
Comparative Analysis of Gut Microbiota between Captive and Wild Long-Tailed Gorals for Ex Situ Conservation


Abstract
:1. Introduction
2. Materials and Methods
2.1. Fecal Sample Collection
2.2. Bioinformatics Analysis
3. Results
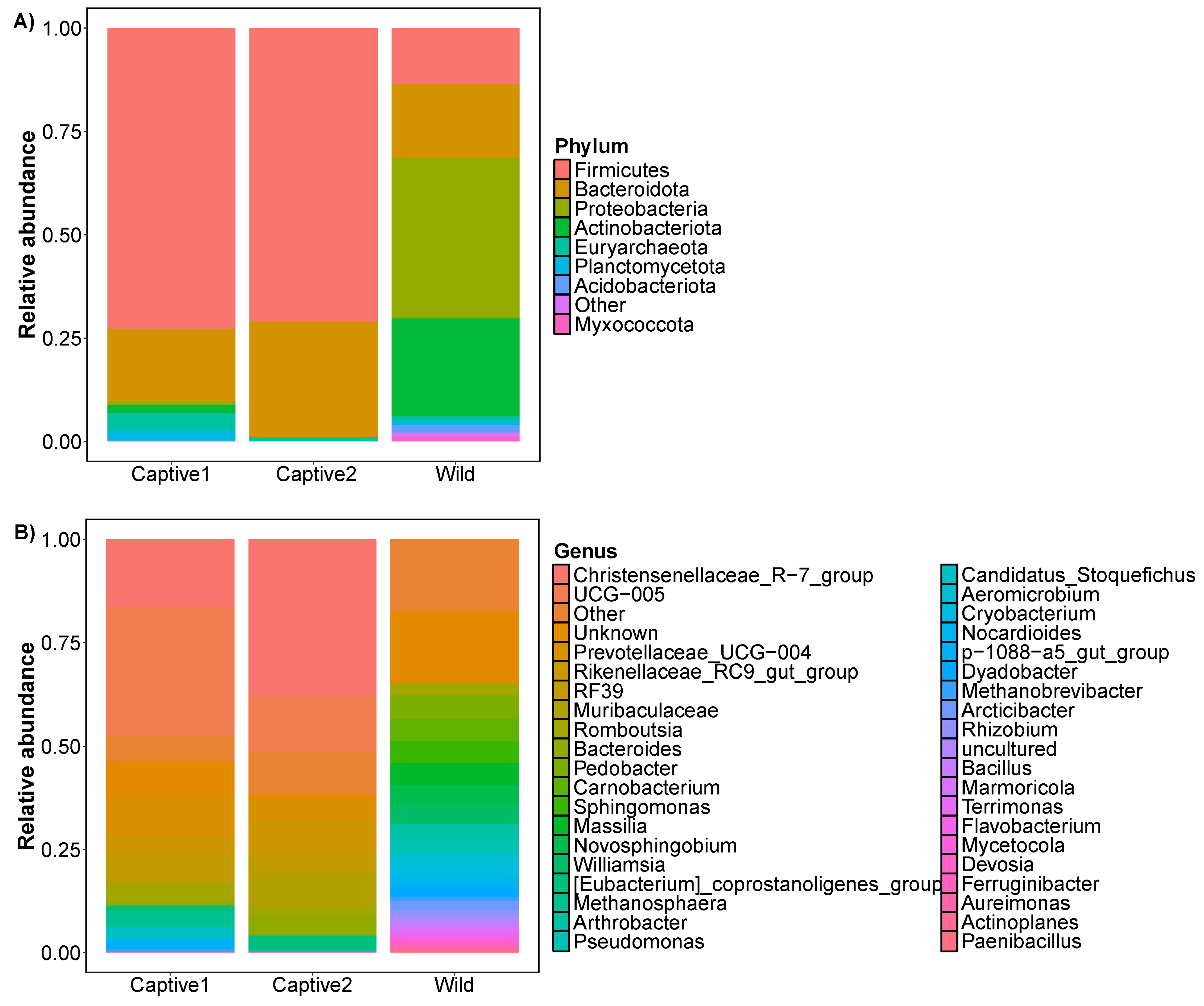
3.1. Relative Abundance
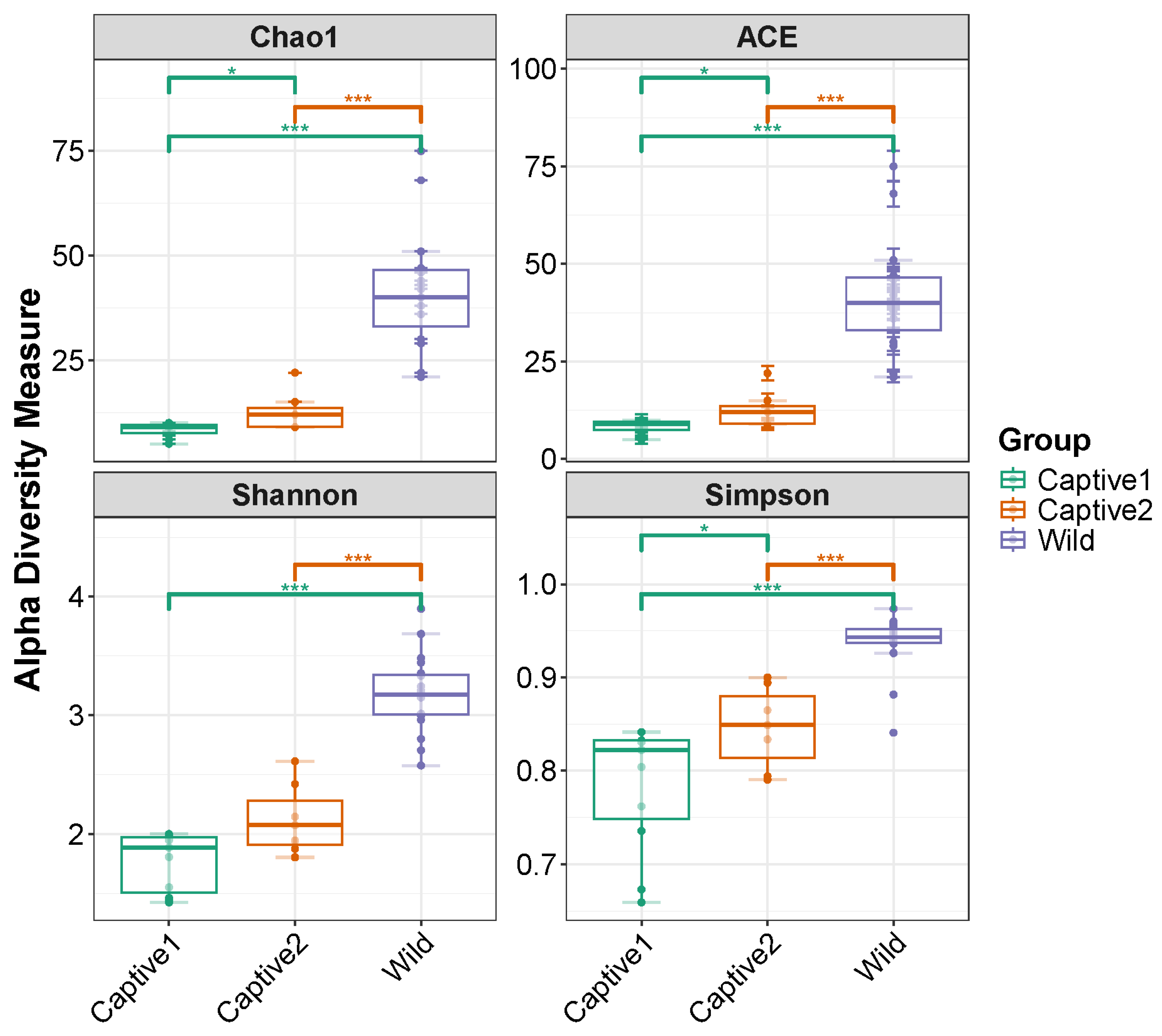
3.2. Alpha Diversity
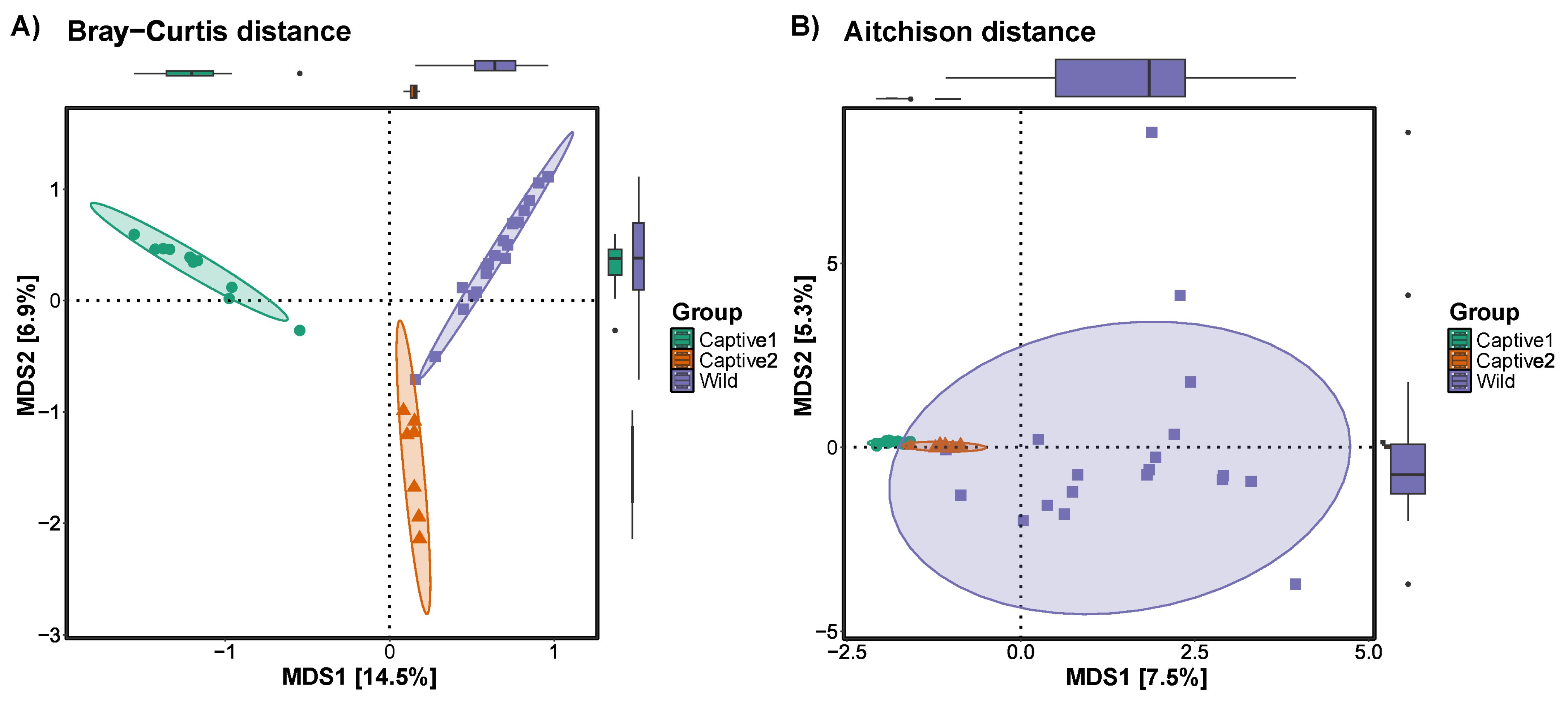
3.3. Beta Diversity
3.4. LEfSe
3.5. Redundancy Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Clark, F.E.; Greggor, A.L.; Montgomery, S.H.; Plotnik, J.M. The endangered brain: Actively preserving ex-situ animal behaviour and cognition will benefit in-situ conservation. R. Soc. Open Sci. 2023, 10, 230707. [Google Scholar] [CrossRef]
- Kaplan, G. Casting the net widely for change in animal welfare: The plight of birds in zoos, ex situ conservation, and conservation fieldwork. Animals 2021, 12, 31. [Google Scholar] [CrossRef]
- Mestanza-Ramón, C.; Henkanaththegedara, S.M.; Vásconez Duchicela, P.; Vargas Tierras, Y.; Sánchez Capa, M.; Constante Mejía, D.; Jimenez Gutierrez, M.; Charco Guamán, M.; Mestanza Ramón, P. In-situ and ex-situ biodiversity conservation in ecuador: A review of policies, actions and challenges. Diversity 2020, 12, 315. [Google Scholar] [CrossRef]
- Moloney, D.J.F.; Collins, C.; Holloway, P.; O’Riordan, R. The Conservationist’s Toolkit: A critical review of the need for a conceptual framework of both in-situ and ex-situ conservation strategies to ensure the success of restoration ecology. Biol. Conserv. 2023, 287, 110345. [Google Scholar] [CrossRef]
- Lewton, J.; Rose, P.E. Social networks research in ex situ populations: Patterns, trends, and future directions for conservation-focused behavioral research. Zoo Biol. 2021, 40, 493–502. [Google Scholar] [CrossRef]
- Leiss, L.; Rauhaus, A.; Rakotoarison, A.; Fusari, C.; Vences, M.; Ziegler, T. Review of threatened Malagasy freshwater fishes in zoos and aquaria: The necessity of an ex situ conservation network—A call for action. Zoo Biol. 2022, 41, 244–262. [Google Scholar] [CrossRef]
- Schwartz, K.R.; Byers, O.; Miller, P.; Blessington, J.; Smith, B. The role of zoos in tree kangaroo conservation: Connecting ex situ and in situ conservation action. In Tree Kangaroos; Academic Press: Cambridge, UK, 2021; pp. 329–361. [Google Scholar] [CrossRef]
- Sanders, K.; Fernandez, E.J. Behavioral implications of enrichment for golden lion tamarins: A tool for ex situ conservation. J. Appl. Anim. Welf. Sci. 2022, 25, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Feliciano, R.; de Barros Leite, A.; Garbeloto, M.C.; Silveira, L.F.; Francisco, M.R. A critical assessment of ex situ conservation based on the Brazilian avifauna: Are we focusing on what is easier? Perspect. Ecol. Conserv. 2023, 21, 62–70. [Google Scholar] [CrossRef]
- Campos, C.I.; Martinez, M.A.; Acosta, D.; Diaz-Luque, J.A.; Berkunsky, I.; Lamberski, N.L.; Cruz-Nieto, J.; Russello, M.A.; Wright, T.F. Genetic diversity and population structure of two endangered Neotropical parrots inform in situ and ex situ conservation strategies. Diversity 2021, 13, 386. [Google Scholar] [CrossRef]
- Hu, T.; Chitnis, N.; Monos, D.; Dinh, A. Next-generation sequencing technologies: An overview. Hum. Immunol. 2021, 82, 801–811. [Google Scholar] [CrossRef]
- Rodriguez, R.; Krishnan, Y. The chemistry of next-generation sequencing. Nat. Biotechnol. 2023, 41, 1709–1715. [Google Scholar] [CrossRef] [PubMed]
- Wensel, C.R.; Pluznick, J.L.; Salzberg, S.L.; Sears, C.L. Next-generation sequencing: Insights to advance clinical investigations of the microbiome. J. Clin. Investig. 2022, 132. [Google Scholar] [CrossRef] [PubMed]
- Pereira, R.; Oliveira, J.; Sousa, M. Bioinformatics and computational tools for next-generation sequencing analysis in clinical genetics. J. Clin. Med. 2020, 9, 132. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Quake, S.R. Sequential sequencing by synthesis and the next-generation sequencing revolution. Trends Biotechnol. 2023, 41, 1565–1572. [Google Scholar] [CrossRef] [PubMed]
- De Vos, W.M.; Tilg, H.; Van Hul, M.; Cani, P.D. Gut microbiome and health: Mechanistic insights. Gut 2022, 71, 1020–1032. [Google Scholar] [CrossRef] [PubMed]
- Shanahan, F.; Ghosh, T.S.; O’Toole, P.W. The healthy microbiome—what is the definition of a healthy gut microbiome? Gastroenterology 2021, 160, 483–494. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.S.; Koller, K.R.; Ramaboli, M.C.; Nesengani, L.T.; Ocvirk, S.; Chen, C.; Flanagan, C.A.; Sapp, F.R.; Merritt, Z.T.; Bhatti, F.; et al. Diet and the human gut microbiome: An international review. Dig. Dis. Sci. 2020, 65, 723–740. [Google Scholar] [CrossRef]
- Kuziel, G.A.; Rakoff-Nahoum, S. The gut microbiome. Curr. Biol. 2022, 32, R257–R264. [Google Scholar] [CrossRef] [PubMed]
- Gacesa, R.; Kurilshikov, A.; Vich Vila, A.; Sinha, T.; Klaassen, M.A.Y.; Bolte, L.A.; Andreu-Sánchez, S.; Chen, L.; Collij, V.; Hu, S.; et al. Environmental factors shaping the gut microbiome in a Dutch population. Nature 2022, 604, 732–739. [Google Scholar] [CrossRef]
- Schneider, T. Different meaning in different sizes: Ecology in size scales. Philos. Sci. 2022, 89, 1134–1144. [Google Scholar] [CrossRef]
- Xia, Y.; Sun, J. Applied Microbiome Statistics: Correlation, Association, Interaction and Composition; CRC Press: Boca Raton, FL, USA, 2024. [Google Scholar]
- Davidson, G.L.; Raulo, A.; Knowles, S.C.L. Identifying microbiome-mediated behaviour in wild vertebrates. Trends Ecol. Evol. 2020, 35, 972–980. [Google Scholar] [CrossRef]
- Trevelline, B.K.; Kohl, K.D. The gut microbiome influences host diet selection behavior. Proc. Natl. Acad. Sci. USA 2022, 119, e2117537119. [Google Scholar] [CrossRef] [PubMed]
- Clauss, M.; Gérard, P.; Mosca, A.; Leclerc, M. Interplay between exercise and gut microbiome in the context of human health and performance. Front. Nutr. 2021, 8, 637010. [Google Scholar] [CrossRef]
- Ramos, C.; Gibson, G.R.; Walton, G.E.; Magistro, D.; Kinnear, W.; Hunter, K. Systematic review of the effects of exercise and physical activity on the gut microbiome of older adults. Nutrients 2022, 14, 674. [Google Scholar] [CrossRef]
- Bycura, D.; Santos, A.C.; Shiffer, A.; Kyman, S.; Winfree, K.; Sutliffe, J.; Pearson, T.; Sonderegger, D.; Cope, E.; Caporaso, J.G. Impact of different exercise modalities on the human gut microbiome. Sports 2021, 9, 14. [Google Scholar] [CrossRef]
- De Filippis, F.; Pasolli, E.; Ercolini, D. The food-gut axis: Lactic acid bacteria and their link to food, the gut microbiome and human health. FEMS Microbiol. Rev. 2020, 44, 454–489. [Google Scholar] [CrossRef]
- Pasolli, E.; De Filippis, F.; Mauriello, I.E.; Cumbo, F.; Walsh, A.M.; Leech, J.; Cotter, P.D.; Segata, N.; Ercolini, D. Large-scale genome-wide analysis links lactic acid bacteria from food with the gut microbiome. Nat. Commun. 2020, 11, 2610. [Google Scholar] [CrossRef]
- Wolter, M.; Grant, E.T.; Boudaud, M.; Steimle, A.; Pereira, G.V.; Martens, E.C.; Desai, M.S. Leveraging diet to engineer the gut microbiome. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 885–902. [Google Scholar] [CrossRef]
- Zhang, P. Influence of foods and nutrition on the gut microbiome and implications for intestinal health. Int. J. Mol. Sci. 2022, 23, 9588. [Google Scholar] [CrossRef] [PubMed]
- Trakman, G.L.; Fehily, S.; Basnayake, C.; Hamilton, A.L.; Russell, E.; Wilson-O’Brien, A.; Kamm, M.A. Diet and gut microbiome in gastrointestinal disease. J. Gastroenterol. Hepatol. 2022, 37, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Cho, C.U.; Kim, K.C.; Kwon, G.H.; Kim, K.Y.; Lee, B.K.; Son, J.I. Habitat use of reintroduced long-tailed gorals (Naemorhedus caudatus) in Woraksan (Mt.) National Park in Korea. Korean J. Environ. Ecol. 2015, 29, 184–191. [Google Scholar] [CrossRef]
- Lim, M.Y.; Park, Y.S.; Kim, J.H.; Nam, Y.D. Evaluation of fecal DNA extraction protocols for human gut microbiome studies. BMC Microbiol. 2020, 20, 212. [Google Scholar] [CrossRef]
- Srirungruang, S.; Mahajindawong, B.; Nimitpanya, P.; Bunkasem, U.; Ayuyoe, P.; Nuchprayoon, S.; Sanprasert, V. Comparative study of DNA extraction methods for the PCR detection of intestinal parasites in human stool samples. Diagnostics 2022, 12, 2588. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.; Hoffbeck, C.; West, A.G.; Pas, A.; Taylor, M.W. 16S rRNA gene-based microbiota profiles from diverse avian faeces are largely independent of DNA preservation and extraction method. Front. Microbiol. 2023, 14, 1239167. [Google Scholar] [CrossRef]
- Lee, H.B.; Jeong, D.H.; Cho, B.C.; Park, J.S. Comparative analyses of eight primer sets commonly used to target the bacterial 16S rRNA gene for marine metabarcoding-based studies. Front. Mar. Sci. 2023, 10, 1199116. [Google Scholar] [CrossRef]
- Zhao, J.; Rodriguez, J.; Martens-Habbena, W. Fine-scale evaluation of two standard 16S rRNA gene amplicon primer pairs for analysis of total prokaryotes and archaeal nitrifiers in differently managed soils. Front. Microbiol. 2023, 14, 1140487. [Google Scholar] [CrossRef]
- Yuan, J.; Deng, X.; Xie, X.; Chen, L.; Wei, C.; Feng, C.; Qiu, G. Blind spots of universal primers and specific FISH probes for functional microbe and community characterization in EBPR systems. ISME Commun. 2024, 4, ycae011. [Google Scholar] [CrossRef]
- Fadeev, E.; Cardozo-Mino, M.G.; Rapp, J.Z.; Bienhold, C.; Salter, I.; Salman-Carvalho, V.; Molari, M.; Tegetmeyer, H.E.; Buttigieg, P.L.; Boetius, A. Comparison of two 16S rRNA primers (V3-V4 and V4-V5) for studies of arctic microbial communities. Front. Microbiol. 2021, 12, 637526. [Google Scholar] [CrossRef]
- Umbach, A.K.; Stegelmeier, A.A.; Neufeld, J.D. Archaea are rare and uncommon members of the mammalian skin microbiome. mSystems 2021, 6, e0064221. [Google Scholar] [CrossRef]
- Fentie, E.G.; Jeong, M.; Emire, S.A.; Demsash, H.D.; Kim, M.A.; Jeon, H.J.; Lee, S.E.; Tagele, S.B.; Park, Y.J.; Shin, J.H. Microbiome dataset of spontaneously fermented Ethiopian honey wine, Tej. Data Brief 2022, 42, 108022. [Google Scholar] [CrossRef]
- Kumaishi, K.; Usui, E.; Suzuki, K.; Kobori, S.; Sato, T.; Toda, Y.; Takanashi, H.; Shinozaki, S.; Noda, M.; Takakura, A.; et al. High throughput method of 16S rRNA gene sequencing library preparation for plant root microbial community profiling. Sci. Rep. 2022, 12, 19289. [Google Scholar] [CrossRef]
- Severn-Ellis, A.A.; Scheben, A.; Neik, T.X.; Saad, N.S.M.; Pradhan, A.; Batley, J. Genotyping for species identification and diversity assessment using double-digest restriction site-associated DNA sequencing (ddRAD-seq). Legume Genom. Methods Protoc. 2020, 2107, 159–187. [Google Scholar] [CrossRef]
- Liu, D.; Li, Q.; Luo, J.; Huang, Q.; Zhang, Y. An SPRI beads-based DNA purification strategy for flexibility and cost-effectiveness. BMC Genom. 2023, 24, 125. [Google Scholar] [CrossRef]
- Kawakatsu, T. Whole-genome bisulfite sequencing and epigenetic variation in cereal methylomes. Cereal Genom. Methods Protoc. 2020, 2072, 119–128. [Google Scholar] [CrossRef]
- Oldoni, F.; Bader, D.; Fantinato, C.; Wootton, S.C.; Lagacé, R.; Kidd, K.K.; Podini, D. A sequence-based 74plex microhaplotype assay for analysis of forensic DNA mixtures. Forensic Sci. Int. Genet. 2020, 49, 102367. [Google Scholar] [CrossRef] [PubMed]
- Paniccia, A.; Polanco, P.M.; Boone, B.A.; Wald, A.I.; McGrath, K.; Brand, R.E.; Khalid, A.; Kubiliun, N.; O’Broin-Lennon, A.M.; Park, W.G.; et al. Prospective, multi-institutional, real-time next-generation sequencing of pancreatic cyst fluid reveals diverse genomic alterations that improve the clinical management of pancreatic cysts. Gastroenterology 2023, 164, 117–133.e7. [Google Scholar] [CrossRef]
- Niu, D.; Li, L.; Yu, Y.; Zang, W.; Li, Z.; Zhou, L.; Jia, L.; Rao, G.; Gao, L.; Cheng, G.; et al. Evaluation of next generation sequencing for detecting HER2 copy number in breast and gastric cancers. Pathol. Oncol. Res. 2020, 26, 2577–2585. [Google Scholar] [CrossRef]
- Wang, H.; Qian, Y.; Lu, Y.; Qin, Q.; Lu, G.; Cheng, G.; Zhang, P.; Yang, L.; Wu, B.; Zhou, W. Clinical utility of 24-h rapid trio-exome sequencing for critically ill infants. npj Genom. Med. 2020, 5, 20. [Google Scholar] [CrossRef] [PubMed]
- Cihlar, J.C.; Strobl, C.; Lagacé, R.; Muenzler, M.; Parson, W.; Budowle, B. Distinguishing mitochondrial DNA and NUMT sequences amplified with the precision ID mtDNA whole genome panel. Mitochondrion 2020, 55, 122–133. [Google Scholar] [CrossRef]
- Tong, J.; Niu, Y.; Chen, Z.J.; Zhang, C. Comparison of the transcriptional profile in the decidua of early-onset and late-onset pre-eclampsia. J. Obstet. Gynaecol. Res. 2020, 46, 1055–1066. [Google Scholar] [CrossRef]
- Salvianti, F.; Gelmini, S.; Mancini, I.; Pazzagli, M.; Pillozzi, S.; Giommoni, E.; Brugia, M.; Di Costanzo, F.; Galardi, F.; De Luca, F.; et al. Circulating tumour cells and cell-free DNA as a prognostic factor in metastatic colorectal cancer: The OMITERC prospective study. Br. J. Cancer 2021, 125, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Isaka, K.; Sugawara, D.; Yamazaki, H.; Kimura, Y.; Osaka, T.; Tsuneda, S. Long-term limitation effects of Se(VI), Zn(II), and Ni(II) on start-up of the anammox process using gel carrier. Front. Bioeng. Biotechnol. 2022, 10, 851617. [Google Scholar] [CrossRef]
- Toscano, A.; Giannuzzi, D.; Malgwi, I.H.; Deb, S.; Broccanello, C.; Squartini, A.; Stevanato, P.; Cecchinato, A.; Gallo, L.; Schiavon, S.; et al. Characterization of dry-cured ham microbiota at 12 months of seasoning obtained from different rearing strategies using 16S rRNA profiling. Food Microbiol. 2024, 122, 104558. [Google Scholar] [CrossRef] [PubMed]
- Berg Luecke, L.; Gundry, R.L. Assessment of streptavidin bead binding capacity to improve quality of streptavidin-based enrichment studies. J. Proteome Res. 2021, 20, 1153–1164. [Google Scholar] [CrossRef]
- Strobel, E.J.; Kelly, S.L.; Szyjka, C.E. Isolation of synchronized E. coli elongation complexes for solid-phase and solution-based in vitro transcription assays. In Methods Enzymol; Academic Press: Cambridge, UK, 2022; p. 675. [Google Scholar] [CrossRef]
- Kelly, S.L.; Szyjka, C.E.; Strobel, E.J. Purification of synchronized E. coli transcription elongation complexes by reversible immobilization on magnetic beads. bioRxiv 2022, 2022-01. [Google Scholar] [CrossRef]
- Santos-Barriopedro, I.; van Mierlo, G.; Vermeulen, M. Off-the-shelf proximity biotinylation for interaction proteomics. Nat. Commun. 2021, 12, 5015. [Google Scholar] [CrossRef]
- Chen, Q.F.; Kang, K.L.; Song, J.J.; Zhang, C.; Yu, Z.L.; Zhao, G.B.; Wu, H.; Ji, A.Q.; Ye, J.; Wang, L. Allelic diversity and forensic estimations of the Beijing Hans: Comparative data on sequence-based and length-based STRs. Forensic Sci. Int. Genet. 2021, 51, 102424. [Google Scholar] [CrossRef]
- Yu, C.; Wang, Y.; Liu, T.; Sha, K.; Song, Z.; Zhao, M.; Wang, X. The microRNA miR-3174 suppresses the expression of ADAM15 and inhibits the proliferation of patient-derived bladder cancer cells. OncoTargets Ther. 2020, 13, 4157–4168. [Google Scholar] [CrossRef] [PubMed]
- Nageeb, A.M.; Mohamed, M.M.; Ezz El Arab, L.R.; Khalifa, M.K.; Swellam, M. Next generation sequencing of BRCA genes in glioblastoma multiform Egyptian patients: A pilot study. Arch. Physiol. Biochem. 2022, 128, 809–817. [Google Scholar] [CrossRef]
- Marani, C.; Akaev, I.; Yeoh, C.C.; Walsh, E.; Rahimi, S. Cervical malignant mixed mesonephric tumour: A case report with local recurrence after six-years and next-generation sequencing analysis with particular reference to the ataxia telangiectasia mutated gene. Exp. Ther. Med. 2021, 21, 394. [Google Scholar] [CrossRef]
- Domenis, R.; Cifù, A.; Mio, C.; Fabris, M.; Curcio, F. Pro-inflammatory microenvironment modulates the transfer of mutated TP53 mediated by tumor exosomes. Int. J. Mol. Sci. 2021, 22, 6258. [Google Scholar] [CrossRef] [PubMed]
- Romano, C.; Di Gregorio, S.; Pennisi, M.S.; Tirrò, E.; Broggi, G.; Caltabiano, R.; Manzella, L.; Ruggieri, M.; Vigneri, P.; Di Cataldo, A. Multiple primary malignances managed with surgical excision: A case report with next generation sequencing analysis. Mol. Biol. Rep. 2022, 49, 9059–9064. [Google Scholar] [CrossRef]
- El Kadiri, Y.; Ratbi, I.; Laarabi, F.Z.; Kriouile, Y.; Sefiani, A.; Lyahyai, J. Identification of a novel LAMA2 c.2217G > A, p.(Trp739*) mutation in a Moroccan patient with congenital muscular dystrophy: A case report. BMC Med. Genom. 2021, 14, 113. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, M.; Tanaka, T.; Murata, S.; Miyabe, A.; Ishige, T.; Kawasaki, K.; Yokoyama, M.; Hashimoto, N.; Yamagata, K.; Nagano, H.; et al. Extension of bacterial rDNA sequencing for simultaneous methylation detection and its application in microflora analysis. Sci. Rep. 2023, 13, 5731. [Google Scholar] [CrossRef] [PubMed]
- Man, M.A.; Ungur, R.A.; Motoc, N.S.; Pop, L.A.; Berindan-Neagoe, I.; Ruta, V.M. Lung microbiota in idiopathic pulmonary fibrosis, hypersensitivity pneumonitis, and unclassified interstitial lung diseases: A preliminary pilot study. Diagnostics 2023, 13, 3157. [Google Scholar] [CrossRef] [PubMed]
- Krasnov, Y.M.; Alkhova, Z.V.; Generalov, S.V.; Tuchkov, I.V.; Naryshkina, E.A.; Sharapova, N.A.; Abramova, E.G.; Nikiforov, A.K. Whole genome sequencing and phylogenetic analysis of the rabies virus strain Moscow 3253 adapted to a Vero cell line. Mol. Genet. Microbiol. Virol. 2020, 35, 237–242. [Google Scholar] [CrossRef]
- Lombardi, A.; Russo, M.; Luce, A.; Morgillo, F.; Tirino, V.; Misso, G.; Martinelli, E.; Troiani, T.; Desiderio, V.; Papaccio, G.; et al. Comparative study of NGS platform Ion Torrent personal genome machine and Therascreen Rotor-Gene Q for the detection of somatic variants in cancer. High-Throughput 2020, 9, 4. [Google Scholar] [CrossRef]
- Goodwin, D.; Rathi, V.; Conron, M.; Wright, G.M. Genomic and clinical significance of multiple primary lung cancers as determined by next-generation sequencing. J. Thorac. Oncol. 2021, 16, 1166–1175. [Google Scholar] [CrossRef] [PubMed]
- Brenner, E.D.; Scheid, P.E.; DeGrazia, J.; Geltzeiler, A.R.; Katari, M.S. Using the Integrated Genome Viewer to reveal amplicon-derived polymorphism enriched at the phenylthiocarbamide locus in the teaching lab. Biochem. Mol. Biol. Educ. 2021, 49, 361–371. [Google Scholar] [CrossRef]
- Candelaria, M.; Gutierrez-Hernandez, O.; Diaz-Chavez, J.; Cerrato, D.; Gutierrez-Ramirez, F.J.; Aviles, A. Mutational landscape of diffuse large B-cell lymphoma (DLBCL) in Mexican patients. Blood 2023, 142 (Suppl. 1), 6096. [Google Scholar] [CrossRef]
- Manderstedt, E.; Nilsson, R.; Ljung, R.; Lind-Halldén, C.; Astermark, J.; Halldén, C. Detection of mosaics in hemophilia A by deep Ion Torrent sequencing and droplet digital PCR. Res. Pract. Thromb. Haemost. 2020, 4, 1121–1130. [Google Scholar] [CrossRef]
- Rai, S.N.; Qian, C.; Pan, J.; Rai, J.P.; Song, M.; Bagaitkar, J.; Merchant, M.; Cave, M.; Egilmez, N.K.; McClain, C.J. Microbiome data analysis with applications to pre-clinical studies using QIIME2: Statistical considerations. Genes Dis. 2021, 8, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Odom, A.R.; Faits, T.; Castro-Nallar, E.; Crandall, K.A.; Johnson, W.E. Metagenomic profiling pipelines improve taxonomic classification for 16S amplicon sequencing data. Sci. Rep. 2023, 13, 13957. [Google Scholar] [CrossRef] [PubMed]
- Wen, T.; Niu, G.; Chen, T.; Shen, Q.; Yuan, J.; Liu, Y.X. The best practice for microbiome analysis using R. Protein Cell 2023, 14, 713–725. [Google Scholar] [CrossRef]
- Bajaj, A.; Markandey, M.; Singh, M.; Sahu, P.; Vuyyuru, S.K.; Kante, B.; Kumar, P.; Verma, M.; Makharia, G.; Kedia, S.; et al. Exclusive enteral nutrition mediates beneficial gut microbiome enrichment in acute severe colitis. Inflamm. Bowel Dis. 2024, 30, 641–650. [Google Scholar] [CrossRef]
- Tollefson, M.; Tollefson, M. Graphics with the ggplot2 Package: An Introduction. In Visualizing Data in R 4: Graphics Using the Base, Graphics, Stats, and ggplot2 Packages; Apress: Berkeley, CA, USA, 2021; pp. 281–293. [Google Scholar] [CrossRef]
- Ahlmann-Eltze, C.; Patil, I. ggsignif: R Package for Displaying Significance Brackets for ‘ggplot2’. 2021. Available online: https://www.researchgate.net/profile/Indrajeet-Patil-2/publication/350527292_ggsignif_R_Package_for_Displaying_Significance_Brackets_for_‘ggplot2′/links/608124a0907dcf667bb5cc84/ggsignif-R-Package-for-Displaying-Significance-Brackets-for-ggplot2.pdf (accessed on 8 April 2021).
- Barnett, D.J.; Arts, I.C.; Penders, J. microViz: An R package for microbiome data visualization and statistics. J. Open Source Softw. 2021, 6, 3201. [Google Scholar] [CrossRef]
- Nugawela, N.P.P.S.; Mahaliyana, A.S.; Abhiram, G.; Abeygunawardena, A.P. A meta-analytic review of microplastic pollution in the Indian Ocean: Ecological health and seafood safety risk implications. Mar. Pollut. Bull. 2023, 193, 115213. [Google Scholar] [CrossRef]
- Cao, Y.; Dong, Q.; Wang, D.; Zhang, P.; Liu, Y.; Niu, C. microbiomeMarker: An R/Bioconductor package for microbiome marker identification and visualization. Bioinformatics 2022, 38, 4027–4029. [Google Scholar] [CrossRef]
- Alessandri, G.; Argentini, C.; Milani, C.; Turroni, F.; Cristina Ossiprandi, M.; van Sinderen, D.; Ventura, M. Catching a glimpse of the bacterial gut community of companion animals: A canine and feline perspective. Microb. Biotechnol. 2020, 13, 1708–1732. [Google Scholar] [CrossRef]
- Vasques-Monteiro, I.M.L.; Silva-Veiga, F.M.; Miranda, C.S.; de Andrade Gonçalves, É.C.B.; Daleprane, J.B.; Souza-Mello, V. A rise in proteobacteria is an indicator of gut-liver axis-mediated nonalcoholic fatty liver disease in high-fructose-fed adult mice. Nutr. Res. 2021, 91, 26–35. [Google Scholar] [CrossRef]
- Moon, C.D.; Young, W.; Maclean, P.H.; Cookson, A.L.; Bermingham, E.N. Metagenomic insights into the roles of proteobacteria in the gastrointestinal microbiomes of healthy dogs and cats. MicrobiologyOpen 2018, 7, e00677. [Google Scholar] [CrossRef] [PubMed]
- Grgas, D.; Rukavina, M.; Bešlo, D.; Štefanac, T.; Crnek, V.; Šikić, T.; Habuda-Stanić, M.; Landeka Dragičević, T. The bacterial degradation of lignin—A review. Water 2023, 15, 1272. [Google Scholar] [CrossRef]
- Law, J.W.F.; Letchumanan, V.; Tan, L.T.H.; Ser, H.L.; Goh, B.H.; Lee, L.H. The rising of “modern Actinobacteria” era. Prog. Microbes Mol. Biol. 2020, 3, a0000064. [Google Scholar] [CrossRef]
- Dupont, H.L.; Jiang, Z.D.; Dupont, A.W.; Utay, N.S. The intestinal microbiome in human health and disease. Trans. Am. Clin. Climatol. Assoc. 2020, 131, 178–197. [Google Scholar] [PubMed]
- Kubinyi, E.; Bel Rhali, S.; Sándor, S.; Szabó, A.; Felföldi, T. Gut microbiome composition is associated with age and memory performance in pet dogs. Animals 2020, 10, 1488. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Yang, L.L.; Xin, Y.H. Diversity of the genus Cryobacterium and proposal of 19 novel species isolated from glaciers. Front. Microbiol. 2023, 14, 1115168. [Google Scholar] [CrossRef]
- Liu, Y.; Shen, L.; Zeng, Y.; Xing, T.; Xu, B.; Wang, N. Genomic insights of Cryobacterium isolated from ice core reveal genome dynamics for adaptation in glacier. Front. Microbiol. 2020, 11, 1530. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Tian, J.H.; Liu, H.C.; Zhou, Y.G.; Xin, Y.H. Cryobacterium ruanii sp. nov. and Cryobacterium breve sp. nov., isolated from glaciers. Int. J. Syst. Evol. Microbiol. 2020, 70, 1918–1923. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Singh, S.M.; Segawa, T.; Singh, P.K. Bacterial diversity and biopotentials of Hamtah glacier cryoconites, Himalaya. Front. Microbiol. 2024, 15, 1362678. [Google Scholar] [CrossRef]
- Teoh, C.P.; González-Aravena, M.; Lavin, P.; Wong, C.M.V.L. Cold adaptation and response genes of Antarctic Cryobacterium sp. SO2 from the Fildes Peninsula, King George Island. Polar Biol. 2024, 47, 135–156. [Google Scholar] [CrossRef]
- Han, S.R.; Kim, B.; Jang, J.H.; Park, H.; Oh, T.J. Complete genome sequence of Arthrobacter sp. PAMC25564 and its comparative genome analysis for elucidating the role of CAZymes in cold adaptation. BMC Genom. 2021, 22, 403. [Google Scholar] [CrossRef] [PubMed]
- Vodickova, P.; Suman, J.; Benesova, E.; Strejcek, M.; Neumann-Schaal, M.; Cajthaml, T.; Ridl, J.; Pajer, P.; Ulbrich, P.; Uhlik, O.; et al. Arthrobacter polaris sp. nov., a new cold-adapted member of the family Micrococcaceae isolated from Antarctic fellfield soil. Int. J. Syst. Evol. Microbiol. 2022, 72, 005541. [Google Scholar] [CrossRef]
- Lee, G.L.Y.; Zakaria, N.N.; Futamata, H.; Suzuki, K.; Zulkharnain, A.; Shaharuddin, N.A.; Convey, P.; Zahri, K.N.M.; Ahmad, S.A. Metabolic pathway of phenol degradation of a cold-adapted Antarctic bacteria, Arthrobacter sp. Catalysts 2022, 12, 1422. [Google Scholar] [CrossRef]
- Jiang, C.; Cheng, Y.; Zang, H.; Chen, X.; Wang, Y.; Zhang, Y.; Wang, J.; Shen, X.; Li, C. Biodegradation of lignin and the associated degradation pathway by psychrotrophic Arthrobacter sp. C2 from the cold region of China. Cellulose 2020, 27, 1423–1440. [Google Scholar] [CrossRef]
- Abdulrasheed, M.; Zakaria, N.N.; Ahmad Roslee, A.F.A.; Shukor, M.Y.; Zulkharnain, A.; Napis, S.; Convey, P.; Alias, S.A.; Gonzalez-Rocha, G.; Ahmad, S.A. Biodegradation of diesel oil by cold-adapted bacterial strains of Arthrobacter spp. from Antarctica. Antarct. Sci. 2020, 32, 341–353. [Google Scholar] [CrossRef]
- Amin, A.; Ahmed, I.; Habib, N.; Abbas, S.; Xiao, M.; Hozzein, W.N.; Li, W.J. Nocardioides pakistanensis sp. nov., isolated from a hot water spring of Tatta Pani in Pakistan. Antonie Leeuwenhoek 2016, 109, 1101–1109. [Google Scholar] [CrossRef]
- Dong, L.; Ming, H.; Liu, L.; Zhou, E.M.; Yin, Y.R.; Duan, Y.Y.; Nie, G.X.; Feng, H.G.; Li, W.J. Zhizhongheella caldifontis gen. nov., sp. nov., a novel member of the family Comamonadaceae. Antonie Leeuwenhoek 2014, 105, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.P.; Khan, I.U.; Li, M.M.; Xian, W.D.; Liu, L.; Zhou, E.M.; Salam, N.; Li, W.J. Calidifontimicrobium sediminis gen. nov., sp. nov., a new member of the family Comamonadaceae. Int. J. Syst. Evol. Microbiol. 2019, 69, 434–440. [Google Scholar] [CrossRef]
- Sharma, A.; Paul, D.; Dhotre, D.; Jani, K.; Pandey, A.; Shouche, Y.S. Deep sequencing analysis of bacterial community structure of Soldhar hot spring, India. Microbiology 2017, 86, 136–142. [Google Scholar] [CrossRef]
- Khan, I.U.; Hussain, F.; Tian, Y.; Habib, N.; Xian, W.D.; Jiang, Z.; Amin, A.; Yuan, C.G.; Zhou, E.M.; Zhi, X.Y.; et al. Tibeticola sediminis gen. nov., sp. nov., a thermophilic bacterium isolated from a hot spring. Int. J. Syst. Evol. Microbiol. 2017, 67, 1133–1139. [Google Scholar] [CrossRef]
- Chadraabal, A.; Shinoda, M.; Suzuki, Y.; Komiyama, H. Mitigation of severe wintertime disasters in northern Mongolia through the early implementation of local action. Int. J. Disaster Risk Reduct. 2020, 50, 101739. [Google Scholar] [CrossRef]
- Ozaki, T.; Takakura, H. Introduction: Environmental disaster in Mongolian modern history. J. Contemp. East Asia Stud. 2022, 11, 1–21. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Region | No. of Samples | Type of Samples |
|---|---|---|
| Seoraksan National Park | 19 | Wild |
| Northern Conservation Center | 11 | Captive1 |
| Long-tailed Goral & Musk Deer Center | 7 | Captive2 |
| Total | 37 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, C.-E.; Jo, Y.-J.; Jung, D.-R.; Park, H.-C.; Shin, J.-H. Comparative Analysis of Gut Microbiota between Captive and Wild Long-Tailed Gorals for Ex Situ Conservation. Microorganisms 2024, 12, 1419. https://doi.org/10.3390/microorganisms12071419
Park C-E, Jo Y-J, Jung D-R, Park H-C, Shin J-H. Comparative Analysis of Gut Microbiota between Captive and Wild Long-Tailed Gorals for Ex Situ Conservation. Microorganisms. 2024; 12(7):1419. https://doi.org/10.3390/microorganisms12071419
Chicago/Turabian StylePark, Chang-Eon, Young-Jae Jo, Da-Ryung Jung, Hee-Cheon Park, and Jae-Ho Shin. 2024. "Comparative Analysis of Gut Microbiota between Captive and Wild Long-Tailed Gorals for Ex Situ Conservation" Microorganisms 12, no. 7: 1419. https://doi.org/10.3390/microorganisms12071419