
Shiga-Toxin-Producing Escherichia coli Strains from Romania: A Whole Genome-Based Description
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains
2.2. Whole Genome Sequencing
2.3. In Silico Analyses Based on Whole Genome Sequencing Data
3. Results
3.1. Correlation between Serotypes, Clinical Status, Sequence Types, Phylogroups, and Virulence Genes
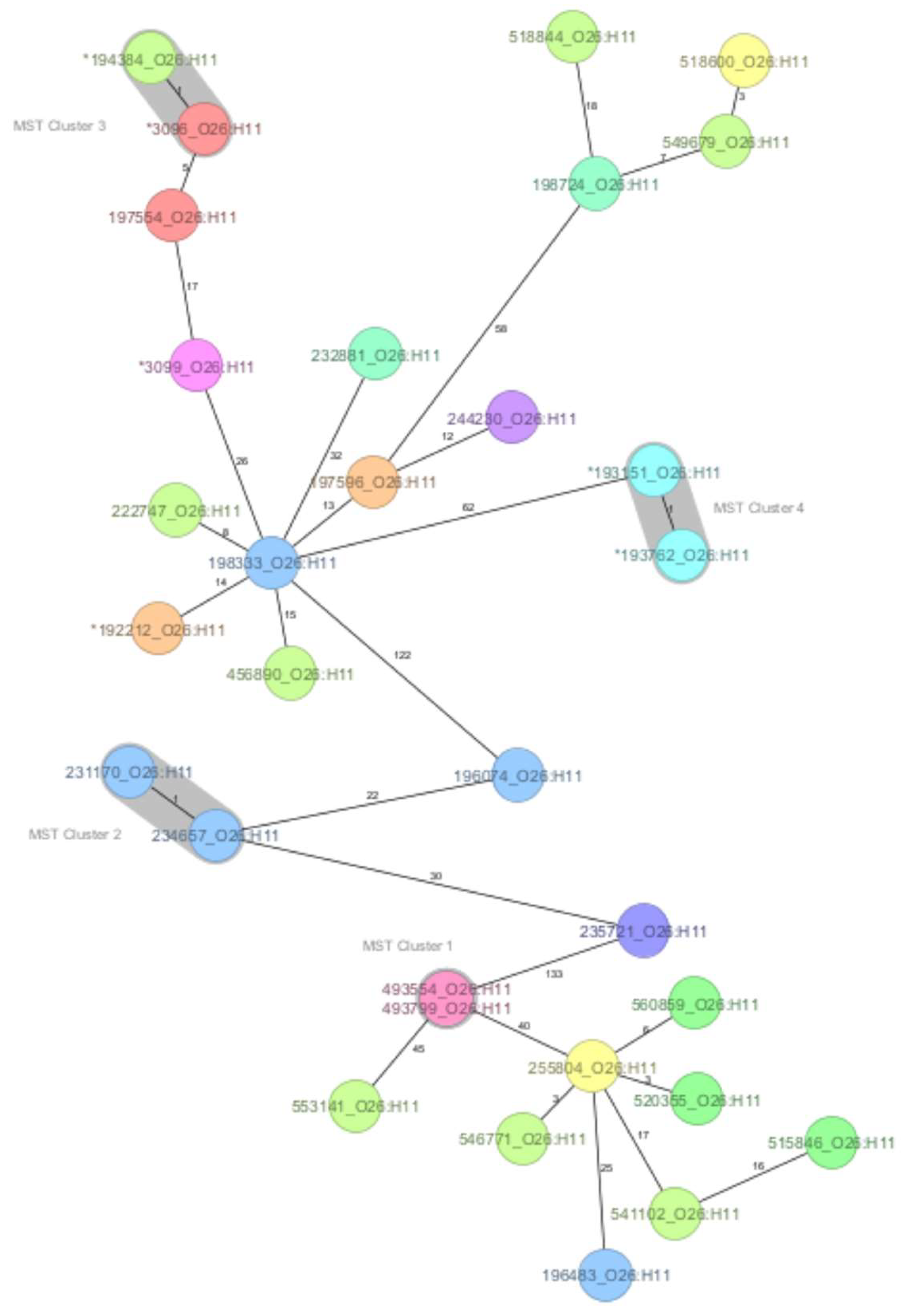
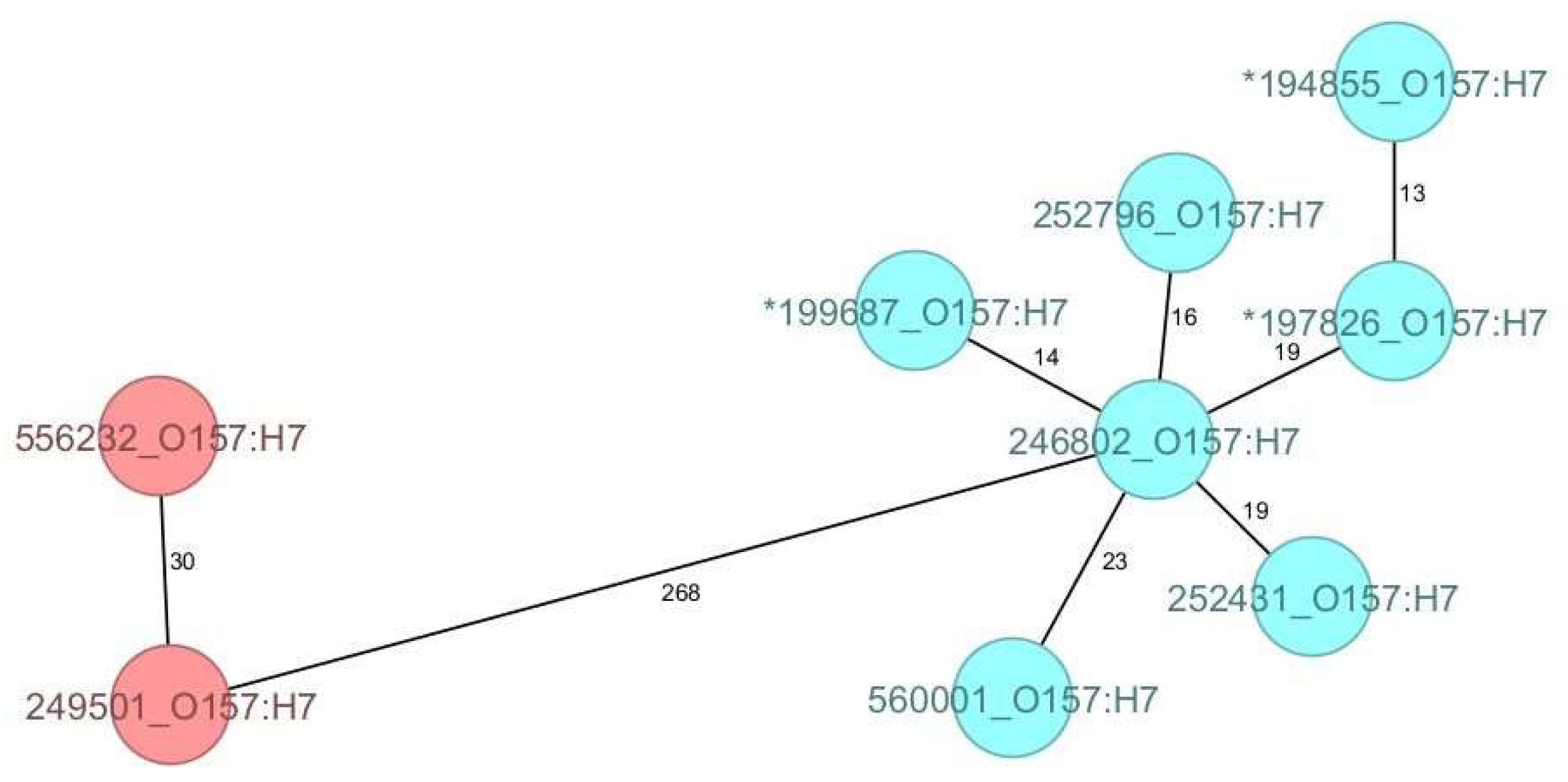
3.2. Core Genome MLST-Derived Relatedness of O26:H11 Strains
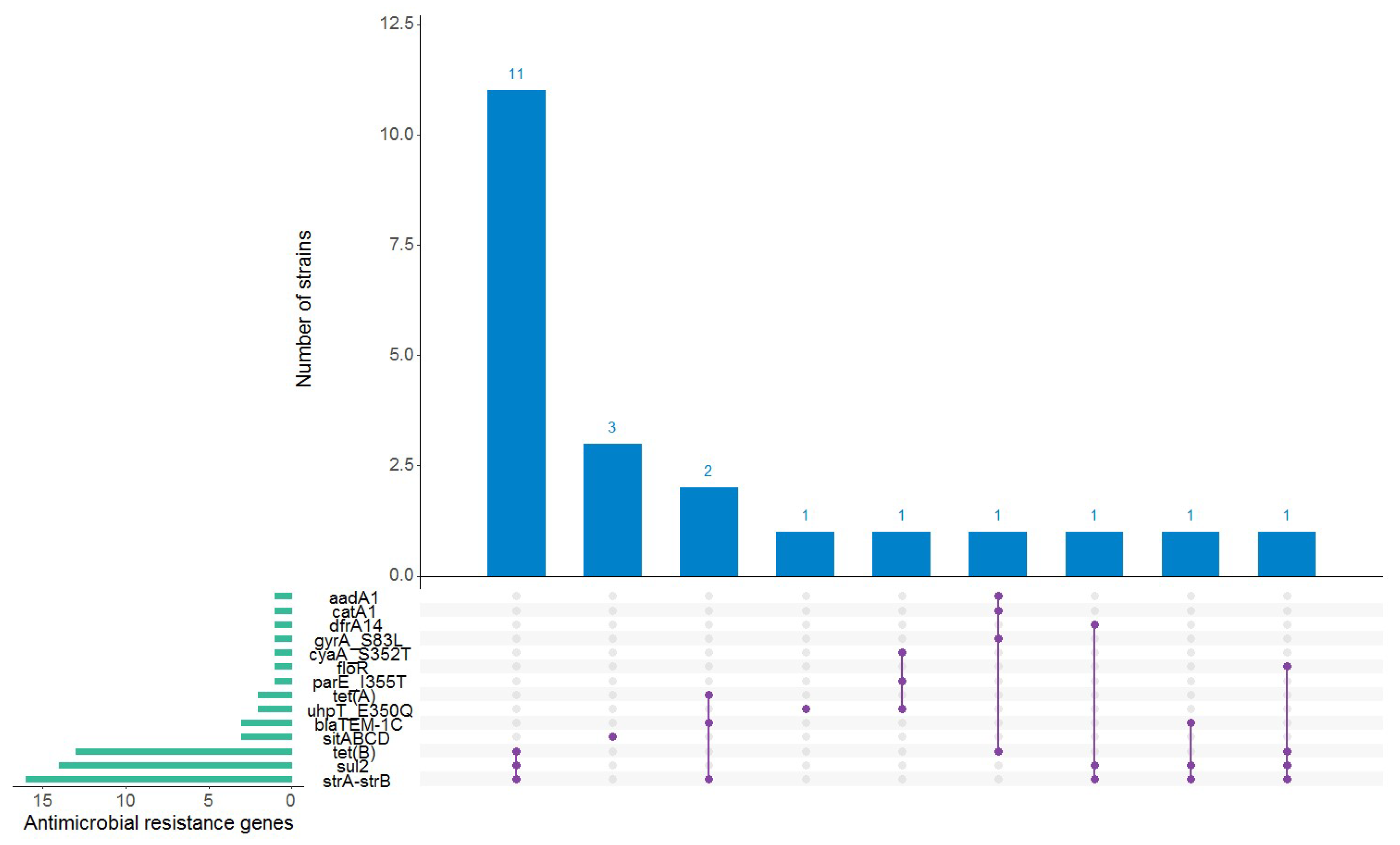
3.3. Antimicrobial Resistance and Plasmid Content
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Mühlen, S.; Dersch, P. Treatment Strategies for Infections with Shiga Toxin-Producing Escherichia coli. Front. Cell Infect. Microbiol. 2020, 10, 169. [Google Scholar] [CrossRef] [PubMed]
- Radosavljevic, V.; Finke, E.J.; Belojevic, G. Escherichia coli O104:H4 outbreak in Germany—Clarification of the origin of the epidemic. Eur. J. Public Health 2015, 25, 125–129. [Google Scholar] [CrossRef] [PubMed]
- EFSA BIOHAZ Panel; Koutsoumanis, K.; Allende, A.; Alvarez-Ordóñez, A.; Bover-Cid, S.; Chemaly, M.; Davies, R.; De Cesare, A.; Herman, L.; Hilbert, F.; et al. Scientific Opinion on the pathogenicity assessment of Shiga toxin-producing Escherichia coli (STEC) and the public health risk posed by contamination of food with STEC. EFSA J. 2020, 18, 5967. [Google Scholar] [CrossRef]
- Available online: https://www.ecdc.europa.eu/en/publications-data/amended-ecdc-strategy-2021-2027 (accessed on 18 July 2024).
- Severi, E.; Vial, F.; Peron, E.; Mardh, O.; Niskanen, T.; Takkinen, J. Community-wide outbreaks of haemolytic uraemic syndrome associated with Shiga toxin-producing Escherichia coli O26 in Italy and Romania: A new challenge for the European Union. Eurosurveillance 2016, 21, 30420. [Google Scholar] [CrossRef] [PubMed]
- European Centre for Disease Prevention and Control. Shigatoxin/verocytotoxin-producing Escherichia coli (STEC/VTEC) infection. In Annual Epidemiological Report for 2015; ECDC: Stockholm, Sweden, 2018. [Google Scholar]
- European Centre for Disease Prevention and Control. STEC infection. In Annual Epidemiological Report for 2021; ECDC: Stockholm, Sweden, 2022. [Google Scholar]
- Scheutz, F.; Teel, L.D.; Beutin, L.; Piérard, D.; Buvens, G.; Karch, H.; Mellmann, A.; Caprioli, A.; Tozzoli, R.; Morabito, S.; et al. Multicenter evaluation of a sequence-based protocol for subtyping Shiga toxins and standardizing Stx nomenclature. J. Clin. Microbiol. 2012, 50, 2951–2963. [Google Scholar] [CrossRef] [PubMed]
- Usein, C.R.; Ciontea, A.S.; Militaru, C.M.; Condei, M.; Dinu, S.; Oprea, M.; Cristea, D.; Michelacci, V.; Scavia, G.; Zota, L.C.; et al. Molecular characterisation of human Shiga toxin-producing Escherichia coli O26 strains: Results of an outbreak investigation, Romania, February to August 2016. Eurosurveillance 2017, 22, 17–00148. [Google Scholar] [CrossRef] [PubMed]
- Oprea, M.; Ciontea, A.S.; Militaru, M.; Dinu, S.; Cristea, D.; Usein, C.R. Molecular Typing of Escherichia coli O157 Isolates from Romanian Human Cases. Jpn. J. Infect. Dis. 2018, 71, 455–461. [Google Scholar] [CrossRef]
- Wirth, T.; Falush, D.; Lan, R.; Colles, F.; Mensa, P.; Wieler, L.H.; Karch, H.; Reeves, P.R.; Maiden, M.C.; Ochman, H.; et al. Sex and virulence in Escherichia coli: An evolutionary perspective. Mol. Microbiol. 2006, 60, 1136–1151. [Google Scholar] [CrossRef]
- Zhou, Z.; Alikhan, N.F.; Mohamed, K.; Fan, Y.; Agama Study Group; Achtman, M. The EnteroBase user’s guide, with case studies on Salmonella transmissions, Yersinia pestis phylogeny, and Escherichia core genomic diversity. Genome Res. 2020, 30, 138–152. [Google Scholar] [CrossRef] [PubMed]
- Joensen, K.G.; Tetzschner, A.M.; Iguchi, A.; Aarestrup, F.M.; Scheutz, F. Rapid and Easy In Silico Serotyping of Escherichia coli Isolates by Use of Whole-Genome Sequencing Data. J. Clin. Microbiol. 2015, 53, 2410–2426. [Google Scholar] [CrossRef] [PubMed]
- Joensen, K.G.; Scheutz, F.; Lund, O.; Hasman, H.; Kaas, R.S.; Nielsen, E.M.; Aarestrup, F.M. Real-time whole-genome sequencing for routine typing, surveillance, and outbreak detection of verotoxigenic Escherichia coli. J. Clin. Microbiol. 2014, 52, 1501–1510. [Google Scholar] [CrossRef] [PubMed]
- Malberg Tetzschner, A.M.; Johnson, J.R.; Johnston, B.D.; Lund, O.; Scheutz, F. In Silico Genotyping of Escherichia coli Isolates for Extraintestinal Virulence Genes by Use of Whole-Genome Sequencing Data. J. Clin. Microbiol. 2020, 58, e01269-20. [Google Scholar] [CrossRef] [PubMed]
- Bortolaia, V.; Kaas, R.S.; Ruppe, E.; Roberts, M.C.; Schwarz, S.; Cattoir, V.; Philippon, A.; Allesoe, R.L.; Rebelo, A.R.; Florensa, A.F.; et al. ResFinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. 2020, 75, 3491–3500. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Carattoli, A.; Zankari, E.; Garcia-Fernandez, A.; Voldby Larsen, M.; Lund, O.; Villa, L.; Aarestrup, F.M.; Hasman, H. PlasmidFinder and pMLST: In silico detection and typing of plasmids. Agents Chemother. 2014, 58, 3895–3903. [Google Scholar] [CrossRef] [PubMed]
- Beghain, J.; Bridier-Nahmias, A.; Le Nagard, H.; Denamur, E.; Clermont, O. ClermonTyping: An easy-to-use and accurate in silico method for Escherichia genus strain phylotyping. Microb. Genom. 2018, 4, e000192. [Google Scholar] [CrossRef] [PubMed]
- Manning, S.D.; Motiwala, A.S.; Springman, A.C.; Qi, W.; Lacher, D.W.; Ouellette, L.M.; Mladonicky, J.M.; Somsel, P.; Rudrik, J.T.; Dietrich, S.E.; et al. Variation in virulence among clades of Escherichia coli O157:H7 associated with disease outbreaks. Proc. Natl. Acad. Sci. USA 2008, 105, 4868–4873. [Google Scholar] [CrossRef] [PubMed]
- Riordan, J.T.; Viswanath, S.B.; Manning, S.D.; Whittam, T.S. Genetic differentiation of Escherichia coli O157:H7 clades associated with human disease by real-time PCR. J. Clin. Microbiol. 2008, 46, 2070–2073. [Google Scholar] [CrossRef]
- Hunter, P.R.; Gaston, M.A. Numerical index of the discriminatory ability of typing systems: An application of Simpson’s index of diversity. J. Clin. Microbiol. 1988, 26, 2465–2466. [Google Scholar] [CrossRef]
- Zhou, Z.; Charlesworth, J.; Achtman, M. HierCC: A multi-level clustering scheme for population assignments based on core genome MLST. Bioinformatics 2021, 37, 3645–3646. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- RStudio Team. RStudio: Integrated Development for R; RStudio, Inc.: Boston, MA, USA, 2019; Available online: http://www.rstudio.com/ (accessed on 18 July 2024).
- Conway, J.R.; Lex, A.; Gehlenborg, N. UpSetR: An R package for the visualization of intersecting sets and their properties. Bioinformatics 2017, 33, 2938–2940. [Google Scholar] [CrossRef] [PubMed]
- European Centre for Disease Prevention and Control. STEC infection. In Annual Epidemiological Report for 2022; ECDC: Stockholm, Sweden, 2024. [Google Scholar]
- Pollock, K.G.J.; Stewart, A.; Beattie, T.J.; Todd, W.T.A.; Ahn, C.K.; Tarr, P.I.; Cowden, J.M. From diarrhoea to haemolytic uraemic syndrome—When to seek advice. J. Med. Microbiol. 2009, 58 Pt 4, 397–398. [Google Scholar] [CrossRef] [PubMed]
- Uelze, L.; Grützke, J.; Borowiak, M.; Hammerl, J.A.; Juraschek, K.; Deneke, C.; Tausch, S.H.; Malorny, B. Typing methods based on whole genome sequencing data. One Health Outlook 2020, 2, 3. [Google Scholar] [CrossRef] [PubMed]
- Bessonov, K.; Laing, C.; Robertson, J.; Yong, I.; Ziebell, K.; Gannon, V.P.J.; Nichani, A.; Arya, G.; Nash, J.H.E.; Christianson, S. ECTyper: In silico Escherichia coli serotype and species prediction from raw and assembled whole-genome sequence data. Microb. Genom. 2021, 7, 000728. [Google Scholar] [CrossRef]
- EFSA and ECDC (European Food Safety Authority and European Centre for Disease Prevention and Control). The European Union One Health 2018 Zoonoses Report. EFSA J. 2019, 17, 5926. [Google Scholar] [CrossRef]
- EFSA and ECDC (European Food Safety Authority and European Centre for Disease Prevention and Control). The European Union One Health 2019 Zoonoses Report. EFSA J. 2021, 19, 6406. [Google Scholar] [CrossRef]
- Valilis, E.; Ramsey, A.; Sidiq, S.; DuPont, H.L. Non-O157 Shiga toxin-producing Escherichia coli—A poorly appreciated enteric pathogen: Systematic review. Int. J. Infect. Dis. 2018, 76, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Zweifel, C.; Cernela, N.; Stephan, R. Detection of the emerging Shiga toxin-producing Escherichia coli O26:H11/H-sequence type 29 (ST29) clone in human patients and healthy cattle in Switzerland. Appl. Environ. Microbiol. 2013, 79, 5411–5413. [Google Scholar] [CrossRef] [PubMed]
- Bielaszewska, M.; Mellmann, A.; Bletz, S.; Zhang, W.; Köck, R.; Kossow, A.; Prager, R.; Fruth, A.; Orth-Höller, D.; Marejková, M.; et al. Enterohemorrhagic Escherichia coli O26:H11/H-: A new virulent clone emerges in Europe. Clin. Infect. Dis. 2013, 56, 1373–1381. [Google Scholar] [CrossRef] [PubMed]
- Ogura, Y.; Gotoh, Y.; Itoh, T.; Sato, M.P.; Seto, K.; Yoshino, S.; Isobe, J.; Etoh, Y.; Kurogi, M.; Kimata, K.; et al. Population structure of Escherichia coli O26:H11 with recent and repeated stx2 acquisition in multiple lineages. Microb. Genom. 2017, 3, e000141. [Google Scholar] [CrossRef] [PubMed]
- Krüger, A.; Lucchesi, P.M.; Sanso, A.M.; Etcheverría, A.I.; Bustamante, A.V.; Burgán, J.; Fernández, L.; Fernández, D.; Leotta, G.; Friedrich, A.W.; et al. Genetic characterization of Shiga toxin-producing Escherichia coli O26:H11 strains isolated from animal, food, and clinical samples. Front. Cell Infect. Microbiol. 2015, 5, 74. [Google Scholar] [CrossRef] [PubMed]
- Leomil, L.; Pestana de Castro, A.F.; Krause, G.; Schmidt, H.; Beutin, L. Characterization of two major groups of diarrheagenic Escherichia coli O26 strains which are globally spread in human patients and domestic animals of different species. FEMS Microbiol. Lett. 2005, 249, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Chirila, F.; Tabaran, A.; Fit, N.; Nadas, G.; Mihaiu, M.; Tabaran, F.; Cătoi, C.; Reget, O.L.; Dan, S.D. Concerning Increase in Antimicrobial Resistance in Shiga Toxin-Producing Escherichia coli Isolated from Young Animals during 1980–2016. Microbes Environ. 2017, 32, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Tabaran, A.; Mihaiu, M.; Tăbăran, F.; Colobatiu, L.; Reget, O.; Borzan, M.M.; Dan, S.D. First study on characterization of virulence and antibiotic resistance genes in verotoxigenic and enterotoxigenic E. coli isolated from raw milk and unpasteurized traditional cheeses in Romania. Folia Microbiol. 2017, 62, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Tabaran, A.; Soulageon, V.; Chirila, F.; Reget, O.L.; Mihaiu, M.; Borzan, M.; Dan, S.D. Pathogenic E. coli from Cattle as a Reservoir of Resistance Genes to Various Groups of Antibiotics. Antibiotics 2022, 11, 404. [Google Scholar] [CrossRef] [PubMed]
- Aarestrup, F.M. The livestock reservoir for antimicrobial resistance: A personal view on changing patterns of risks, effects of interventions and the way forward. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015, 370, 20140085. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, J.; Dudley, E.G.; Sui, B.; Tamboura, B.; Suleman, A.; Nataro, J.P. EilA, a HilA-like regulator in enteroaggregative Escherichia coli. Mol. Microbiol. 2006, 61, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Miajlovic, H.; Smith, S.G. Bacterial self-defence: How Escherichia coli evades serum killing. FEMS Microbiol. Lett. 2014, 354, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Burgos, Y.; Beutin, L. Common origin of plasmid encoded alpha-hemolysin genes in Escherichia coli. BMC Microbiol. 2010, 10, 193. [Google Scholar] [CrossRef] [PubMed]
- Toma, C.; Martínez Espinosa, E.; Song, T.; Miliwebsky, E.; Chinen, I.; Iyoda, S.; Iwanaga, M.; Rivas, M. Distribution of putative adhesins in different seropathotypes of Shiga toxin-producing Escherichia coli. J. Clin. Microbiol. 2004, 42, 4937–4946. [Google Scholar] [CrossRef] [PubMed]
- Okuno, M.; Arimizu, Y.; Miyahara, S.; Wakabayashi, Y.; Gotoh, Y.; Yoshino, S.; Harada, T.; Seto, K.; Yamamoto, T.; Nakamura, K.; et al. Escherichia cryptic clade I is an emerging source of human intestinal pathogens. BMC Biol. 2023, 21, 81. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, A.W.; Bielaszewska, M.; Zhang, W.L.; Pulz, M.; Kuczius, T.; Ammon, A.; Karch, H. Escherichia coli harboring Shiga toxin 2 gene variants: Frequency and association with clinical symptoms. J. Infect. Dis. 2002, 185, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Ethelberg, S.; Olsen, K.E.; Scheutz, F.; Jensen, C.; Schiellerup, P.; Enberg, J.; Petersen, A.M.; Olesen, B.; Gerner-Smidt, P.; Mølbak, K. Virulence factors for hemolytic uremic syndrome, Denmark. Emerg. Infect. Dis. 2004, 10, 842–847. [Google Scholar] [CrossRef] [PubMed]
- De Rauw, K.; Buyl, R.; Jacquinet, S.; Piérard, D. Risk determinants for the development of typical haemolytic uremic syndrome in Belgium and proposition of a new virulence typing algorithm for Shiga toxin-producing Escherichia coli. Epidemiol. Infect. 2018, 147, e6. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yu, D.; Chui, L.; Zhou, T.; Feng, Y.; Cao, Y.; Zhi, S. A Comprehensive Review on Shiga Toxin Subtypes and Their Niche-Related Distribution Characteristics in Shiga-Toxin-Producing E. coli and Other Bacterial Hosts. Microorganisms 2024, 12, 687. [Google Scholar] [CrossRef]
- Escobar-Páramo, P.; Clermont, O.; Blanc-Potard, A.B.; Bui, H.; Le Bouguénec, C.; Denamur, E. A specific genetic background is required for acquisition and expression of virulence factors in Escherichia coli. Mol. Biol. Evol. 2004, 21, 1085–1094. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Escalona, N.; McFarland, M.A.; Rump, L.V.; Payne, J.; Andrzejewski, D.; Brown, E.W.; Evans, P.S.; Croley, T.R. Draft Genome Sequences of Two O104:H21 Escherichia coli Isolates Causing Hemorrhagic Colitis during a 1994 Montana Outbreak Provide Insight into Their Pathogenicity. Genome Announc. 2013, 1, e00805-13. [Google Scholar] [CrossRef] [PubMed]
- Mellmann, A.; Bielaszewska, M.; Köck, R.; Friedrich, A.W.; Fruth, A.; Middendorf, B.; Harmsen, D.; Schmidt, M.A.; Karch, H. Analysis of collection of hemolytic uremic syndrome-associated enterohemorrhagic Escherichia coli. Emerg. Infect. Dis. 2008, 14, 1287–1290. [Google Scholar] [CrossRef] [PubMed]
- Boisen, N.; Østerlund, M.T.; Joensen, K.G.; Santiago, A.E.; Mandomando, I.; Cravioto, A.; Chattaway, M.A.; Gonyar, L.A.; Overballe-Petersen, S.; Stine, O.C.; et al. Redefining enteroaggregative Escherichia coli (EAEC): Genomic characterization of epidemiological EAEC strains. PLoS Negl. Trop. Dis. 2020, 14, e0008613. [Google Scholar] [CrossRef] [PubMed]
- Jouve, M.; Garcia, M.-I.; Courcoux, P.; Labigne, A.; Gounon, P.; Le Bouguénec, C. Adhesion to and invasion of HeLa cells by pathogenic Escherichia coli carrying the afa-3gene cluster are mediated by the AfaE and AfaD proteins, respectively. Infect. Immun. 1997, 65, 4082. [Google Scholar] [CrossRef]
- Grande, L.; Michelacci, V.; Bondì, R.; Gigliucci, F.; Franz, E.; Badouei, M.A.; Schlager, S.; Minelli, F.; Tozzoli, R.; Caprioli, A.; et al. Whole-Genome Characterization and Strain Comparison of VT2f-Producing Escherichia coli Causing Hemolytic Uremic Syndrome. Emerg. Infect. Dis. 2016, 22, 2078–2086. [Google Scholar] [CrossRef] [PubMed]
- Den Ouden, A.; Greig, D.R.; Rodwell, E.V.; Tripodo, F.; Olonade, I.; Swift, C.; Jenkins, C. Escherichia coli encoding Shiga toxin subtype Stx2f causing human infections in England, 2015–2022. J. Med. Microbiol. 2023, 72, 001707. [Google Scholar] [CrossRef] [PubMed]
- Beutin, L.; Krüger, U.; Krause, G.; Miko, A.; Martin, A.; Strauch, E. Evaluation of major types of Shiga toxin 2E-producing Escherichia coli bacteria present in food, pigs, and the environment as potential pathogens for humans. Appl. Environ. Microbiol. 2008, 74, 4806–4816. [Google Scholar] [CrossRef]
- Liu, B.; Furevi, A.; Perepelov, A.V.; Guo, X.; Cao, H.; Wang, Q.; Reeves, P.R.; Knirel, Y.A.; Wang, L.; Widmalm, G. Structure and genetics of Escherichia coli O antigens. FEMS Microbiol. Rev. 2020, 44, 655–683. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wu, Y.; Liu, Q.; Sun, H.; Luo, M.; Xiong, Y.; Matussek, A.; Hu, B.; Bai, X. Genomic Characteristics of Stx2e-Producing Escherichia coli Strains Derived from Humans, Animals, and Meats. Pathogens 2021, 10, 1551. [Google Scholar] [CrossRef] [PubMed]
- Nüesch-Inderbinen, M.; Barmettler, K.; Stevens, M.J.A.; Cernela, N. Shiga toxin-producing Escherichia coli isolated from hunted wild boar (Sus. scrofa) in Switzerland. Schweiz. Arch. Tierheilkd. 2024, 166, 131–140, English. [Google Scholar] [CrossRef] [PubMed]
- Cavalcanti, A.M.F.; Hernandes, R.T.; Takagi, E.H.; Guth, B.E.C.; Ori, É.L.; Pinheiro, S.R.S.; Andrade, T.S.; Oliveira, S.L.; Cergole-Novella, M.C.; Francisco, G.R.; et al. Virulence Profiling and Molecular Typing of Shiga Toxin-Producing E. coli (STEC) from Human Sources in Brazil. Microorganisms 2020, 8, 171. [Google Scholar] [CrossRef] [PubMed]
- Lindstedt, B.A.; Finton, M.D.; Porcellato, D.; Brandal, L.T. High frequency of hybrid Escherichia coli strains with combined Intestinal Pathogenic Escherichia coli (IPEC) and Extraintestinal Pathogenic Escherichia coli (ExPEC) virulence factors isolated from human faecal samples. BMC Infect. Dis. 2018, 18, 544. [Google Scholar] [CrossRef] [PubMed]
- De Rycke, J.; Milon, A.; Oswald, E. Necrotoxic Escherichia coli (NTEC): Two emerging categories of human and animal pathogens. Vet. Res. 1999, 30, 221–233. [Google Scholar] [PubMed]
- Servin, A.L. Pathogenesis of human diffusely adhering Escherichia coli expressing Afa/Dr adhesins (Afa/Dr DAEC): Current insights and future challenges. Clin. Microbiol. Rev. 2014, 27, 823–869. [Google Scholar] [CrossRef] [PubMed]
- Gérardin, J.; Lalioui, L.; Jacquemin, E.; Le Bouguénec, C.; Mainil, J.G. The afa-related gene cluster in necrotoxigenic and other Escherichia coli from animals belongs to the afa-8 variant. Vet. Microbiol. 2000, 76, 175–184. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Serotype | Sequence Type | stx Genotype | No. of Strains |
|---|---|---|---|
| O26:H11 | ST21; ST15140 | stx1a; stx2a; stx1a + stx2a | 25 |
| O157:H7 | ST11; ST1804 | stx2c; stx1a + stx2c | 6 |
| O113:H4 | ST10 | stx1c + stx2b | 3 |
| O91:H14 | ST33 | stx1a + stx2b | 2 |
| O128ac:H2 | ST25 | stx2b | 2 |
| O9a:H10 | ST1791 | stx2e | 1 |
| O10:H45 | ST10716 | stx2a | 1 |
| O43:H2 | ST937 | stx1c | 1 |
| O55:H12 | ST101 | stx1a | 1 |
| O76:H19 | ST675 | stx1c + stx2b | 1 |
| O78:H4 | ST3101 | stx1c | 1 |
| O100:H20 | ST2514 | stx2e | 1 |
| O104:H21 | ST8160 | stx1a + stx2d | 1 |
| O125ac:H6 | ST583 | stx2f | 1 |
| O146:H2 | ST442 | stx1c + stx2b | 1 |
| Ont:H4 | ST10 | stx2a | 1 |
| Ont:H10 | ST34 | stx2a | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Usein, C.-R.; Oprea, M.; Dinu, S.; Popa, L.-I.; Cristea, D.; Militaru, C.-M.; Ghiță, A.; Costin, M.; Popa, I.-L.; Croitoru, A.; et al. Shiga-Toxin-Producing Escherichia coli Strains from Romania: A Whole Genome-Based Description. Microorganisms 2024, 12, 1469. https://doi.org/10.3390/microorganisms12071469
Usein C-R, Oprea M, Dinu S, Popa L-I, Cristea D, Militaru C-M, Ghiță A, Costin M, Popa I-L, Croitoru A, et al. Shiga-Toxin-Producing Escherichia coli Strains from Romania: A Whole Genome-Based Description. Microorganisms. 2024; 12(7):1469. https://doi.org/10.3390/microorganisms12071469
Chicago/Turabian StyleUsein, Codruța-Romanița, Mihaela Oprea, Sorin Dinu, Laura-Ioana Popa, Daniela Cristea, Cornelia-Mădălina Militaru, Andreea Ghiță, Mariana Costin, Ionela-Loredana Popa, Anca Croitoru, and et al. 2024. "Shiga-Toxin-Producing Escherichia coli Strains from Romania: A Whole Genome-Based Description" Microorganisms 12, no. 7: 1469. https://doi.org/10.3390/microorganisms12071469