | prophage integrase IntA | |
| QCK92_06810 | 1,482,955–1,483,173 | 219 | reverse | HEAT repeat domain-containing protein | hypothetical protein | |
| QCK92_10410 | 2,229,236–2,229,460 | 225 | reverse | type I toxin-antitoxin system ptaRNA1 family toxin | hypothetical protein | |
| QCK92_10415 | 2,229,524–2,229,772 | 249 | reverse | hypothetical protein | hypothetical protein | |
| QCK92_10420 | 2,229,777–2,230,790 | 1014 | reverse | P-type conjugative transfer protein TrbL | hypothetical protein | |
| QCK92_10425 | 2,230,694–2,231,203 | 510 | reverse | type IV secretion system protein | hypothetical protein | |
| QCK92_10430 | 2,231,214–2,231,417 | 204 | reverse | entry exclusion lipoprotein TrbK | not annotated | |
| QCK92_10435 | 2,231,461–2,232,240 | 780 | reverse | P-type conjugative transfer protein TrbJ | hypothetical protein | |
| QCK92_10440 | 2,232,381–2,232,569 | 189 | reverse | stabilization protein | hypothetical protein | |
| QCK92_10445 | 2,233,600–2,234,480 | 881 | reverse | replication protein C, IncQ-type | hypothetical protein | |
| QCK92_10450 | 2,234,467–2,235,293 | 827 | reverse | helicase RepA family protein | regulatory protein RepA | |
| QCK92_10455 | 2,235,298–2,235,519 | 222 | reverse | AlpA family phage regulatory protein | hypothetical protein | |
| QCK92_10460 | 2,235,667–2,236,869 | 1203 | reverse | tyrosine-type recombinase/integrase | prophage integrase IntA | |
| QCK92_12525 | 2,679,658–2,679,966 | 309 | reverse | helix-turn-helix domain-containing protein | hypothetical protein | |
| QCK92_12530 | 2,680,018–2,680,206 | 189 | reverse | DNA-binding protein | hypothetical protein | |
| QCK92_12535 | 2,680,233–2,680,589 | 357 | forward | hypothetical protein | hypothetical protein | |
| QCK92_12540 | 2,680,907–2,681,245 | 339 | forward | helix-turn-helix domain-containing protein | hypothetical protein | |
| QCK92_12545 | 2,681,381–2,682,151 | 771 | reverse | DNA adenine methylase | hypothetical protein | |
| QCK92_12550 | 2,682,335–2,682,811 | 477 | reverse | ABC transporter ATPase | hypothetical protein | |
| QCK92_12555 | 2,682,943–2,683,293 | 351 | forward | putative holin | hypothetical protein | |
| QCK92_12645 | 2,695,862–2,696,143 | 282 | forward | hypothetical protein | hypothetical protein | |
| QCK92_12700 | 2,704,416–2,705,507 | 1092 | forward | phage tail protein | hypothetical protein | |
| QCK92_12705 | 2,705,507–2,706,286 | 780 | forward | DUF4376 domain-containing protein | hypothetical protein | |
| QCK92_15815 | 3,431,156–3,431,878 | 723 | reverse | 4′-phosphopantetheinyl transferase superfamily protein | hypothetical protein | HP |
| QCK92_15820 | 3,432,045–3,433,091 | 1047 | reverse | 3-deoxy-7-phosphoheptulonate synthase | phospho-2-dehydro-3-deoxyheptonate aldolase, Trp-sensitive | phzC |
| QCK92_15825 | 3,433,123–3,434,535 | 1413 | reverse | FAD-dependent oxidoreductase | hypothetical protein | HP |
| QCK92_15830 | 3,434,532–3,434,990 | 459 | reverse | hypothetical protein | hypothetical protein | HP |
| QCK92_15835 | 3,435,058–3,436,827 | 1770 | reverse | non-ribosomal peptide synthetase | dimodular nonribosomal peptide synthase | dhbF |
| QCK92_15840 | 3,436,876–3,437,802 | 927 | reverse | hypothetical protein | hypothetical protein | HP |
| QCK92_15845 | 3,437,799–3,439,169 | 1371 | reverse | aldehyde dehydrogenase family protein | hypothetical protein | ehpG |
| QCK92_15850 | 3,439,172–3,440,233 | 1062 | reverse | AMP-binding protein | hypothetical protein | ehpF |
| QCK92_15855 | 3,440,246–3,440,881 | 636 | reverse | pyridoxal 5′-phosphate synthase | phenazine biosynthesis protein PhzG | phzG |
| QCK92_15860 | 3,440,895–3,441,740 | 846 | reverse | PhzF family phenazine biosynthesis protein | trans-2,3-dihydro-3-hydroxyanthranilate isomerase | phzF |
| QCK92_15865 | 3,441,719–3,442,072 | 354 | reverse | hypothetical protein | hypothetical protein | HP |
| QCK92_15870 | 3,442,069–3,443,934 | 1866 | reverse | anthranilate synthase family protein | isochorismate synthase MenF | phzE |
| QCK92_15875 | 3,443,931–3,444,551 | 621 | reverse | isochorismatase family protein | Phenazine biosynthesis protein PhzD | phzD |
| QCK92_15880 | 3,444,627–3,445,085 | 459 | reverse | PhzA/PhzB family protein | phenazine biosynthesis protein PhzB | phzA/B |
| QCK92_15885 | 3,445,132–3,445,914 | 783 | reverse | SDR family NAD(P)-dependent oxidoreductase | 3-oxoacyl-[acyl-carrier-protein] reductase FabG | ehpK, fabG |
| QCK92_15890 | 3,446,019–3,446,405 | 387 | reverse | VOC family protein | phenazine antibiotic resistance protein EhpR | ehpR |
| QCK92_15895 | 3,446,919–3,447,617 | 699 | forward | DUF2461 domain-containing protein | hypothetical protein | HP |
| QCK92_15905 | 3,448,489–3,449,697 | 1209 | forward | IS256 family transposase | IS256 family transposase ISEic2 | tnp_IS256 |
| QCK92_15910 | 3,449,845–3,450,042 | 198 | reverse | hypothetical protein | not annotated | HP |
| QCK92_15915 | 3,450,152–3,450,505 | 354 | reverse | hypothetical protein | hypothetical protein | cdiI-o2 |
| QCK92_15920 | 3,450,518–3,451,501 | 984 | reverse | cysteine peptidase family C39 domain-containing protein | hypothetical protein | cdiA-o2 |
| QCK92_15925 | 3,451,663–3,452,433 | 771 | reverse | VENN motif pre-toxin domain-containing protein | deoxyribonuclease CdiA | cdiA-o2′ |
| QCK92_15930 | 3,452,688–3,453,185 | 498 | reverse | contact-dependent growth inhibition system immunity protein | immunity protein CdiI-o11 | cdiI-o1 |
| QCK92_15935 | 3,453,316–3,453,561 | 246 | reverse | hypothetical protein | deoxyribonuclease CdiA-o11 | cdiA-o1 |
| QCK92_15940 | 3,453,536–3,453,922 | 387 | reverse | hypothetical protein | deoxyribonuclease CdiA-o11 | cdiA-o1′ |
| QCK92_15945 | 3,454,113–3,454,409 | 297 | forward | SymE family type I addiction module toxin | not annotated | symE |
| QCK92_15950 | 3,454,471–3,454,740 | 270 | reverse | hypothetical protein | hypothetical protein | HP |
| QCK92_15955 | 3,454,771–3,455,304 | 534 | reverse | contact-dependent growth inhibition system immunity protein | immunity protein CdiI-YPIII | cdiI |
| QCK92_15960 | 3,455,502–3,455,945 | 444 | reverse | hypothetical protein | hypothetical protein | HP |
| QCK92_15965 | 3,455,942–3,465,757 | 9816 | reverse | polymorphic toxin type 25 domain-containing protein | tRNA nuclease CdiA | cdiA |
| QCK92_16130 | 3,495,436–3,495,606 | 171 | forward | hypothetical protein | hypothetical protein |
| Locus_Tag | Protein Name | Commonly Used Gene Name | Gene Length | Functional Category |
|---|---|---|---|---|
| QCK92_15815 | 4′-phosphopantetheinyl transferase superfamily protein | 723 | B | |
| QCK92_15820 | 3-deoxy-7-phosphoheptulonate synthase | phzC, aroH | 1047 | A |
| QCK92_15825 | FAD-dependent oxidoreductase | 1413 | B | |
| QCK92_15830 | hypothetical protein | 459 | B | |
| QCK92_15835 | non-ribosomal peptide synthetase | dhbF | 1770 | B |
| QCK92_15840 | hypothetical protein | 927 | B | |
| QCK92_15845 | aldehyde dehydrogenase family protein | ehpG | 1371 | B |
| QCK92_15850 | AMP-binding protein | ehpF | 1062 | B |
| QCK92_15855 | pyridoxal 5′-phosphate synthase | phzG | 636 | A |
| QCK92_15860 | PhzF family phenazine biosynthesis protein | phzF | 846 | A |
| QCK92_15865 | hypothetical protein * | 354 | A | |
| QCK92_15870 | anthranilate synthase family protein | phzE, menF2 | 1866 | A |
| QCK92_15875 | isochorismatase family protein | phzD | 621 | A |
| QCK92_15880 | PhzA/PhzB family protein | phzA/B | 459 | A |
| QCK92_15885 | SDR family NAD(P)-dependent oxidoreductase | ehpK, fabG | 783 | B |
| QCK92_15890 | VOC family protein | ehpR | 387 | C |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tatarenkov, A.; Muñoz-Gutiérrez, I.; Vargas, I.; Behnsen, J.; Mota-Bravo, L. Pangenome Analysis Reveals Novel Contact-Dependent Growth Inhibition System and Phenazine Biosynthesis Operons in Proteus mirabilis BL95 That Are Located in An Integrative and Conjugative Element. Microorganisms 2024, 12, 1321. https://doi.org/10.3390/microorganisms12071321
Tatarenkov A, Muñoz-Gutiérrez I, Vargas I, Behnsen J, Mota-Bravo L. Pangenome Analysis Reveals Novel Contact-Dependent Growth Inhibition System and Phenazine Biosynthesis Operons in Proteus mirabilis BL95 That Are Located in An Integrative and Conjugative Element. Microorganisms. 2024; 12(7):1321. https://doi.org/10.3390/microorganisms12071321
Chicago/Turabian StyleTatarenkov, Andrey, Iván Muñoz-Gutiérrez, Isabel Vargas, Judith Behnsen, and Luis Mota-Bravo. 2024. "Pangenome Analysis Reveals Novel Contact-Dependent Growth Inhibition System and Phenazine Biosynthesis Operons in Proteus mirabilis BL95 That Are Located in An Integrative and Conjugative Element" Microorganisms 12, no. 7: 1321. https://doi.org/10.3390/microorganisms12071321
APA StyleTatarenkov, A., Muñoz-Gutiérrez, I., Vargas, I., Behnsen, J., & Mota-Bravo, L. (2024). Pangenome Analysis Reveals Novel Contact-Dependent Growth Inhibition System and Phenazine Biosynthesis Operons in Proteus mirabilis BL95 That Are Located in An Integrative and Conjugative Element. Microorganisms, 12(7), 1321. https://doi.org/10.3390/microorganisms12071321