

Long-Term Contaminant Exposure Alters Functional Potential and Species Composition of Soil Bacterial Communities in Gulf Coast Prairies

Abstract
:1. Introduction
2. Materials and Methods
2.1. Site and Soil Collection
2.2. Bacterial Community Assessment
2.3. Bacterial ASV Diversity and Composition Analyses
2.4. Bacterial Functional Potential Analyses
2.5. Soil Property Analyses
3. Results
3.1. Soil Property Analysis
3.2. Summary Statistics of Microbial Sequences
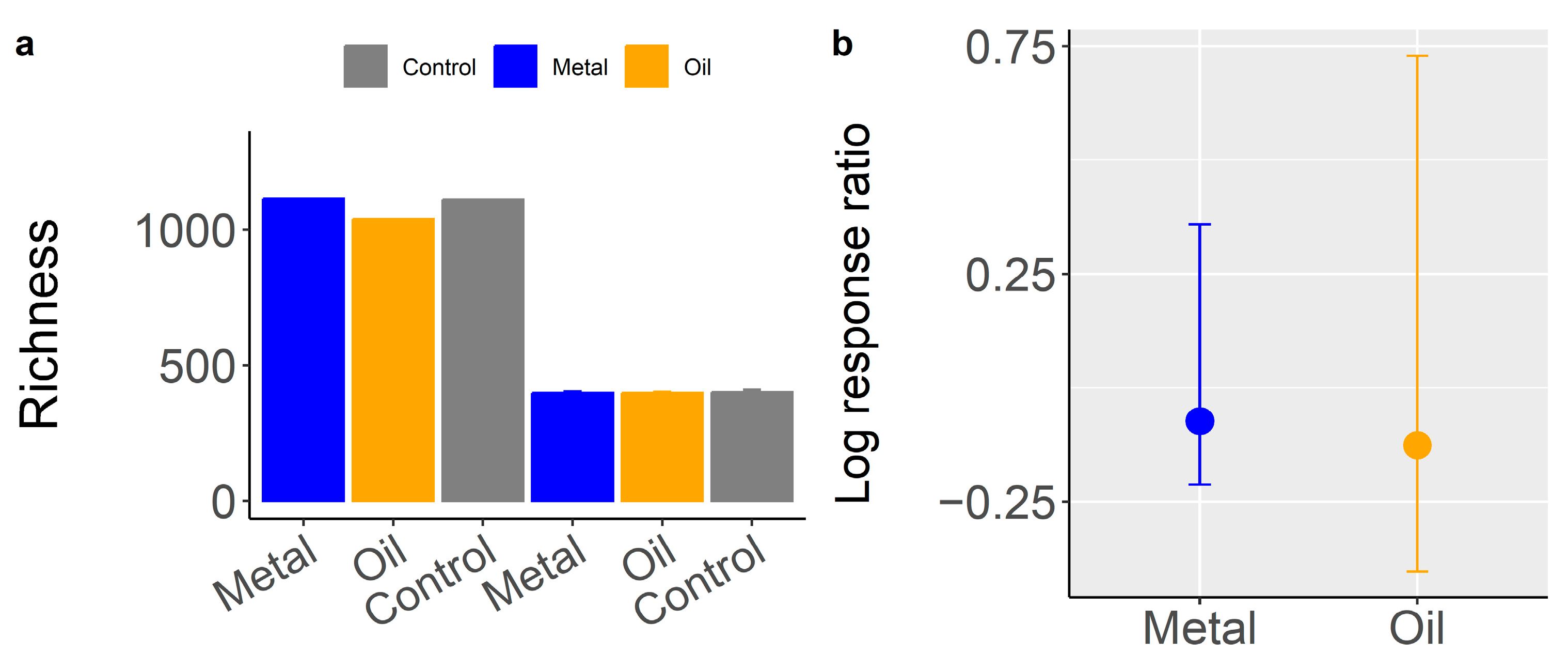
3.3. Contaminant Impacts on Species Diversity of Bacterial Communities (H1)
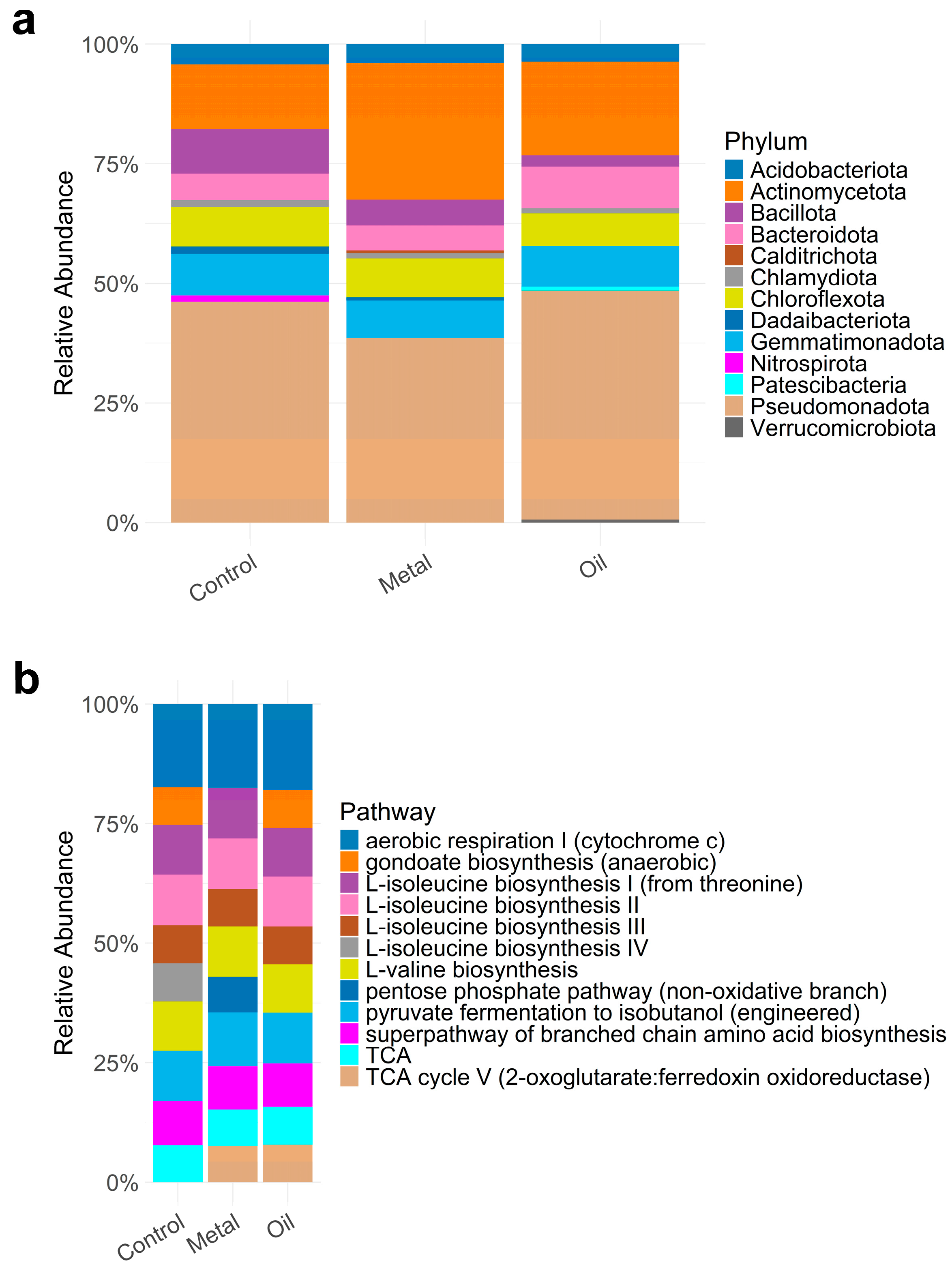
3.4. Contaminant Impacts of the Functional Potential of Bacterial Communities (H1)
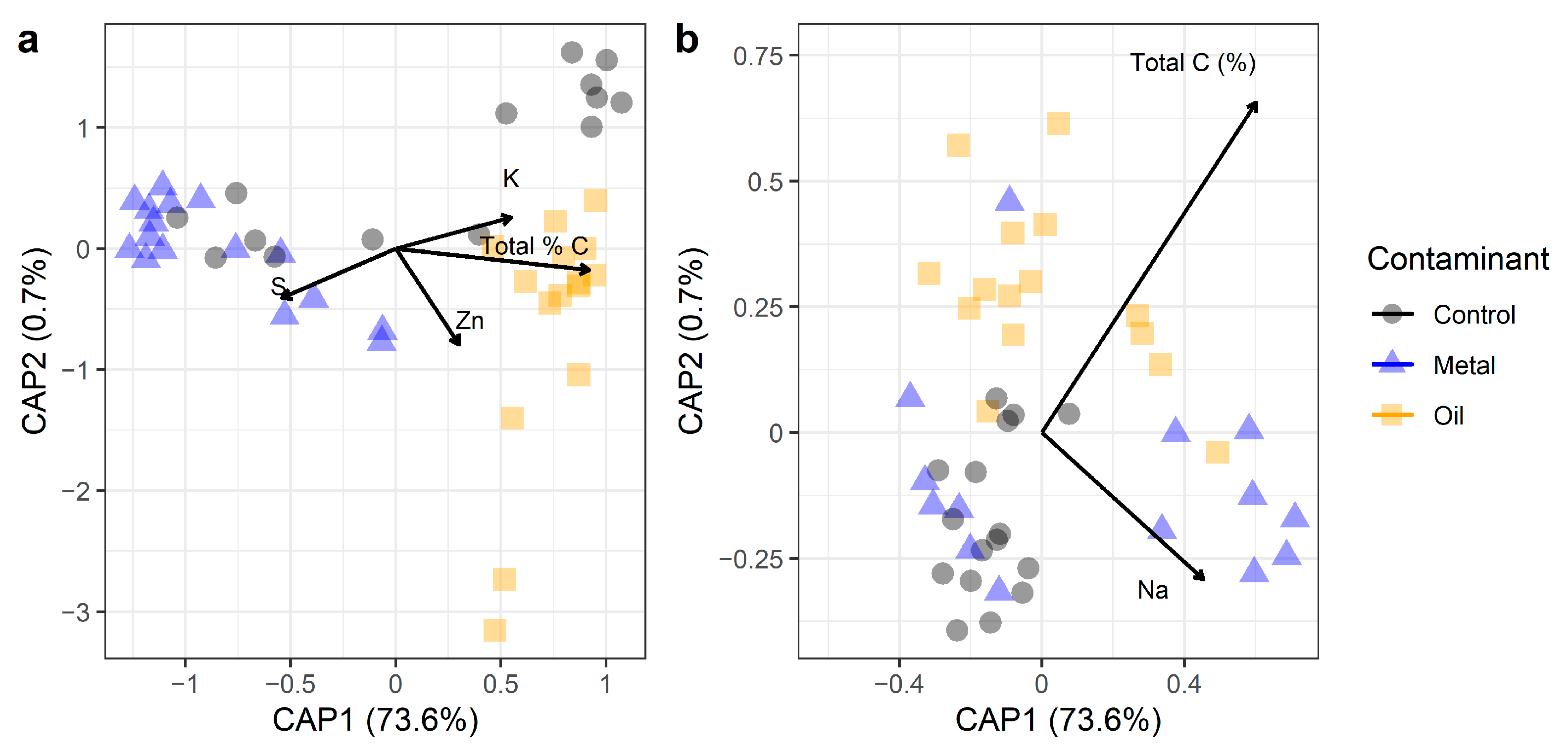
3.5. ASV Diversity vs. Functional Potential According to Contamination History (H2)
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, C.; Wang, H.; Liao, X.; Xiao, R.; Liu, K.; Bai, J.; Li, B.; He, Q. Heavy Metal Pollution in Coastal Wetlands: A Systematic Review of Studies Globally over the Past Three Decades. J. Hazard. Mater. 2022, 424, 127312. [Google Scholar] [CrossRef] [PubMed]
- Lumibao, C.Y.; Formel, S.; Elango, V.; Pardue, J.H.; Blum, M.; Van Bael, S.A. Persisting Responses of Salt Marsh Fungal Communities to the Deepwater Horizon Oil Spill. Sci. Total Environ. 2018, 642, 904–913. [Google Scholar] [CrossRef] [PubMed]
- Sweet, W.V.; Kopp, R.E.; Weaver, C.P.; Obeysekera, J.; Horton, R.M.; Thieler, E.R.; Zervas, C. Global and Regional Sea-Level Rise Scenarios for the United States; National Oceanic and Atmospheric Administration: Silver Spring, MD, USA, 2017. [Google Scholar]
- Feagin, R.A.; Smith, W.K.; Psuty, N.P.; Young, D.R.; Martnez, M.L.; Carter, G.A.; Lucas, K.L.; Gibeaut, J.C.; Gemma, J.N.; Koske, R.E. Barrier Islands: Coupling Anthropogenic Stability with Ecological Sustainability. J. Coast. Res. 2010, 26, 987–992. [Google Scholar] [CrossRef]
- Petrolia, D.R.; Kim, T.-G. What Are Barrier Islands Worth? Estimates of Willingness to Pay for Restoration. Mar. Resour. Econ. 2009, 24, 42731376. [Google Scholar] [CrossRef]
- Joos, L.; De Tender, C.; Holderbeke, A.; Clement, L.; Vandecasteele, B.; Debode, J. Exploring the Microbial Response as a Potential Bio-Indicator for Soil Health: Insights from a Controlled Incubator Experiment. Agric. Ecosyst. Environ. 2023, 356, 108634. [Google Scholar] [CrossRef]
- Chen, H.; Ma, K.; Huang, Y.; Yao, Z.; Chu, C. Stable Soil Microbial Functional Structure Responding to Biodiversity Loss Based on Metagenomic Evidences. Front. Microbiol. 2021, 12, 716764. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Whalen, J.K. A New Perspective on Functional Redundancy and Phylogenetic Niche Conservatism in Soil Microbial Communities. Pedosphere 2020, 30, 18–24. [Google Scholar] [CrossRef]
- Fierer, N.; Ladau, J.; Clemente, J.C.; Leff, J.W.; Owens, S.M.; Pollard, K.S.; Knight, R.; Gilbert, J.A.; McCulley, R.L. Reconstructing the Microbial Diversity and Function of Pre-Agricultural Tallgrass Prairie Soils in the United States. Science 2013, 342, 621–624. [Google Scholar] [CrossRef]
- Louca, S.; Polz, M.F.; Mazel, F.; Albright, M.B.N.; Huber, J.A.; O’Connor, M.I.; Ackermann, M.; Hahn, A.S.; Srivastava, D.S.; Crowe, S.A.; et al. Function and Functional Redundancy in Microbial Systems. Nat. Ecol. Evol. 2018, 2, 936–943. [Google Scholar] [CrossRef]
- Müller, A.K.; Rasmussen, L.D.; Sorensen, S.J. Adaptation of the Bacterial Community to Mercury Contamination. FEMS Microbiol. Lett. 2001, 204, 49–53. [Google Scholar] [CrossRef]
- Chen, H.; Ma, K.; Lu, C.; Fu, Q.; Qiu, Y.; Zhao, J.; Huang, Y.; Yang, Y.; Schadt, C.W.; Chen, H. Functional Redundancy in Soil Microbial Community Based on Metagenomics Across the Globe. Front. Microbiol. 2022, 13, 878978. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; McIlroy, S.E.; Archana, A.; Baker, D.M.; Panagiotou, G. A Pollution Gradient Contributes to the Taxonomic, Functional, and Resistome Diversity of Microbial Communities in Marine Sediments. Microbiome 2019, 7, 104. [Google Scholar] [CrossRef]
- Wang, X.; Wang, X.; Wu, F.; Zhang, J.; Ai, S.; Liu, Z. Microbial Community Composition and Degradation Potential of Petroleum-Contaminated Sites under Heavy Metal Stress. J. Hazard. Mater. 2023, 457, 131814. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-C.; Liang, C.-M.; Yang, T.-I.; Chen, J.-L.; Wang, W.-K. Shift of Bacterial Communities in Heavy Metal-Contaminated Agricultural Land during a Remediation Process. PLoS ONE 2021, 16, e0255137. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Jiang, Y.; Huang, H.; Mou, L.; Ru, J.; Zhao, J.; Xiao, S. Long-Term and High-Concentration Heavy-Metal Contamination Strongly Influences the Microbiome and Functional Genes in Yellow River Sediments. Sci. Total Environ. 2018, 637–638, 1400–1412. [Google Scholar] [CrossRef] [PubMed]
- Gong, W.J.; Niu, Z.F.; Wang, X.R.; Zhao, H.P. How the Soil Microbial Communities and Activities Respond to Long-Term Heavy Metal Contamination in Electroplating Contaminated Site. Microorganisms 2021, 9, 362. [Google Scholar] [CrossRef] [PubMed]
- Klimek, B.; Sitarz, A.; Choczyński, M.; Niklińska, M. The Effects of Heavy Metals and Total Petroleum Hydrocarbons on Soil Bacterial Activity and Functional Diversity in the Upper Silesia Industrial Region (Poland). Water Air Soil Pollut. 2016, 227, 265. [Google Scholar] [CrossRef] [PubMed]
- Gran-Scheuch, A.; Ramos-Zuñiga, J.; Fuentes, E.; Bravo, D.; Pérez-Donoso, J.M. Effect of Co-Contamination by PAHs and Heavy Metals on Bacterial Communities of Diesel Contaminated Soils of South Shetland Islands, Antarctica. Microorganisms 2020, 8, 1749. [Google Scholar] [CrossRef]
- Rezaei Somee, M.; Dastgheib, S.M.M.; Shavandi, M.; Ghanbari Maman, L.; Kavousi, K.; Amoozegar, M.A.; Mehrshad, M. Distinct Microbial Community along the Chronic Oil Pollution Continuum of the Persian Gulf Converge with Oil Spill Accidents. Sci. Rep. 2021, 11, 11316. [Google Scholar] [CrossRef]
- Lumibao, C.Y.; Torres Martínez, L.; Megonigal, J.P.; Van Bael, S.A.; Blum, M.J. Microbial Mediation of Salinity Stress Response Varies by Plant Genotype and Provenance over Time. Mol. Ecol. 2022, 31, 4571–4585. [Google Scholar] [CrossRef]
- Lumibao, C.Y.; Kimbrough, E.R.; Day, R.H.; Conner, W.H.; Krauss, K.W.; van Bael, S.A. Divergent Biotic and Abiotic Filtering of Root Endosphere and Rhizosphere Soil Fungal Communities along Ecological Gradients. FEMS Microbiol. Ecol. 2021, 96, fiaa124. [Google Scholar] [CrossRef] [PubMed]
- Lumibao, C.Y.; Bernik, B.M.; Formel, S.K.; Kandalepas, D.; Mighell, K.L.; Pardue, J.; Van Bael, S.A.; Blum, M.J. Rhizosphere Microbial Communities Reflect Genotypic and Trait Variation in a Salt Marsh Ecosystem Engineer. Am. J. Bot. 2020, 107, 941–949. [Google Scholar] [CrossRef] [PubMed]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet. J. 2013, 17, 10–12. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic Acids Res. 2013, 41, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; James, R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for Prediction of Metagenome Functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef] [PubMed]
- van den Boogaart, K.G.; Tolosana-Delgado, R. “Compositions”: A Unified R Package to Analyze Compositional Data. Comput. Geosci. 2008, 34, 320–338. [Google Scholar] [CrossRef]
- Lenth, R.; Singmann, H.; Love, J.; Buerkner, P.; Herve, M. Package “Emmeans”, R Package version 4.0-3; 2018. [Google Scholar]
- Pustejovsky, J.E.; Chen, M.; Grekov, P.; Swan, D.M. SingleCaseES: A Calculator for Single-Case Effect Size Indices, version 0.7.2 [R Package]; 2023. Available online: https://jepusto.github.io/SingleCaseES/ (accessed on 24 May 2024).
- De Cáceres, M.; Legendre, P. Associations between Species and Groups of Sites: Indices and Statistical Inference. Ecology 2009, 90, 3566–3574. [Google Scholar] [CrossRef]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer-Verlag: New York, NY, USA, 2016. [Google Scholar]
- Valenzuela-Soto, E.M.; Figueroa-Soto, C.G. Biosynthesis and Degradation of Glycine Betaine and Its Potential to Control Plant Growth and Development BT. In Osmoprotectant-Mediated Abiotic Stress Tolerance in Plants: Recent Advances and Future Perspectives; Hossain, M.A., Kumar, V., Burritt, D.J., Fujita, M., Mäkelä, P.S.A., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 123–140. ISBN 978-3-030-27423-8. [Google Scholar]
- González Henao, S.; Ghneim-Herrera, T. Heavy Metals in Soils and the Remediation Potential of Bacteria Associated With the Plant Microbiome. Front. Environ. Sci. 2021, 9, 604216. [Google Scholar] [CrossRef]
- Madrova, P.; Vetrovsky, T.; Omelka, M.; Grunt, M.; Smutna, Y.; Rapoport, D.; Vach, M.; Baldrian, P.; Kopecky, J.; Sagova-Mareckova, M. A Short-Term Response of Soil Microbial Communities to Cadmium and Organic Substrate Amendment in Long-Term Contaminated Soil by Toxic Elements. Front. Microbiol. 2018, 9, 2807. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.R.; Gommeaux, M.; Duclercq, J.; Fanin, N.; Conreux, A.; Alahmad, A.; Lacoux, J.; Roger, D.; Spicher, F.; Ponthieu, M.; et al. Response of Bacterial Communities to Pb Smelter Pollution in Contrasting Soils. Sci. Total Environ. 2017, 605–606, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Azarbad, H.; Niklińska, M.; Laskowski, R.; van Straalen, N.M.; van Gestel, C.A.M.; Zhou, J.; He, Z.; Wen, C.; Röling, W.F.M. Microbial Community Composition and Functions Are Resilient to Metal Pollution along Two Forest Soil Gradients. FEMS Microbiol. Ecol. 2015, 91, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sharma, J.; Shamim, K.; Dubey, S.K.; Meena, R.M. Metallothionein Assisted Periplasmic Lead Sequestration as Lead Sulfite by Providencia Vermicola Strain SJ2A. Sci. Total Environ. 2017, 579, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Barra Caracciolo, A.; Terenzi, V. Rhizosphere Microbial Communities and Heavy Metals. Microorganisms 2021, 9, 1462. [Google Scholar] [CrossRef]
- Allen, C.R.; Boyd, D.R.; Hempenstall, F.; Larkin, M.J.; Sharma, N.D. Contrasting Effects of a Nonionic Surfactant on the Biotransformation of Polycyclic Aromatic Hydrocarbons Tocis-Dihydrodiols by Soil Bacteria. Appl. Environ. Microbiol. 1999, 65, 1335–1339. [Google Scholar] [CrossRef] [PubMed]
- Kanaly, R.A.; Harayama, S. Advances in the Field of High-Molecular-Weight Polycyclic Aromatic Hydrocarbon Biodegradation by Bacteria. Microb. Biotechnol. 2010, 3, 136–164. [Google Scholar] [CrossRef]
- Markova, Y.A.; Petrushin, I.S.; Belovezhets, L.A. Detection of Gene Clusters for Biodegradation of Alkanes and Aromatic Compounds in the Rhodococcus Qingshengii VKM Ac-2784D Genome. Vavilovskii Zhurnal Genet. Sel. 2023, 27, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Ivshina, I.B.; Krivoruchko, A.V.; Kuyukina, M.S.; Peshkur, T.A.; Cunningham, C.J. Adhesion of Rhodococcus Bacteria to Solid Hydrocarbons and Enhanced Biodegradation of These Compounds. Sci. Rep. 2022, 12, 21559. [Google Scholar] [CrossRef]
- Thi Mo, L.; Irina, P.; Natalia, S.; Irina, N.; Lenar, A.; Andrey, F.; Ekaterina, A.; Sergey, A.; Olga, P. Hydrocarbons Biodegradation by Rhodococcus: Assimilation of Hexadecane in Different Aggregate States. Microorganisms 2022, 10, 1594. [Google Scholar] [CrossRef]
- Huang, L.; Ye, J.; Jiang, K.; Wang, Y.; Li, Y. Oil Contamination Drives the Transformation of Soil Microbial Communities: Co-Occurrence Pattern, Metabolic Enzymes and Culturable Hydrocarbon-Degrading Bacteria. Ecotoxicol. Environ. Saf. 2021, 225, 112740. [Google Scholar] [CrossRef] [PubMed]
- Neethu, C.S.; Saravanakumar, C.; Purvaja, R.; Robin, R.S.; Ramesh, R. Oil-Spill Triggered Shift in Indigenous Microbial Structure and Functional Dynamics in Different Marine Environmental Matrices. Sci. Rep. 2019, 9, 1354. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Zhang, Y.; Zhao, N.; Guo, J.; Xu, W.; Ma, M.; Li, X. Remediation of Crude Oil-Polluted Soil by the Bacterial Rhizosphere Community of Suaeda Salsa Revealed by 16S RRNA Genes. Int. J. Environ. Res. Public Health 2020, 17, 1471. [Google Scholar] [CrossRef] [PubMed]
- Galand, P.E.; Pereira, O.; Hochart, C.; Auguet, J.C.; Debroas, D. A Strong Link between Marine Microbial Community Composition and Function Challenges the Idea of Functional Redundancy. ISME J. 2018, 12, 2470–2478. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Factors | ASV ENSPIE | ASV Shannon Diversity | Functional Richness Potential | |||
|---|---|---|---|---|---|---|
| Estimate | p-Value | Estimate | p-Value | Estimate | p-Value | |
| Intercept | 6.434 | <0.001 | 1.964 | 0.000 | 6.071 | <0.001 |
| Control vs. Metal | 0.255 | 0.726 | 0.023 | 0.836 | 0.000 | 1.000 |
| Control vs. Oil | −0.366 | 0.748 | 0.002 | 0.999 | −0.006 | 0.986 |
| Metal vs. Oil | −0.111 | 0.963 | 0.022 | 0.829 | −0.006 | 0.976 |
| Total C (%) | −0.091 | 0.502 | −0.018 | 0.203 | 0.000 | 0.980 |
| Total N (%) | −3.470 | 0.671 | 0.344 | 0.633 | 0.190 | 0.708 |
| Cu | 0.390 | 0.012 | 0.026 | 0.135 | 0.001 | 0.955 |
| Zn | −0.025 | 0.044 | −0.001 | 0.266 | 0.000 | 0.915 |
| K | −0.001 | 0.376 | 0.000 | 0.531 | 0.000 | 0.727 |
| S | 0.001 | 0.652 | 0.000 | 0.305 | 0.000 | 0.588 |
| Na | 0.000 | 0.828 | 0.000 | 0.521 | 0.000 | 0.980 |
| pH | −0.094 | 0.576 | −0.015 | 0.442 | −0.011 | 0.403 |
| ASV Composition | |||||
| Predictor | Df | Sum of Squares | R2 | F | p Values |
| Contaminant | 2 | 2.669 | 0.141 | 3.842 | 0.001 |
| Total C (%) | 1 | 0.939 | 0.050 | 2.704 | 0.001 |
| Total N (%) | 1 | 0.425 | 0.022 | 1.224 | 0.108 |
| Zn | 1 | 0.521 | 0.028 | 1.499 | 0.014 |
| Cu | 1 | 0.456 | 0.024 | 1.314 | 0.030 |
| K | 1 | 0.540 | 0.029 | 1.553 | 0.025 |
| S | 1 | 0.634 | 0.034 | 1.826 | 0.003 |
| Na | 1 | 0.385 | 0.020 | 1.107 | 0.232 |
| pH | 1 | 0.530 | 0.028 | 1.525 | 0.034 |
| Residual | 34 | 11.811 | 0.625 | ||
| Functional potential composition | |||||
| Contaminant | 2 | 0.022 | 0.151 | 5.177 | 0.001 |
| Total C (%) | 1 | 0.026 | 0.184 | 12.582 | 0.001 |
| Total N (%) | 1 | 0.004 | 0.025 | 1.719 | 0.126 |
| Zn | 1 | 0.002 | 0.014 | 0.947 | 0.377 |
| Cu | 1 | 0.002 | 0.015 | 1.044 | 0.328 |
| K | 1 | 0.004 | 0.027 | 1.877 | 0.114 |
| S | 1 | 0.006 | 0.042 | 2.891 | 0.030 |
| Na | 1 | 0.004 | 0.026 | 1.758 | 0.126 |
| pH | 1 | 0.003 | 0.020 | 1.374 | 0.206 |
| Residual | 34 | 0.071 | 0.496 | ||
| Pathway | Stat | p-Value |
|---|---|---|
| Heavy metal history | ||
| fatty acid beta-oxidation I (generic) | 0.475 | 0.0021 |
| fatty acid salvage | 0.471 | 0.0034 |
| 4-aminobutanoate degradation V | 0.445 | 0.0043 |
| protocatechuate degradation II | 0.420 | 0.0089 |
| pyruvate fermentation to propanoate I | 0.418 | 0.0047 |
| Control | ||
| hexitol fermentation to lactate, formate, ethanol and acetate | 0.580 | 0.0001 |
| L-methionine salvage cycle III | 0.577 | 0.0001 |
| S-methyl-5-thio-α-D-ribose 1-phosphate degradation I | 0.577 | 0.0001 |
| formaldehyde assimilation II (RuMP Cycle) | 0.573 | 0.0001 |
| formaldehyde oxidation I | 0.569 | 0.0001 |
| Oil history | ||
| androstenedione degradation I (aerobic) | 0.614 | 0.0001 |
| glycine betaine degradation I | 0.556 | 0.0003 |
| beta-alanine biosynthesis II | 0.538 | 0.0001 |
| superpathway of hexuronide and hexuronate degradation | 0.518 | 0.0003 |
| creatinine degradation II | 0.500 | 0.0012 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lumibao, C.Y.; Liu, Y. Long-Term Contaminant Exposure Alters Functional Potential and Species Composition of Soil Bacterial Communities in Gulf Coast Prairies. Microorganisms 2024, 12, 1460. https://doi.org/10.3390/microorganisms12071460
Lumibao CY, Liu Y. Long-Term Contaminant Exposure Alters Functional Potential and Species Composition of Soil Bacterial Communities in Gulf Coast Prairies. Microorganisms. 2024; 12(7):1460. https://doi.org/10.3390/microorganisms12071460
Chicago/Turabian StyleLumibao, Candice Y., and Yue Liu. 2024. "Long-Term Contaminant Exposure Alters Functional Potential and Species Composition of Soil Bacterial Communities in Gulf Coast Prairies" Microorganisms 12, no. 7: 1460. https://doi.org/10.3390/microorganisms12071460