
Characterization of the Hoof Bacterial Communities of Active Digital Dermatitis Lesions in Feedlot Cattle
 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
3. Results
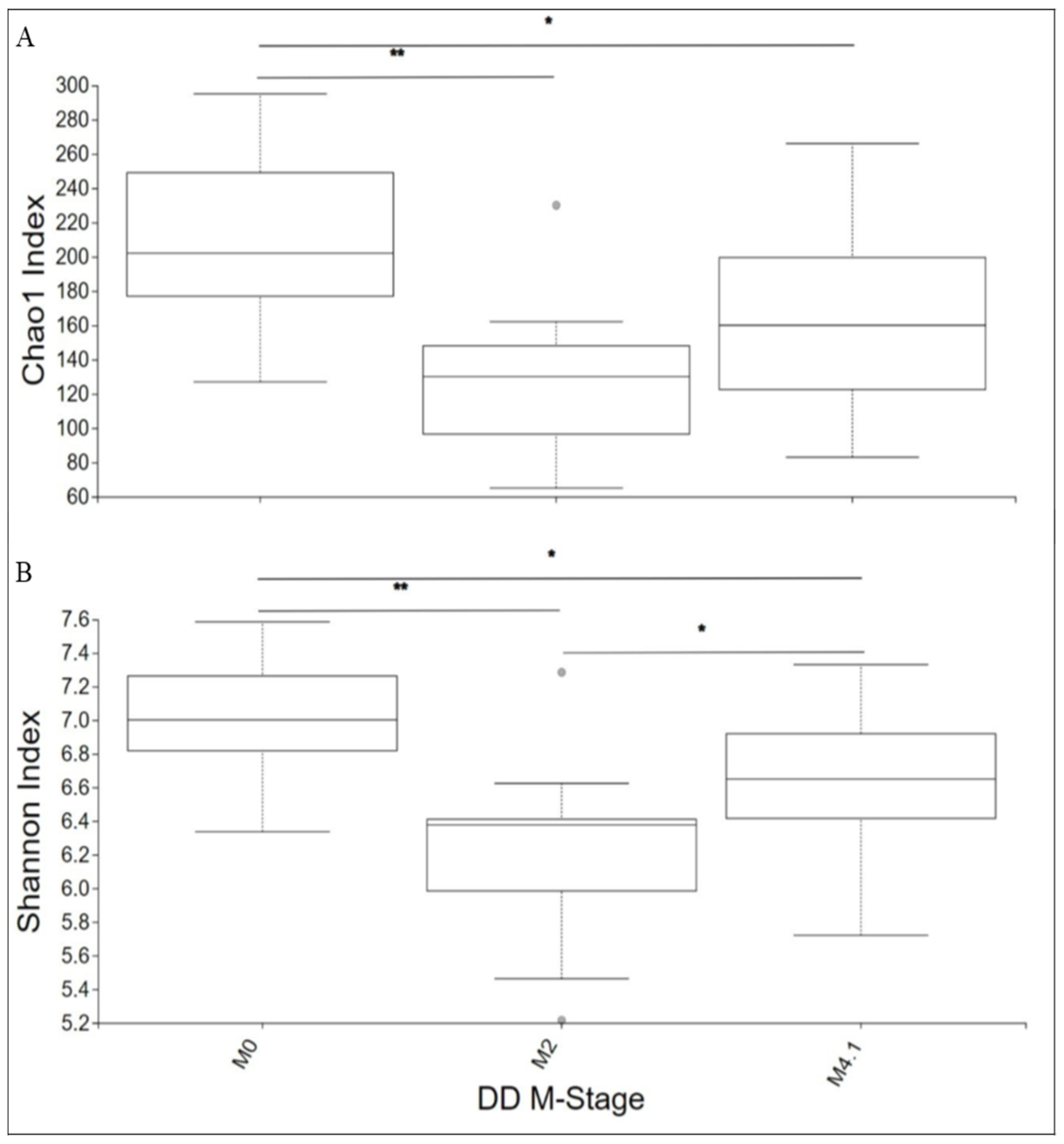
3.1. Diversity Analyses
3.2. Phylogenetic Analysis
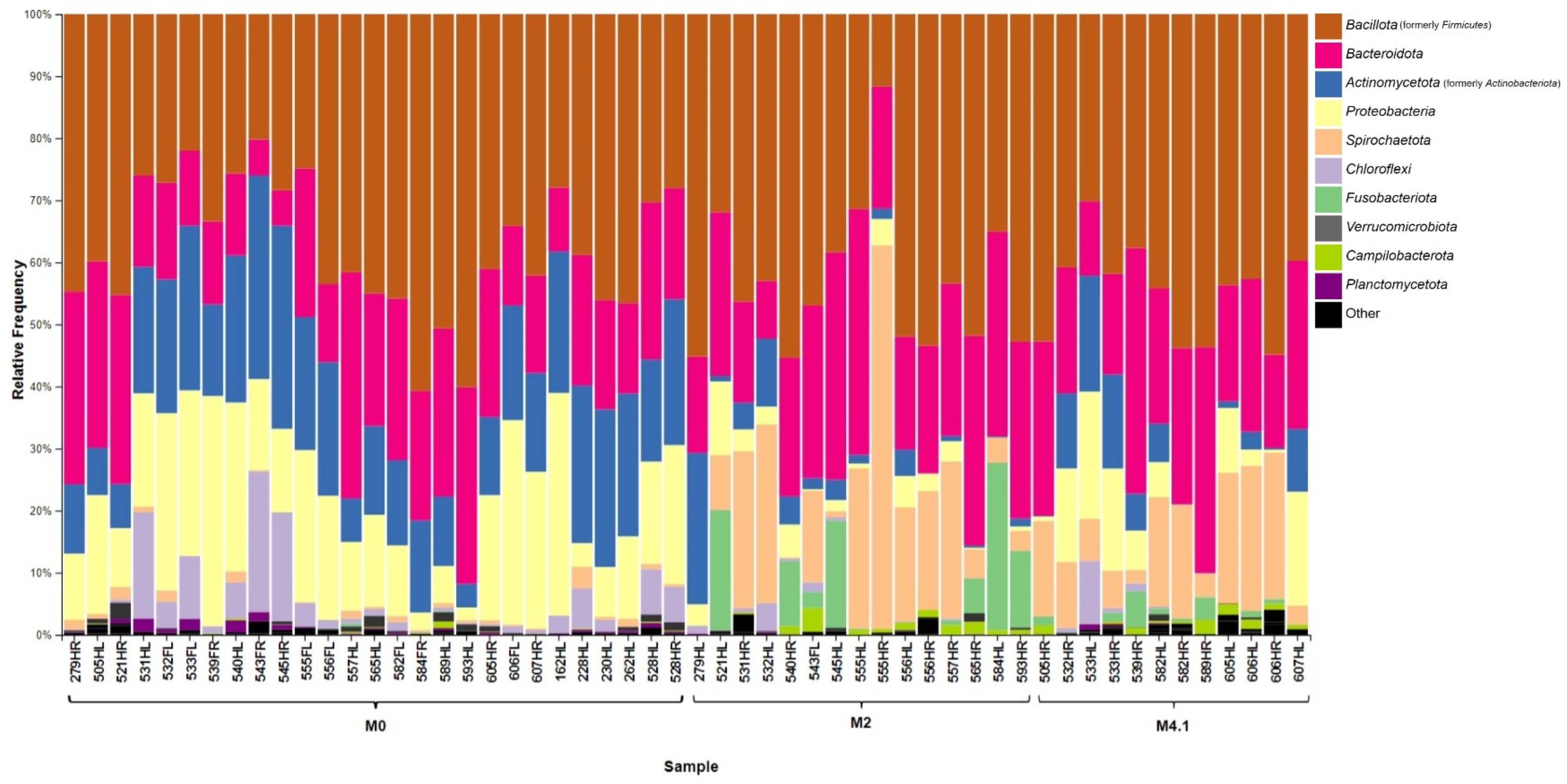
3.3. Taxonomic Summary
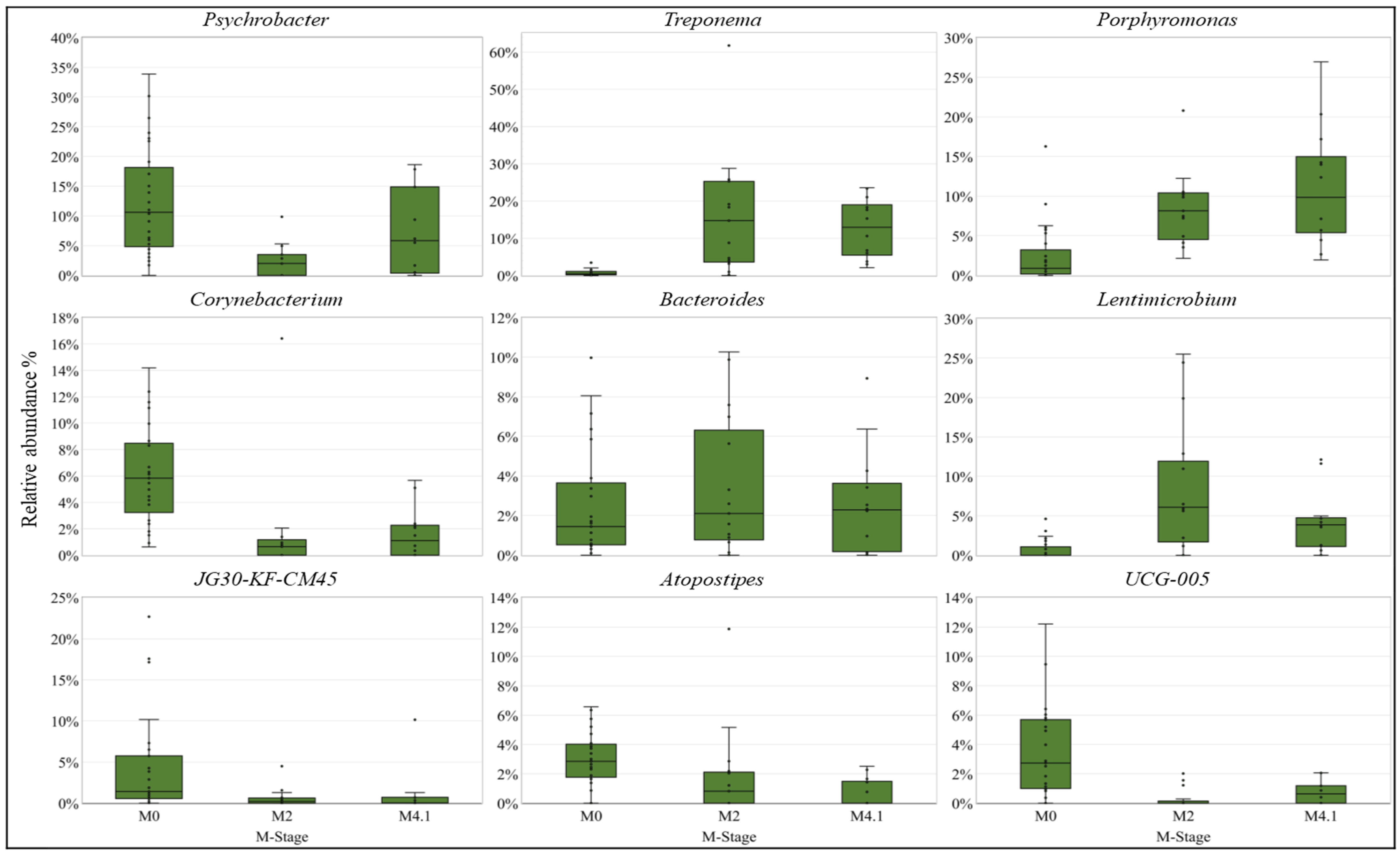
3.4. Differentially Abundant (DA) Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cortes, J.A.; Hendrick, S.; Janzen, E.; Pajor, E.A.; Orsel, K. Economic impact of digital dermatitis, foot rot, and bovine respiratory disease in feedlot cattle. Transl. Anim. Sci. 2021, 5, txab076. [Google Scholar] [CrossRef] [PubMed]
- Plummer, P.J.; Krull, A. Clinical Perspectives of Digital dermatitis in dairy and beef cattle. Vet. Clin. N. Am. Food Anim. Pract. 2017, 33, 165–181. [Google Scholar] [CrossRef]
- Sullivan, L.; Evans, N.; Blowey, R.; Grove-White, D.; Clegg, S.; Duncan, J.; Carter, S. A molecular epidemiology of treponemes in beef cattle digital dermatitis lesions and comparative analyses with sheep contagious ovine digital dermatitis and dairy cattle digital dermatitis lesions. Vet. Microbiol. 2015, 178, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Caddey, B.; Orsel, K.; Naushad, S.; Derakhshani, H.; De Buck, J. Identification and quantification of bovine digital dermatitis-associated microbiota across lesion stages in feedlot beef cattle. mSystems 2021, 6, e00708-21. [Google Scholar] [CrossRef]
- Wilson-Welder, J.H.; Alt, D.P.; Nally, J.E. Digital dermatitis in cattle: Current bacterial and immunological findings. Animals 2015, 5, 1114–1135. [Google Scholar] [CrossRef]
- Orsel, K.; Plummer, P.; Shearer, J.; De Buck, J.; Carter, S.D.; Guatteo, R.; Barkema, H.W. Missing pieces of the puzzle to effectively control digital dermatitis. Transbound. Emerg. Dis. 2018, 65 (Suppl. S1), 186–198. [Google Scholar] [CrossRef]
- Beninger, C.; Naqvi, S.A.; Naushad, S.; Orsel, K.; Luby, C.; Derakhshani, H.; Khafipour, E.; De Buck, J. Associations between digital dermatitis lesion grades in dairy cattle and the quantities of four Treponema species. Vet. Res. 2018, 49, 111. [Google Scholar] [CrossRef] [PubMed]
- Krull, A.C.; Shearer, J.K.; Gorden, P.J.; Cooper, V.L.; Phillips, G.J.; Plummer, P.J. Deep sequencing analysis reveals temporal microbiota changes associated with development of bovine digital dermatitis. Infect. Immun. 2014, 82, 3359–3373. [Google Scholar] [CrossRef]
- Nielsen, M.W.; Strube, M.L.; Isbrand, A.; Al-Medrasi, W.D.; Boye, M.; Jensen, T.K.; Klitgaard, K. Potential bacterial core species associated with digital dermatitis in cattle herds identified by molecular profiling of interdigital skin samples. Vet. Microbiol. 2016, 186, 139–149. [Google Scholar] [CrossRef]
- Moreira, T.F.; Filho, E.J.F.; Carvalho, A.U.; Strube, M.L.; Nielsen, M.W.; Klitgaard, K.; Jensen, T.K. Pathology and bacteria related to digital dermatitis in dairy cattle in all year round grazing system in Brazil. PLoS ONE 2018, 13, e0193870. [Google Scholar] [CrossRef]
- Caddey, B.; De Buck, J. Meta-analysis of bovine digital dermatitis microbiota reveals distinct microbial community structures associated with lesions. Front. Cell. Infect. Microbiol. 2021, 11, 685861. [Google Scholar] [CrossRef]
- Hoby, S.; Jensen, T.K.; Brodard, I.; Gurtner, C.; Eicher, R.; Steiner, A.; Kuhnert, P.; Alsaaod, M. Detection of treponemes in digital dermatitis lesions of captive European bison (Bison bonasus). PLoS ONE 2021, 16, e0255921. [Google Scholar] [CrossRef] [PubMed]
- Döpfer, D.; Koopmans, A.; Meijer, F.A.; Szakáll, I.; Schukken, Y.H.; Klee, W.; Bosma, R.B.; Cornelisse, J.L.; van Asten, A.J.; Huurne, A.A.T. Histological and bacteriological evaluation of digital dermatitis in cattle, with special reference to spirochaetes and Campylobacter faecalis. Vet. Rec. 1997, 140, 620–623. [Google Scholar] [CrossRef] [PubMed]
- Berry, S.L.; Read, D.H.; Famula, T.R.; Mongini, A.; Döpfer, D. Long-term observations on the dynamics of bovine digital dermatitis lesions on a California dairy after topical treatment with lincomycin HCl. Vet. J. 2012, 193, 654–658. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.A.; Laven, R.A.; Müller, K.R.; Gates, M.C. Modelling the transmission dynamics of bovine digital dermatitis in New Zealand pastoral dairy production systems. Vet. Res. 2020, 51, 16. [Google Scholar] [CrossRef] [PubMed]
- Watts, K.M.; Lahiri, P.; Arrazuria, R.; De Buck, J.; Knight, C.G.; Orsel, K.; Barkema, H.W.; Cobo, E.R. Oxytetracycline reduces inflammation and treponeme burden whereas vitamin D3 promotes β-defensin expression in bovine infectious digital dermatitis. Cell Tissue Res. 2020, 379, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Ariza, J.M.; Relun, A.; Bareille, N.; Oberle, K.; Guatteo, R. Effectiveness of collective treatments in the prevention and treatment of bovine digital dermatitis lesions: A systematic review. J. Dairy Sci. 2017, 100, 7401–7418. [Google Scholar] [CrossRef] [PubMed]
- Berry, S.L.; Read, D.H.; Walker, R.L.; Famula, T.R. Clinical, histologic, and bacteriologic findings in dairy cows with digital dermatitis (footwarts) one month after topical treatment with lincomycin hydrochloride or oxytetracycline hydrochloride. J. Am. Vet.—Med. Assoc. 2010, 237, 555–560. [Google Scholar] [CrossRef] [PubMed]
- Wong, N.S.T.; Malmuthge, N.; Gellatly, D.; Nordi, W.M.; Alexander, T.W.; Polo, R.O.; Janzen, E.; Schwartzkopf-Genswein, K.; Jelinski, M. Characterization of the hoof bacterial communities in feedlot cattle affected with digital dermatitis, foot rot or both using a surface swab technique. Anim. Microb. 2024, 6, 2. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 108, 4516–4522. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Caporaso, J.G. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef]
- Robeson, M.S.; O’rourke, D.R.; Kaehler, B.D.; Ziemski, M.; Dillon, M.R.; Foster, J.T.; Bokulich, N.A. RESCRIPt: Reproducible sequence taxonomy reference database management. PLoS Comput. Biol. 2021, 17, e1009581. [Google Scholar] [CrossRef]
- Shannon, C.E. A mathematical theory of communication. Bell Syst. Tech. J. 1948, 27, 379–423. [Google Scholar] [CrossRef]
- Chao, A. Nonparametric estimation of the number of classes in a population. Scand. J. Stat. 1984, 11, 265–270. [Google Scholar]
- Kruskal, W.H.; Wallis, W.A. Use of ranks in one-criterion variance analysis. J. Am. Stat. Assoc. 1952, 47, 583–621. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Lozupone, C.A.; Hamady, M.; Kelley, S.T.; Knight, R. Quantitative and qualitative β diversity measures lead to different insights into factors that structure microbial communities. Appl. Environ. Microbiol. 2007, 73, 1576–1585. [Google Scholar] [CrossRef]
- Vázquez-Baeza, Y.; Gonzalez, A.; Smarr, L.; McDonald, D.; Morton, J.T.; Navas-Molina, J.A.; Knight, R. Bringing the dynamic microbiome to life with animations. Cell Host Microbe 2017, 1, 7–10. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.J. Permutational Multivariate Analysis of Variance (PERMANOVA). In Wiley StatsRef: Statistics Reference Online, 1st ed.; Balakrishnan, N., Colton, T., Everitt, B., Piegorsch, W., Ruggeri, F., Teugels, J.L., Eds.; Wiley: New Jersey, NJ, USA, 2017; pp. 1–15. [Google Scholar]
- Cantrell, K.; Fedarko, M.W.; Rahman, G.; McDonald, D.; Yang, Y.; Zaw, T.; Gonzalez, A.; Janssen, S.; Estaki, M.; Haiminen, N.; et al. EMPress enables tree-guided, interactive, and exploratory analyses of multi-omic data sets. MSystems 2021, 6, e01216-20. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, A.D.; Macklaim, J.M.; Linn, T.G.; Reid, G.; Gloor, G.B. ANOVA-like differential expression (ALDEx) analysis for mixed population RNA-seq. PLoS ONE 2013, 8, e67019. [Google Scholar] [CrossRef] [PubMed]
- Morton, J.T.; Marotz, C.; Washburne, A.; Silverman, J.; Zaramela, L.S.; Edlund, A.; Zengler, K.; Knight, R. Establishing microbial composition measurement standards with reference frames. Nat. Commun. 2019, 10, 2719. [Google Scholar] [CrossRef] [PubMed]
- Hesseling, J.; Legione, A.R.; Stevenson, M.; McCowan, C.; Pyman, M.; Finochio, C.; Nguyen, D.; Roic, C.; Thiris, O.; Zhang, A.; et al. Bovine digital dermatitis in Victoria, Australia. Aust. Vet. J. 2019, 97, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Espiritu, H.M.; Mamuad, L.L.; Kim, S.-H.; Jin, S.-J.; Lee, S.-S.; Kwon, S.-W.; Cho, Y.-I. Microbiome shift, diversity, and overabundance of opportunistic pathogens in bovine digital dermatitis revealed by 16S rRNA amplicon sequencing. Animals 2020, 10, 1798. [Google Scholar] [CrossRef] [PubMed]
- Bay, V.; Gillespie, A.; Ganda, E.; Evans, N.J.; Carter, S.D.; Lenzi, L.; Lucaci, A.; Haldenby, S.; Barden, M.; Griffiths, B.E.; et al. The bovine foot skin microbiota is associated with host genotype and the development of infectious digital dermatitis lesions. Microbiome 2023, 11, 4. [Google Scholar] [CrossRef] [PubMed]
- Borsanelli, A.C.; Lappin, D.F.; Viora, L.; Bennett, D.; Dutra, I.S.; Brandt, B.W.; Riggio, M.P. Microbiomes associated with bovine periodontitis and oral health. Vet. Microbiol. 2018, 218, 1–6. [Google Scholar] [CrossRef]
- Charalambous, E.G.; Mériaux, S.B.; Guebels, P.; Muller, C.P.; Leenen, F.A.D.; Elwenspoek, M.M.C.; Thiele, I.; Hertel, J.; Turner, J.D. Early-life adversity leaves its imprint on the oral microbiome for more than 20 years and is associated with long-term immune changes. Int. J. Mol. Sci. 2021, 22, 12682. [Google Scholar] [CrossRef]
- Grzeczka, A.; Lech, M.; Wozniak, G.; Graczyk, S.; Kordowitzki, P.; Olejnik, M.; Gehrke, M.; Jaśkowski, J.M. Periodontitis disease in farmed ruminants—Current state of research. Int. J. Mol. Sci. 2023, 24, 9763. [Google Scholar] [CrossRef]
- Antezack, A.; Boxberger, M.; La Scola, B.; Monnet-Corti, V. Isolation and description of Catonella massiliensis sp. nov., A novel Catonella species, isolated from a stable periodontitis subject. Pathogens 2021, 10, 367. [Google Scholar] [CrossRef]
- Schlafer, S.; Riep, B.; Griffen, A.L.; Petrich, A.; Hübner, J.; Berning, M.; Friedmann, A.; Göbel, U.B.; Moter, A. Filifactor alocis—Involvement in periodontal biofilms. BMC Microbiol. 2010, 10, 66. [Google Scholar] [CrossRef]
- Khemwong, T.; Kobayashi, H.; Ikeda, Y.; Matsuura, T.; Sudo, T.; Kano, C.; Mikami, R.; Izumi, Y. Fretibacterium sp. human oral taxon 360 is a novel biomarker for periodontitis screening in the Japanese population. PLoS ONE 2019, 14, e0218266. [Google Scholar] [CrossRef]
- Riggio, M.P.; Lennon, A. Identification of oral Peptostreptococcus isolates by PCR-restriction fragment length polymorphism analysis of 16S rRNA genes. J. Clin. Microbiol. 2003, 41, 4475–4479. [Google Scholar] [CrossRef]
- Bortolotti, F.; Innocentini, L.M.A.R.; de Macedo, M.D.; Ferrari, T.C.; Kashima, S.; Covas, D.T.; Ricz, H.M.A.; Simoes, B.P.; Cunha, R.L.G.; de Macedo, L.D. Oral mucosa infection by Mycoplasma salivarium in a patient with chronic graft-versus-host disease: A diagnostic challenge. Hematol. Transfus. Cell Ther. 2022, 44, 259–263. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CH Controls | DD Controls | DD Lesions | ||
|---|---|---|---|---|
| M Stage | M0 | M0 | M2 | M4.1 |
| Feedlot A | 6 | 3 | 3 | 0 |
| Feedlot B | 0 | 12 * | 8 | 10 |
| Feedlot C | 0 | 6 | 4 | 2 |
| Total (n = 54) | 6 | 21 | 15 | 12 |
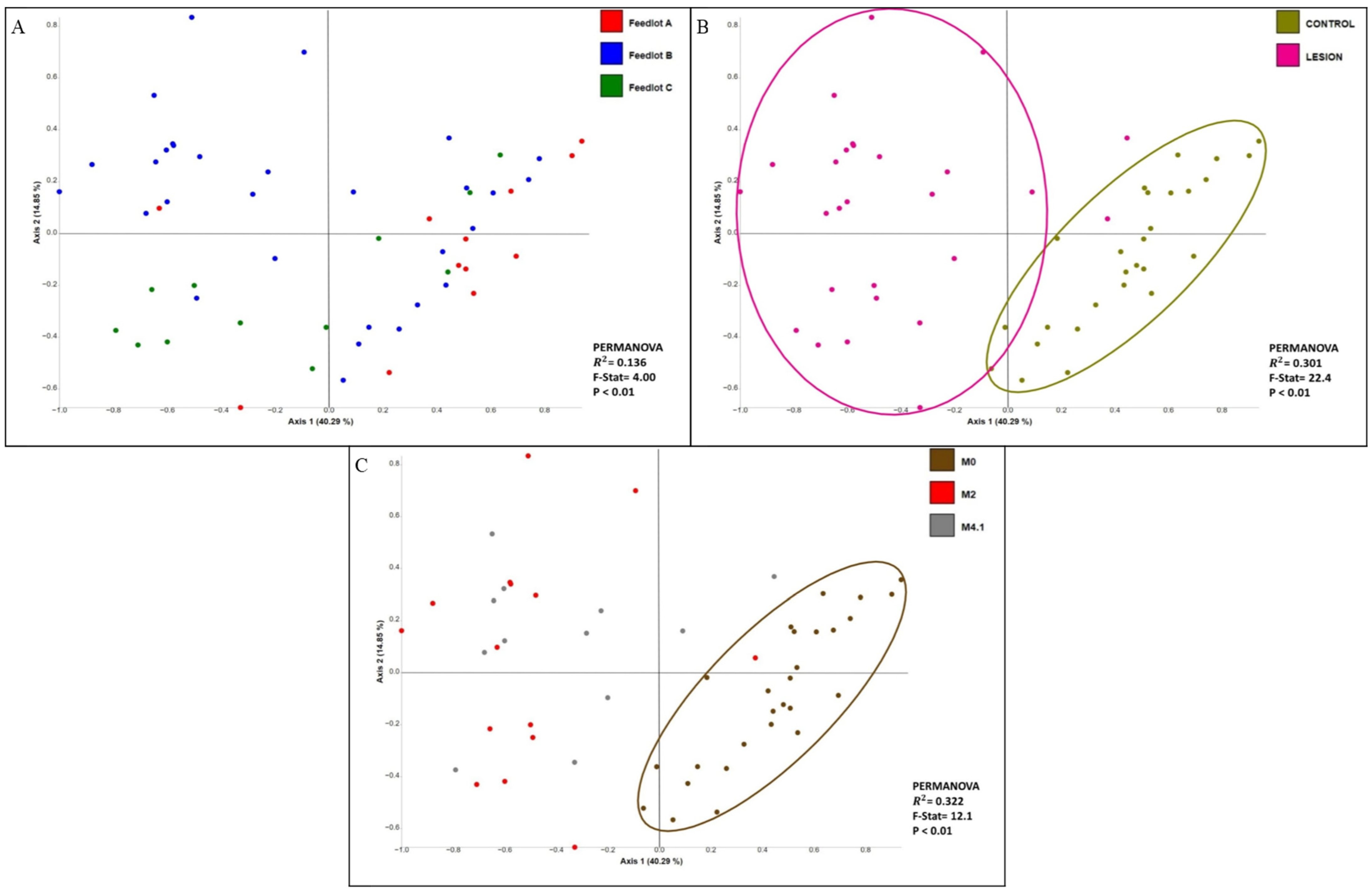
| Feedlot | Pairwise Comparison | PERMANOVA | ||||
|---|---|---|---|---|---|---|
| Group 1 | Group 2 | Sample Size | F-Stat | R2 | Adj p-Value * | |
| All | M0 | M2 | 42 | 18.2 | 0.313 | <0.01 |
| M4.1 | 39 | 14.4 | 0.280 | <0.01 | ||
| M2 | M4.1 | 27 | 1.32 | 0.050 | 0.228 | |
| A | M0 | M2 | 12 | 3.80 | 0.275 | <0.01 |
| B | M0 | M2 | 20 | 15.0 | 0.455 | <0.01 |
| M4.1 | 22 | 11.1 | 0.358 | <0.01 | ||
| M2 | M4.1 | 18 | 1.62 | 0.092 | 0.108 | |
| C | M0 | M2 | 10 | 6.59 | 0.451 | <0.01 |
| M4.1 | 8 | 3.74 | 0.383 | 0.037 | ||
| M2 | M4.1 | 6 | 2.15 | 0.349 | 0.133 | |
| Taxon | M-Stage | Control CLR * | Lesion CLR * | Effect Size | Adj p-Value | Pairwise Comparison | Phylogenetic Level |
|---|---|---|---|---|---|---|---|
| Spirochaetota | M2 | 1.76 | 6.25 | 0.96 | <0.01 | M0 vs. M2 | Phylum |
| Fusobacteriota | M2 | −5.74 | 2.84 | 0.86 | 0.01 | M0 vs. M2 | Phylum |
| Proteobacteria | M0 | 6.90 | 4.19 | −1.05 | <0.01 | M0 vs. M2 | Phylum |
| Actinomycetota | M0 | 7.09 | 3.71 | −1.18 | <0.01 | M0 vs. M2 | Phylum |
| Spirochaetota | M4.1 | 1.76 | 5.77 | 1.39 | <0.01 | M0 vs. M4.1 | Phylum |
| Fusobacteriota | M4.1 | −5.76 | 1.24 | 1.12 | <0.01 | M0 vs. M4.1 | Phylum |
| Synergistota | M4.1 | −6.44 | −0.63 | 0.86 | 0.03 | M0 vs. M4.1 | Phylum |
| Planctomycetota | M0 | 0.18 | −5.04 | −0.81 | 0.02 | M0 vs. M4.1 | Phylum |
| Chloroflexi | M0 | 3.55 | −2.46 | −0.93 | <0.01 | M0 vs. M4.1 | Phylum |
| Actinomycetota | M0 | 7.08 | 2.81 | −1.18 | <0.01 | M0 vs. M4.1 | Phylum |
| Porphyromonas | M2 | 5.18 | 9.14 | 1.38 | <0.01 | M0 vs. M2 | Genus |
| Treponema | M2 | 3.94 | 9.46 | 1.30 | <0.01 | M0 vs. M2 | Genus |
| Amnipila | M2 | −4.01 | 6.76 | 1.14 | <0.01 | M0 vs. M2 | Genus |
| Mycoplasma | M2 | −2.43 | 7.43 | 1.12 | <0.01 | M0 vs. M2 | Genus |
| Catonella | M2 | −4.01 | 5.86 | 1.11 | <0.01 | M0 vs. M2 | Genus |
| Peptostreptococcaceae | M2 | −0.55 | 6.28 | 1.10 | <0.01 | M0 vs. M2 | Genus |
| Fusobacterium | M2 | −3.57 | 5.91 | 0.95 | 0.02 | M0 vs. M2 | Genus |
| Parvimonas | M2 | −4.23 | 0.95 | 0.89 | 0.02 | M0 vs. M2 | Genus |
| Lentimicrobium | M2 | 1.92 | 8.52 | 0.84 | 0.01 | M0 vs. M2 | Genus |
| Falsiporphyromonas | M2 | −4.22 | −0.04 | 0.83 | 0.03 | M0 vs. M2 | Genus |
| Anaerovibrio | M2 | 1.97 | 7.09 | 0.81 | 0.02 | M0 vs. M2 | Genus |
| Jeotgalibaca | M0 | 5.02 | −2.13 | −0.91 | 0.02 | M0 vs. M2 | Genus |
| Olsenella | M0 | 4.97 | −0.81 | −1.27 | <0.01 | M0 vs. M2 | Genus |
| Amnipila | M4.1 | −4.03 | 7.04 | 2.50 | <0.01 | M0 vs. M4.1 | Genus |
| Peptostreptococcaceae | M4.1 | −0.44 | 6.57 | 1.77 | <0.01 | M0 vs. M4.1 | Genus |
| Treponema | M4.1 | 3.94 | 8.67 | 1.69 | <0.01 | M0 vs. M4.1 | Genus |
| Mycoplasma | M4.1 | −2.57 | 7.23 | 1.61 | <0.01 | M0 vs. M4.1 | Genus |
| Clostridia_vadinBB60_group | M4.1 | 2.12 | 6.66 | 1.38 | <0.01 | M0 vs. M4.1 | Genus |
| Fusobacterium | M4.1 | −3.41 | 4.62 | 1.28 | <0.01 | M0 vs. M4.1 | Genus |
| Lachnospiraceae_AC2044_group | M4.1 | −4.04 | 5.12 | 1.24 | <0.01 | M0 vs. M4.1 | Genus |
| Roseburia | M4.1 | −3.00 | 5.86 | 1.23 | <0.01 | M0 vs. M4.1 | Genus |
| Murdochiella | M4.1 | −3.52 | 3.92 | 1.16 | <0.01 | M0 vs. M4.1 | Genus |
| Acholeplasma | M4.1 | −0.72 | 6.21 | 1.07 | <0.01 | M0 vs. M4.1 | Genus |
| Fretibacterium | M4.1 | −4.21 | 3.21 | 1.02 | 0.03 | M0 vs. M4.1 | Genus |
| Porphyromonas | M4.1 | 5.22 | 8.81 | 1.00 | <0.01 | M0 vs. M4.1 | Genus |
| Campylobacter | M4.1 | −0.87 | 5.36 | 0.91 | 0.01 | M0 vs. M4.1 | Genus |
| Anaerovibrio | M4.1 | 1.92 | 6.41 | 0.89 | 0.02 | M0 vs. M4.1 | Genus |
| Filifactor | M4.1 | −3.97 | 2.14 | 0.88 | 0.04 | M0 vs. M4.1 | Genus |
| Olsenella | M0 | 4.98 | 1.97 | −0.80 | 0.03 | M0 vs. M4.1 | Genus |
| Clostridium_sensu_stricto_1 | M0 | 5.51 | −0.18 | −0.99 | 0.02 | M0 vs. M4.1 | Genus |
| Rank | M0 Associated Taxon | M2 Associated Taxon | M4.1 Associated Taxon |
|---|---|---|---|
| 1 | Psychrobacter | ¶ Fusobacterium | † Amnipila |
| 2 | * Corynebacterium | † Amnipila | † Lachnospiraceae_AC2044_group |
| 3 | JG30-KF-CM45 | † Catonella | † Filifactor |
| 4 | † UCG-005 | § Falsiporphyromonas | ‡ Fretibacterium |
| 5 | § Porphyromonas | † Filifactor | † Defluviitaleaceae_UCG-011 |
| 6 | † [Eubacterium]_coprostanoligenes_group | † Lachnospiraceae_AC2044_group | § Falsiporphyromonas |
| 7 | § Bacteroides | † Parvimonas | † Catonella |
| 8 | Atopostipes | † Mycoplasma | † Parvimonas |
| 9 | § Prevotella | ‡ Fretibacterium | † Acholeplasma |
| 10 | † W5053 | # Treponema | † Peptostreptococcaceae |
| 11 | § Prevotellaceae_UCG-003 | † Peptostreptococcaceae | # Treponema |
| 12 | * Dietzia | † Peptostreptococcus | Absconditabacteriales_(SR1) |
| 13 | * Paeniglutamicibacter | † Anaerovibrio | † Murdochiella |
| 14 | † Turicibacter | † Roseburia | † Roseburia |
| 15 | † Tissierella | † Acholeplasma | † [Eubacterium]_brachy_group |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 Nicholas Wong, Desiree Gellatly, Wiolene Nordi, Eugene Janzen, Murray Jelinski, and His Majesty the King in Right of Canada, as represented by the Minister of Agriculture and Agri-Food Canada for the contribution of Nilusha Malmuthuge, Trevor Alexander, Rodrigo Ortega Polo and Karen Schwartzkopf-Genswein. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wong, N.S.T.; Malmuthuge, N.; Gellatly, D.; Nordi, W.M.; Alexander, T.W.; Ortega-Polo, R.; Janzen, E.; Jelinski, M.; Schwartzkopf-Genswein, K. Characterization of the Hoof Bacterial Communities of Active Digital Dermatitis Lesions in Feedlot Cattle. Microorganisms 2024, 12, 1470. https://doi.org/10.3390/microorganisms12071470
Wong NST, Malmuthuge N, Gellatly D, Nordi WM, Alexander TW, Ortega-Polo R, Janzen E, Jelinski M, Schwartzkopf-Genswein K. Characterization of the Hoof Bacterial Communities of Active Digital Dermatitis Lesions in Feedlot Cattle. Microorganisms. 2024; 12(7):1470. https://doi.org/10.3390/microorganisms12071470
Chicago/Turabian StyleWong, Nicholas S. T., Nilusha Malmuthuge, Désirée Gellatly, Wiolene M. Nordi, Trevor W. Alexander, Rodrigo Ortega-Polo, Eugene Janzen, Murray Jelinski, and Karen Schwartzkopf-Genswein. 2024. "Characterization of the Hoof Bacterial Communities of Active Digital Dermatitis Lesions in Feedlot Cattle" Microorganisms 12, no. 7: 1470. https://doi.org/10.3390/microorganisms12071470