
Domestication and Genetic Improvement Alter the Symbiotic Microbiome Structure and Function of Tomato Leaf and Fruit Pericarp

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Genomic and Transcriptomic Data of Tomato
2.2. Kraken Pipeline for Microbial Detection
2.3. Quantification of 16S from Abundant Genera
2.4. Analysis and Comparison of Microbiome Structure
2.5. Tomato Germplasm Differentiation Using Machine Learning
2.6. Identification of Regulatory Sites
3. Results
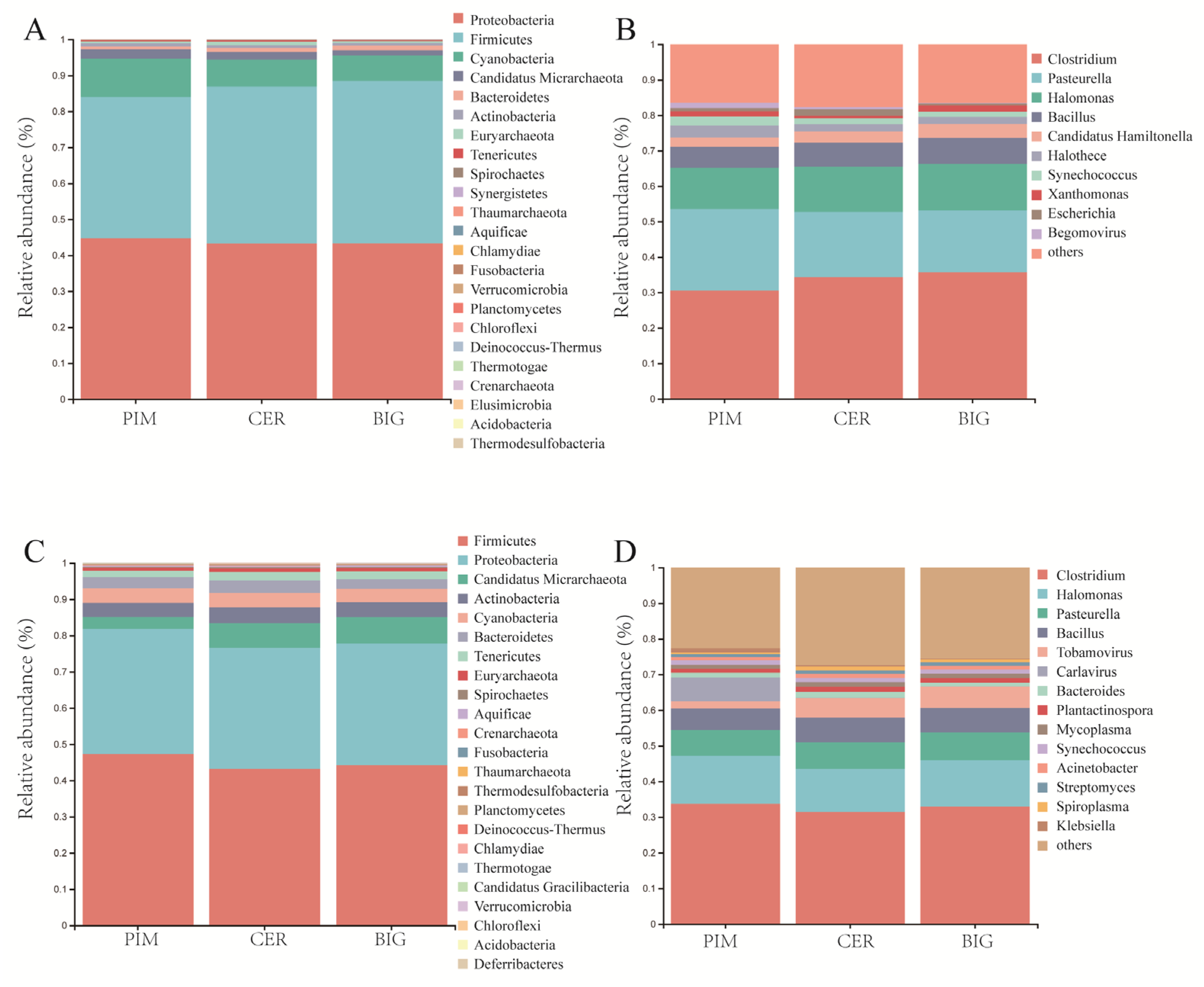
3.1. Microbiome Structure of Tomato Leaf and Fruit Pericarp
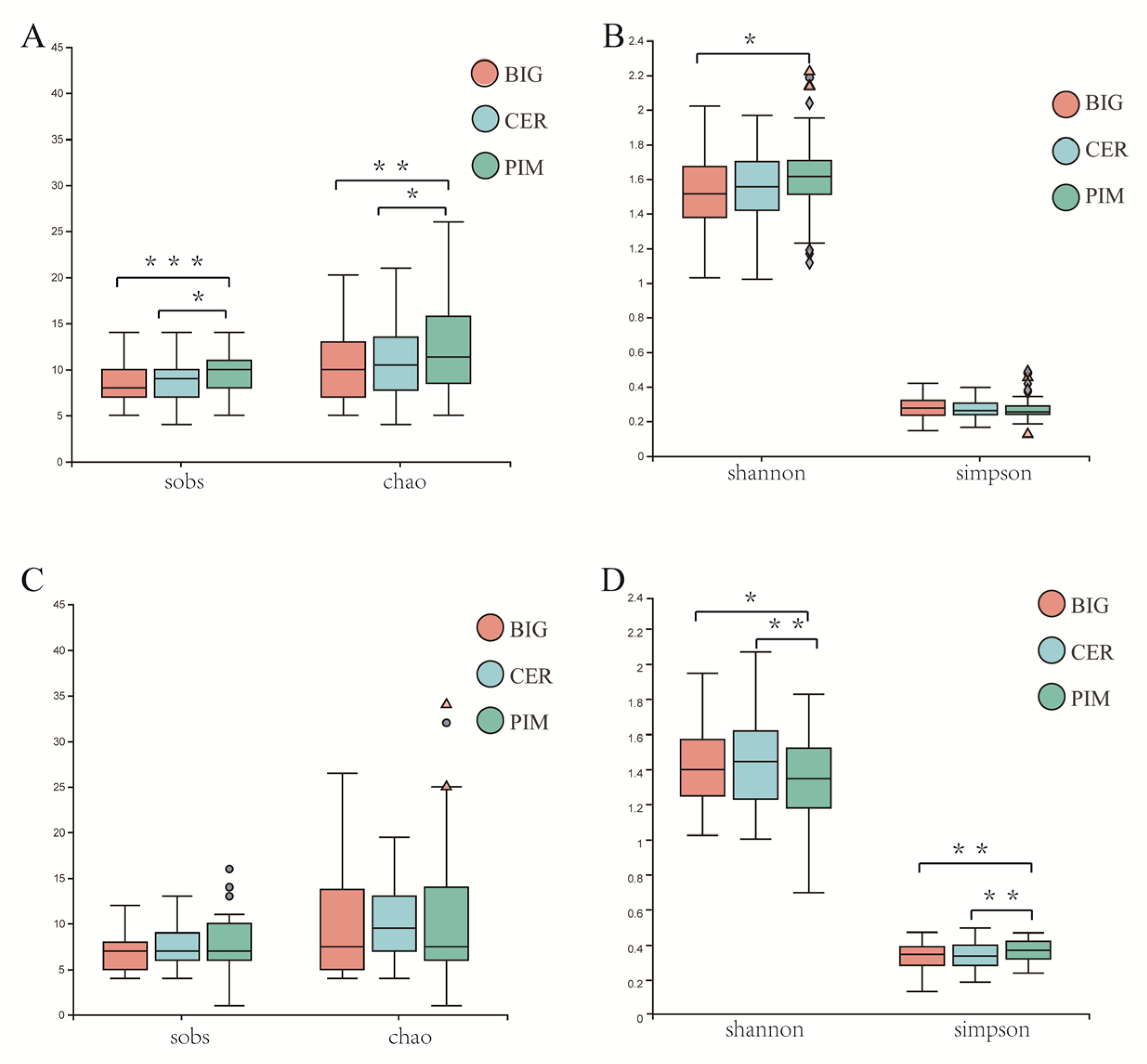
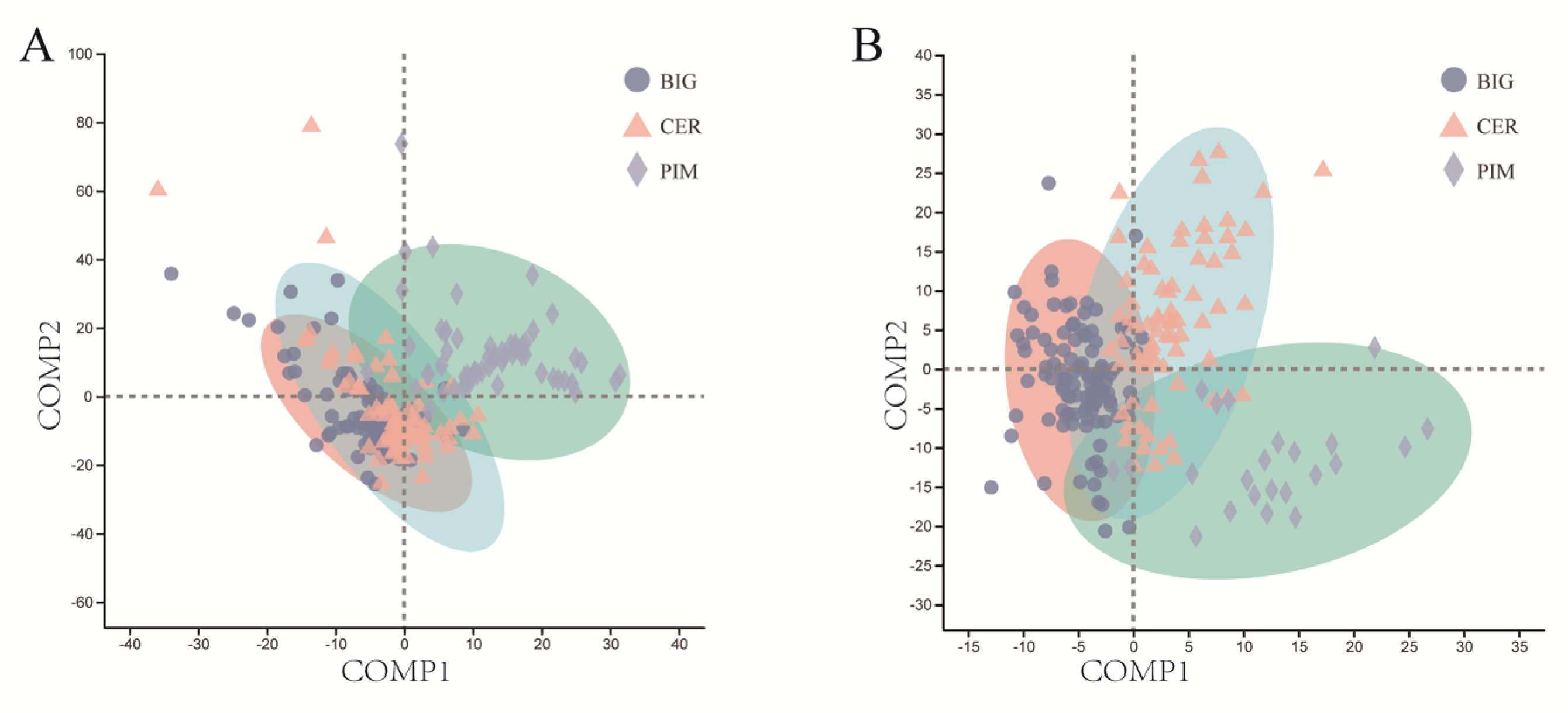
3.2. Differences in Leaf and Fruit Pericarp Microbiome Structure among PIM, CER, and BIG
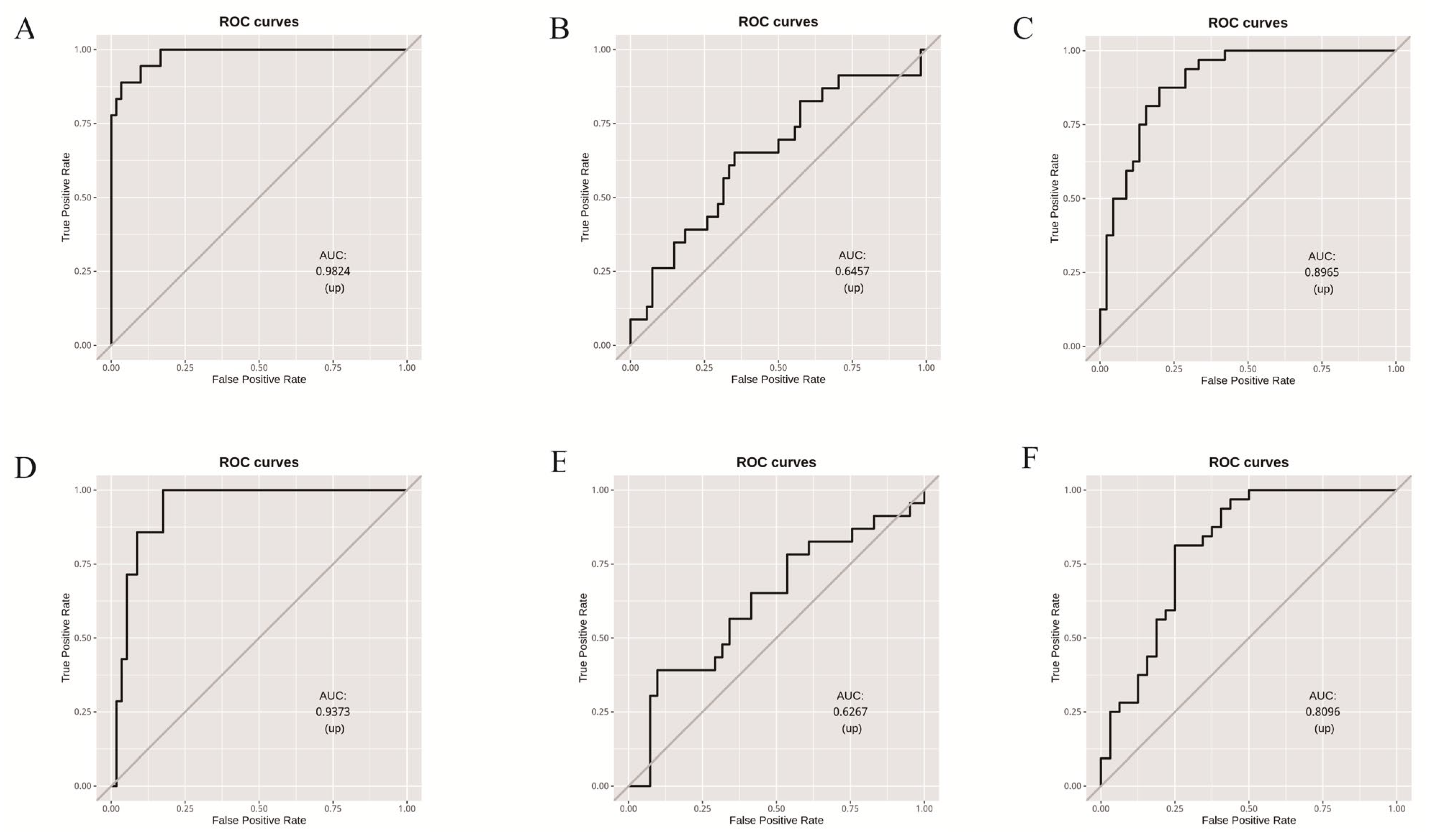
3.3. Random Forest Model Based on Microbial Community Composition Accurately Predicted Tomato Clade
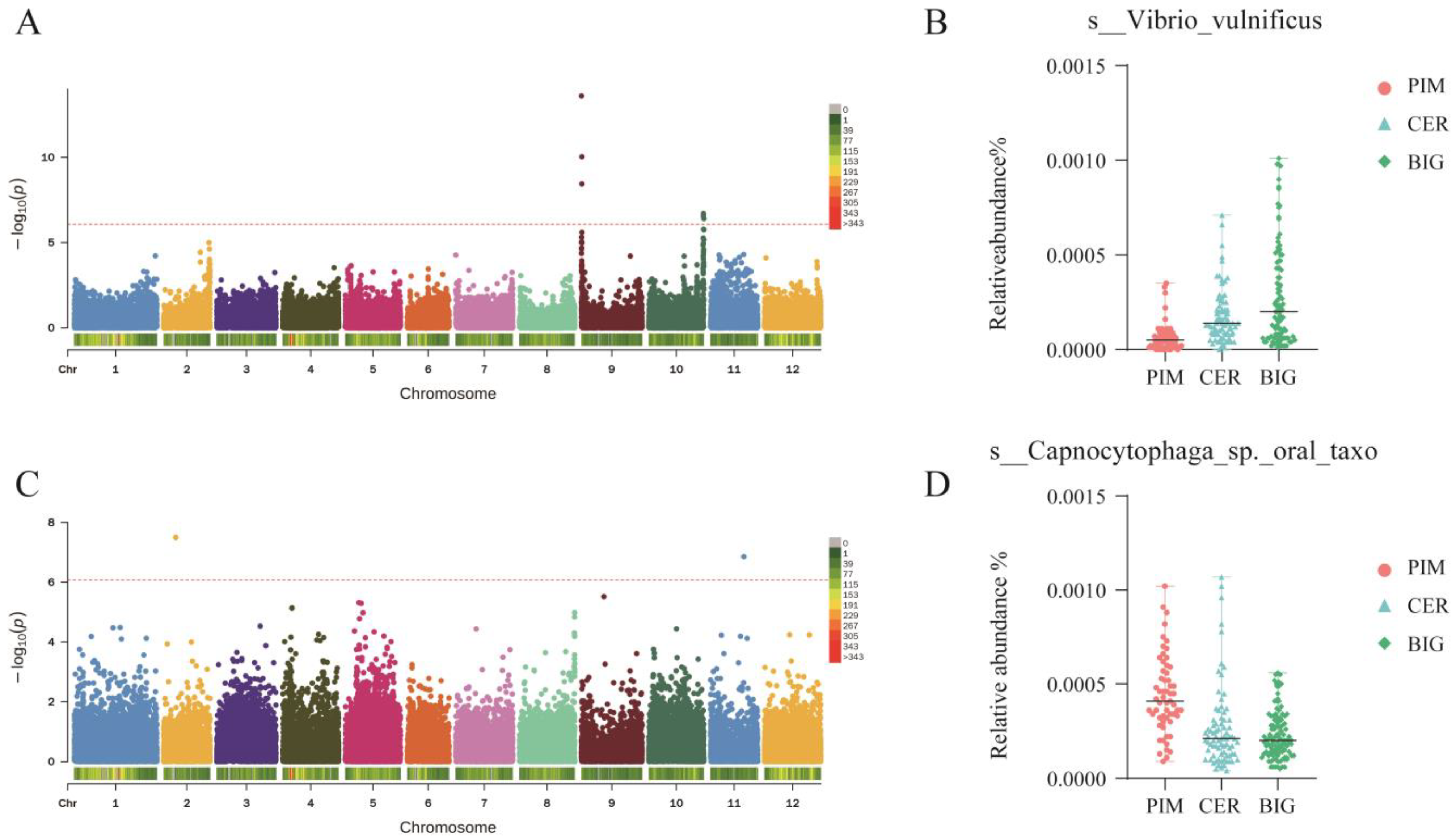
3.4. Differential Microbes in Leaf and Fruit Microbiomes of PIM, CER, and BIG
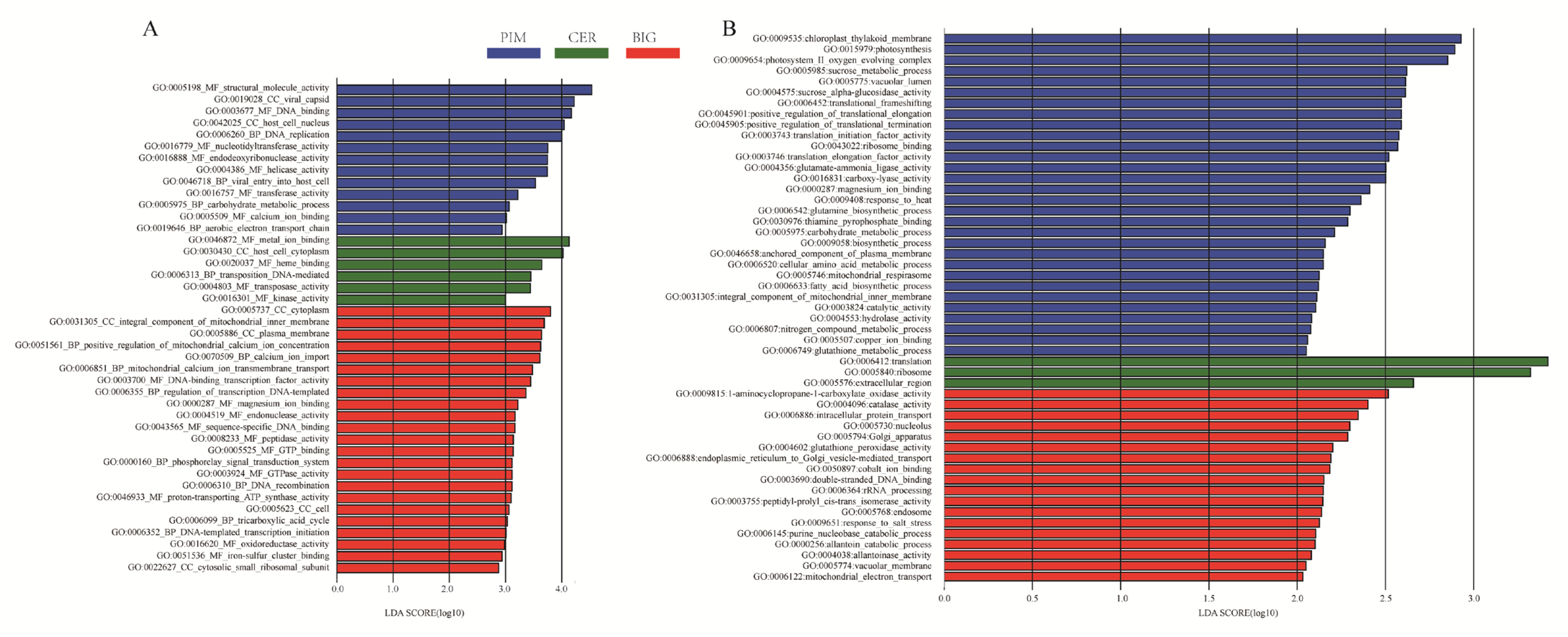
3.5. Functional Profiling of the Tomato Leaf and Fruit Microbiomes
3.6. GWAS Analysis Identifies Loci Regulating Tomato Symbiotic Microbiomes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Berg, G.; Rybakova, D.; Fischer, D.; Cernava, T.; Vergès, M.-C.C.; Charles, T.; Chen, X.; Cocolin, L.; Eversole, K.; Corral, G.H.; et al. Microbiome definition re-visited: Old concepts and new challenges. Microbiome 2020, 8, 103. [Google Scholar]
- Rodriguez, P.A.; Rothballer, M.; Chowdhury, S.P.; Nussbaumer, T.; Gutjahr, C.; Falter-Braun, P. Systems biology of plant-microbiome interactions. Mol. Plant 2019, 12, 804–821. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Brettell, L.E.; Qiu, Z.; Singh, B.K. Microbiome-mediated stress resistance in plants. Trends Plant Sci. 2020, 25, 733–743. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.M.; Potthoff, D.B.; Schäfer, M.; Barandun, N.; Vorholt, J.A. Protective role of the Arabidopsis leaf microbiota against a bacterial pathogen. Nat. Microbiol. 2021, 6, 1537–1548. [Google Scholar] [CrossRef] [PubMed]
- Soldan, R.; Fusi, M.; Cardinale, M.; Daffonchio, D.; Preston, G.M. The effect of plant domestication on host control of the microbiota. Commun. Biol. 2021, 4, 936. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Lee, K.K.; Jeon, J.; Harris, W.A.; Lee, Y.H. Domestication of Oryza species eco-evolutionarily shapes bacterial and fungal communities in rice seed. Microbiome 2020, 8, 20. [Google Scholar] [CrossRef] [PubMed]
- Favela, A.; Bohn, M.O.; Kent, A.D. Maize germplasm chronosequence shows crop breeding history impacts recruitment of the rhizosphere microbiome. ISME J. 2021, 15, 2454–2464. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.; Zhu, G.; Zhang, J.; Xu, X.Y.; Yu, Q.H.; Zheng, Z.; Zhang, Z.H.; Lun, Y.Y.; Li, S.; Wang, X.X.; et al. Genomic analyses provide insights into the history of tomato breeding. Nat. Genet. 2014, 46, 1220–1226. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Wang, S.; Huang, Z.; Zhang, S.B.; Liao, Q.G.; Zhang, C.Z.; Lin, T.; Qin, M.; Peng, M.; Yang, C.K.; et al. Rewiring of the fruit metabolome in tomato breeding. Cell 2018, 172, 249–261.e12. [Google Scholar] [CrossRef]
- Louca, S.; Doebeli, M.; Parfrey, L.W. Correcting for 16S rRNA gene copy numbers in microbiome surveys remains an unsolved problem. Microbiome 2018, 6, 41. [Google Scholar] [CrossRef]
- Roman-Reyna, V.; Pinili, D.; Borja, F.N.; Quibod, I.L.; Groen, S.C.; Alexandrov, N.; Mauleon, R.; Oliva, R. Characterization of the leaf microbiome from whole-genome sequencing data of the 3000 rice genomes project. Rice 2020, 13, 72. [Google Scholar] [CrossRef] [PubMed]
- Kwak, M.J.; Kong, H.G.; Choi, K.; Kwon, S.-K.; Song, J.Y.; Lee, J.; Lee, P.A.; Choi, S.Y.; Seo, M.; Lee, H.J.; et al. Rhizosphere microbiome structure alters to enable wilt resistance in tomato. Nat. Biotechnol. 2018, 36, 1100–1109. [Google Scholar] [CrossRef] [PubMed]
- French, E.; Ghaste, M.; Widhalm, J.R.; Iyer-Pascuzzi, A.S. Defense hormones modulate root microbiome diversity and composition in tomato. bioRxiv 2019. [Google Scholar] [CrossRef]
- Lee, S.A.; Kim, Y.; Kim, J.M.; Chu, B.; Jo, J.-H.; Sang, M.K.; Song, J.; Weon, H.-Y. A preliminary examination of bacterial, archaeal, and fungal communities inhabiting different rhizocompartments of tomato plants under real-world environments. Sci. Rep. 2019, 9, 9300. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wei, Z.; Yang, K.; Wang, J.; Jousset, A.; Xu, Y.; Shen, Q.; Friman, V.-P. Phage combination therapies for bacterial wilt disease in tomato. Nat. Biotechnol. 2019, 37, 1513–1520. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.Q.; Lei, S.N.; Li, Y.; Huang, W.; Ma, R.Q.; Xiong, J.; Zhang, T.; Jin, L.Y.; Haq, H.U.; Xu, X.H.; et al. Revealing the variation and stability of bacterial communities in tomato rhizosphere microbiota. Microorganisms 2020, 8, 170. [Google Scholar] [CrossRef]
- Choi, K.; Choi, J.; Lee, P.A.; Roy, N.; Khan, R.; Lee, H.J.; Weon, H.Y.; Kong, H.G.; Lee, S.-W. Alteration of bacterial wilt resistance in tomato plant by microbiota transplant. Front. Plant Sci. 2020, 11, 1186. [Google Scholar] [CrossRef] [PubMed]
- Wen, T.; Zhao, M.; Liu, T.; Huang, Q.W.; Yuan, J.; Shen, Q.R. High abundance of Ralstonia solanacearum changed tomato rhizosphere microbiome and metabolome. BMC Plant Biol. 2020, 20, 166. [Google Scholar] [CrossRef] [PubMed]
- Ottesen, A.R.; González Peña, A.; White, J.R.; Pettengill, J.B.; Li, C.; Allard, S.; Rideout, S.; Allard, M.; Hill, T.; Evans, P.; et al. Baseline survey of the anatomical microbial ecology of an important food plant: Solanum lycopersicum (tomato). BMC Microbiol. 2013, 13, 114. [Google Scholar] [CrossRef]
- Bergna, A.; Cernava, T.; Rändler, M.; Randler, M.; Grosch, R.; Zachow, C.; Berg, G. Tomato seeds preferably transmit plant beneficial endophytes. Phytobiomes J. 2018, 2, 183–193. [Google Scholar] [CrossRef]
- Rodriguez, R.M.; Khadka, V.S.; Menor, M.; Hernandez, B.Y.; Deng, Y. Tissue-associated microbial detection in cancer using human sequencing data. BMC Bioinform. 2020, 21, 523. [Google Scholar] [CrossRef] [PubMed]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed]
- Poore, G.D.; Kopylova, E.; Zhu, Q.; Carpenter, C.; Fraraccio, S.; Wandro, S.; Kosciolek, T.; Janssen, S.; Metcalf, J.; Song, S.J.; et al. Microbiome analyses of blood and tissues suggest cancer diagnostic approach. Nature 2020, 579, 567–574. [Google Scholar] [CrossRef]
- Reusch, T.B.H.; Schubert, P.R.; Marten, S.M.; Gill, D.; Karez, R.; Busch, K.; Hentschel, U. Lower Vibrio spp. abundances in Zostera marina leaf canopies suggest a novel ecosystem function for temperate seagrass beds. Mar. Biol. 2021, 168, 149. [Google Scholar] [CrossRef]
- Park, Y.S.; Kim, S.K.; Kim, S.Y.; Kim, K.M.; Ryu, C.-M. The transcriptome analysis of the Arabidopsis thaliana in response to the Vibrio vulnificus by RNA-sequencing. PLoS ONE 2019, 14, e0225976. [Google Scholar] [CrossRef] [PubMed]
- Morella, N.M.; Zhang, X.; Koskella, B. Tomato seed-associated bacteria confer protection of seedlings against foliar disease caused by Pseudomonas syringae. Phytobiomes J. 2019, 3, 177–190. [Google Scholar] [CrossRef]
- Huang, J.; Li, Y.F.; Li, Y.S.; Jian, J.I.; Lian, T.X. The rhizospheric microbiome becomes more diverse with maize domestication and genetic improvement. J. Integr. Agric. 2022, 21, 1188–1202. [Google Scholar] [CrossRef]
- Shenton, M.; Iwamoto, C.; Kurata, N.; Ikeo, K. Effect of wild and cultivated rice genotypes on rhizosphere bacterial community composition. Rice 2016, 9, 42. [Google Scholar] [CrossRef]
- Liu, F.; Hewezi, T.; Lebeis, S.L.; Pantalone, V.; Grewal, P.S.; Staton, M.E. Soil indigenous microbiome and plant genotypes cooperatively modify soybean rhizosphere microbiome assembly. BMC Microbiol. 2019, 19, 201. [Google Scholar] [CrossRef]
- Leff, J.W.; Lynch, R.C.; Kane, N.C.; Fierer, N.F. Plant domestication and the assembly of bacterial and fungal communities associated with strains of the common sunflower, Helianthus annuus. New Phytol. 2017, 214, 412–423. [Google Scholar] [CrossRef]
- Perreault, R.; Laforest-Lapointe, I. Plant-microbe interactions in the phyllosphere: Facing challenges of the anthropocene. ISME J. 2022, 16, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Allard, S.M.; Walsh, C.S.; Wallis, A.E.; Ottesen, A.R.; Brown, E.W.; Micallef, S.A. Solanum lycopersicum (tomato) hosts robust phyllosphere and rhizosphere bacterial communities when grown in soil amended with various organic and synthetic fertilizers. Sci. Total Environ. 2016, 573, 555–563. [Google Scholar] [CrossRef] [PubMed]
- WoldemariamYohannes, K.; Wan, Z.; Yu, Q.; Li, H.; Wei, X.; Liu, Y.; Wang, J.; Sun, B. Prebiotic, probiotic, antimicrobial, and functional food applications of Bacillus amyloliquefaciens. J. Agric. Food Chem. 2020, 68, 14709–14727. [Google Scholar] [CrossRef] [PubMed]
- Khalaf, E.M.; Raizada, M.N. Bacterial seed endophytes of domesticated cucurbits antagonize fungal and oomycete pathogens including powdery mildew. Front. Microbiol. 2018, 9, 295117. [Google Scholar]
- Carlström, C.I.; Field, C.M.; Bortfeld-Miller, M.; Müller, B.; Sunagawa, S.; Vorholt, J.A. Synthetic microbiota reveal priority effects and keystone strains in the Arabidopsis phyllosphere. Nat. Ecol. Evol. 2019, 3, 1445–1454. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; He, X.M.; Baer, M.; Beirinckx, S.; Tian, T.; Moya, Y.A.T.; Zhang, X.; Deichmann, M.; Frey, F.P.; Bresgen, V.; et al. Plant flavones enrich rhizosphere Oxalobacteraceae to improve maize performance under nitrogen deprivation. Nat. Plants 2021, 7, 481–499. [Google Scholar] [CrossRef]
- Cai, Q.; He, B.; Wang, S.; Fletcher, S.; Niu, D.; Mitter, N.; Birch, P.R.J.; Jin, H. Message in a bubble: Shuttling small RNAs and proteins between cells and interacting organisms using extracellular vesicles. Annu. Rev. Plant Biol. 2021, 72, 497–524. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, F.; Lyu, H.; Li, H.; Xi, K.; Yi, Y.; Zhang, Y. Domestication and Genetic Improvement Alter the Symbiotic Microbiome Structure and Function of Tomato Leaf and Fruit Pericarp. Microorganisms 2024, 12, 1351. https://doi.org/10.3390/microorganisms12071351
Li F, Lyu H, Li H, Xi K, Yi Y, Zhang Y. Domestication and Genetic Improvement Alter the Symbiotic Microbiome Structure and Function of Tomato Leaf and Fruit Pericarp. Microorganisms. 2024; 12(7):1351. https://doi.org/10.3390/microorganisms12071351
Chicago/Turabian StyleLi, Fei, Hongjun Lyu, Henan Li, Kuanling Xi, Yin Yi, and Yubin Zhang. 2024. "Domestication and Genetic Improvement Alter the Symbiotic Microbiome Structure and Function of Tomato Leaf and Fruit Pericarp" Microorganisms 12, no. 7: 1351. https://doi.org/10.3390/microorganisms12071351