
Identification and Characterization of an Alphacoronavirus in Rhinolophus sinicus and a Betacoronavirus in Apodemus ilex in Yunnan, China
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Samples Collection and Host Identification
2.3. RNA Extraction and CoVs RT-PCR Screening
2.4. Complete Genome Sequencing
2.5. Genomic and Phylogenetic Analysis
2.6. Identification of Positive Selection Sites
2.7. Estimation of Divergence Dates
3. Results
3.1. Identification of Bat and Rodent CoVs
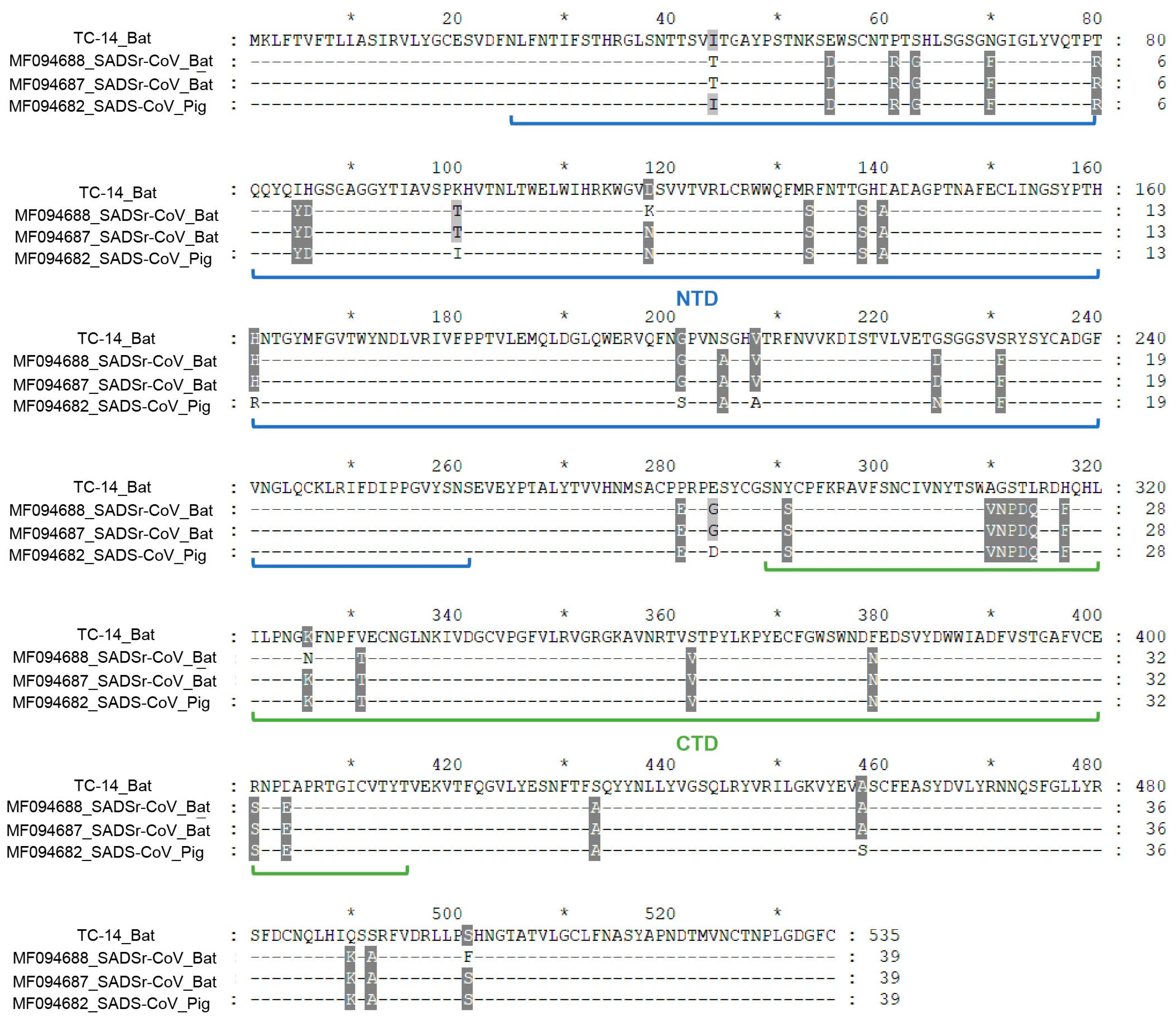
3.2. Genome Analysis of TC-14 and GS-56
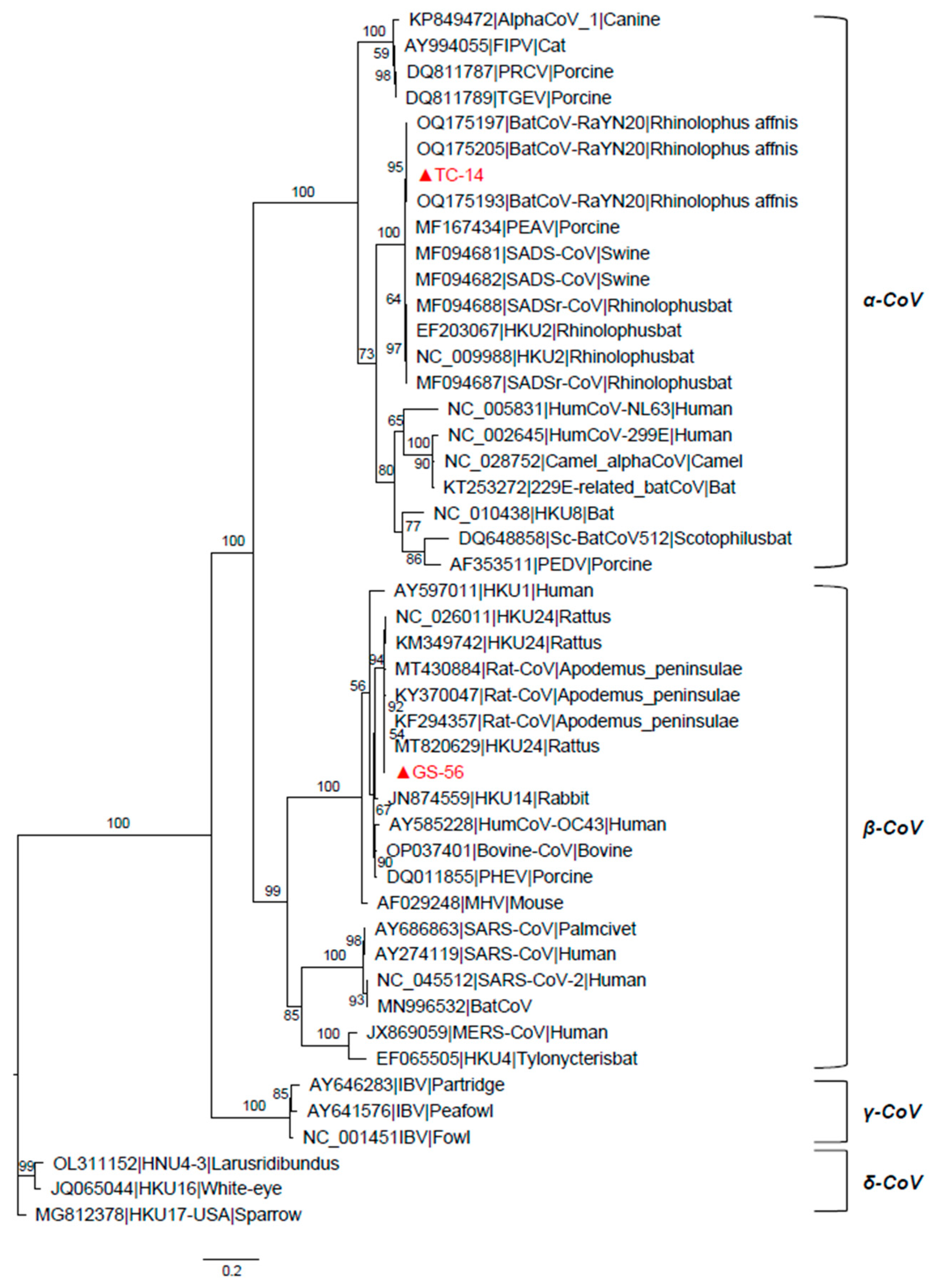
3.3. Phylogenetic Analyses of the TC-14 and GS-56
3.4. Positive Selection Analysis
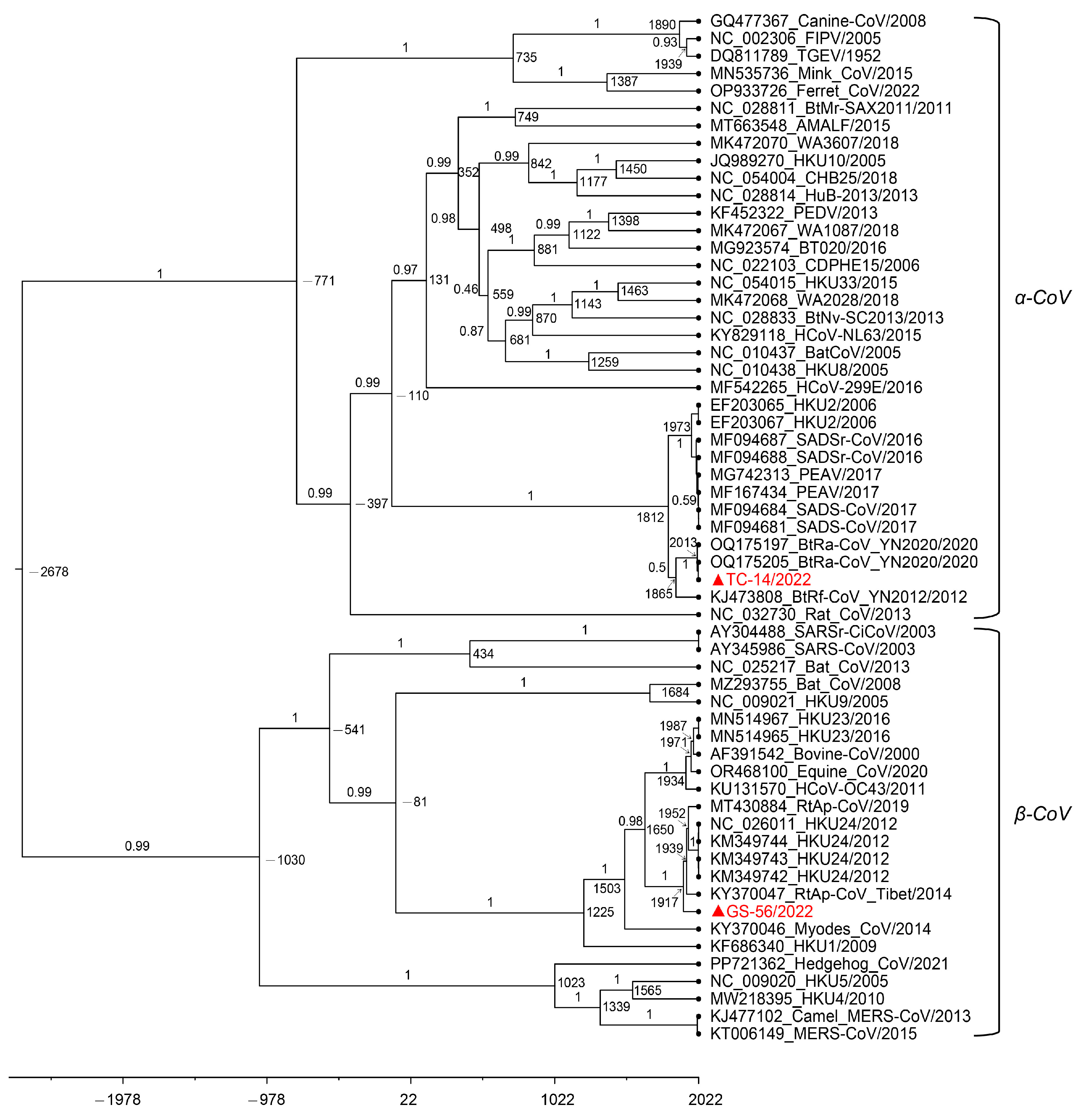
3.5. Estimation of Divergence Dates
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Li, G.; Fan, Y.; Lai, Y.; Han, T.; Li, Z.; Zhou, P.; Pan, P.; Wang, W.; Hu, D.; Liu, X.; et al. Coronavirus infections and immune responses. J. Med. Virol. 2020, 92, 424–432. [Google Scholar] [CrossRef]
- Cui, J.; Li, F.; Shi, Z.L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Qiu, Y.; Ge, X. The taxonomy, host range and pathogenicity of coronaviruses and other viruses in the Nidovirales order. Anim. Dis. 2021, 1, 5. [Google Scholar] [CrossRef]
- Su, S.; Wong, G.; Shi, W.; Liu, J.; Lai, A.C.K.; Zhou, J.; Liu, W.; Bi, Y.; Gao, G.F. Epidemiology, Genetic Recombination, and Pathogenesis of Coronaviruses. Trends Microbiol. 2016, 24, 490–502. [Google Scholar] [CrossRef] [PubMed]
- Forni, D.; Cagliani, R.; Clerici, M.; Sironi, M. Molecular Evolution of Human Coronavirus Genomes. Trends Microbiol. 2017, 25, 35–48. [Google Scholar] [CrossRef]
- Lednicky, J.A.; Tagliamonte, M.S.; White, S.K.; Elbadry, M.A.; Alam, M.M.; Stephenson, C.J.; Bonny, T.S.; Loeb, J.C.; Telisma, T.; Chavannes, S.; et al. Independent infections of porcine deltacoronavirus among Haitian children. Nature 2021, 600, 133–137. [Google Scholar] [CrossRef]
- Li, W.; Shi, Z.; Yu, M.; Ren, W.; Smith, C.; Epstein, J.H.; Wang, H.; Crameri, G.; Hu, Z.; Zhang, H.; et al. Bats are natural reservoirs of SARS-like coronaviruses. Science 2005, 310, 676–679. [Google Scholar] [CrossRef] [PubMed]
- Azhar, E.I.; El-Kafrawy, S.A.; Farraj, S.A.; Hassan, A.M.; Al-Saeed, M.S.; Hashem, A.M.; Madani, T.A. Evidence for camel-to-human transmission of MERS coronavirus. N. Engl. J. Med. 2014, 370, 2499–2505. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021, 19, 141–154. [Google Scholar] [CrossRef]
- Lin, C.M.; Saif, L.J.; Marthaler, D.; Wang, Q. Evolution, antigenicity and pathogenicity of global porcine epidemic diarrhea virus strains. Virus Res. 2016, 226, 20–39. [Google Scholar] [CrossRef]
- Turlewicz-Podbielska, H.; Pomorska-Mól, M. Porcine Coronaviruses: Overview of the State of the Art. Virol. Sin. 2021, 36, 833–851. [Google Scholar] [CrossRef]
- Liu, C.; Tang, J.; Ma, Y.; Liang, X.; Yang, Y.; Peng, G.; Qi, Q.; Jiang, S.; Li, J.; Du, L.; et al. Receptor usage and cell entry of porcine epidemic diarrhea coronavirus. J. Virol. 2015, 89, 6121–6125. [Google Scholar] [CrossRef]
- Woo, P.C.; Lau, S.K.; Lam, C.S.; Lau, C.C.; Tsang, A.K.; Lau, J.H.; Bai, R.; Teng, J.L.; Tsang, C.C.; Wang, M.; et al. Discovery of seven novel Mammalian and avian coronaviruses in the genus deltacoronavirus supports bat coronaviruses as the gene source of alphacoronavirus and betacoronavirus and avian coronaviruses as the gene source of gammacoronavirus and deltacoronavirus. J. Virol. 2012, 86, 3995–4008. [Google Scholar] [PubMed]
- Zhou, P.; Fan, H.; Lan, T.; Yang, X.L.; Shi, W.F.; Zhang, W.; Zhu, Y.; Zhang, Y.W.; Xie, Q.M.; Mani, S.; et al. Fatal swine acute diarrhoea syndrome caused by an HKU2-related coronavirus of bat origin. Nature 2018, 556, 255–258. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Huang, W.; He, X.; Feng, Z.; Chen, Q. Research Advances on Swine Acute Diarrhea Syndrome Coronavirus. Animals 2024, 14, 448. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.W.; Dickerman, A.W.; Piñeyro, P.; Li, L.; Fang, L.; Kiehne, R.; Opriessnig, T.; Meng, X.J. Origin, evolution, and genotyping of emergent porcine epidemic diarrhea virus strains in the United States. mBio 2013, 4, e00737-13. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Liu, B.; Yang, J.; Jin, Q. DBatVir: The database of bat-associated viruses. Database 2014, 2014, bau021. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Sun, J.; Li, D.; Wang, N.; Wang, L.; Zhang, C.; Meng, X.; Ji, X.; Suchard, M.A.; Zhang, X.; et al. Emerging viruses: Cross-species transmission of coronaviruses, filoviruses, henipaviruses, and rotaviruses from bats. Cell Rep. 2022, 39, 110969. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Aravena, M.; McKee, C.; Gamble, A.; Lunn, T.; Morris, A.; Snedden, C.E.; Yinda, C.K.; Port, J.R.; Buchholz, D.W.; Yeo, Y.Y.; et al. Ecology, evolution and spillover of coronaviruses from bats. Nat. Rev. Microbiol. 2022, 20, 299–314. [Google Scholar] [CrossRef]
- Irving, A.T.; Ahn, M.; Goh, G.; Anderson, D.E.; Wang, L.F. Lessons from the host defences of bats, a unique viral reservoir. Nature 2021, 589, 363–370. [Google Scholar] [CrossRef]
- Liu, P.; Li, L.; Keane, S.C.; Yang, D.; Leibowitz, J.L.; Giedroc, D.P. Mouse hepatitis virus stem-loop 2 adopts a uYNMG(U)a-like tetraloop structure that is highly functionally tolerant of base substitutions. J. Virol. 2009, 83, 12084–12093. [Google Scholar] [CrossRef] [PubMed]
- Moya, A.; Holmes, E.C.; González-Candelas, F. The population genetics and evolutionary epidemiology of RNA viruses. Nat. Rev. Microbiol. 2004, 2, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Grange, Z.L.; Goldstein, T.; Johnson, C.K.; Anthony, S.; Gilardi, K.; Daszak, P.; Olival, K.J.; O’Rourke, T.; Murray, S.; Olson, S.H.; et al. Ranking the risk of animal-to-human spillover for newly discovered viruses. Proc. Natl. Acad. Sci. USA 2021, 118, e2002324118. [Google Scholar] [CrossRef] [PubMed]
- Stout, A.E.; Millet, J.K.; Stanhope, M.J.; Whittaker, G.R. Furin cleavage sites in the spike proteins of bat and rodent coronaviruses: Implications for virus evolution and zoonotic transfer from rodent species. One Health 2021, 13, 100282. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.Y.; Li, J.L.; Yang, X.L.; Chmura, A.A.; Zhu, G.; Epstein, J.H.; Mazet, J.K.; Hu, B.; Zhang, W.; Peng, C.; et al. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature 2013, 503, 535–538. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Mayer, F.; von Helversen, O. Cryptic diversity in European bats. Proc. Biol. Sci. 2001, 268, 1825–1832. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Masangkay, J.S.; Nagata, N.; Morikawa, S.; Mizutani, T.; Fukushi, S.; Alviola, P.; Omatsu, T.; Ueda, N.; Iha, K.; et al. Bat coronaviruses and experimental infection of bats, the Philippines. Emerg. Infect. Dis. 2010, 16, 1217–1223. [Google Scholar] [CrossRef]
- Zhou, Z.J.; Qiu, Y.; Pu, Y.; Huang, X.; Ge, X.Y. BioAider: An efficient tool for viral genome analysis and its application in tracing SARS-CoV-2 transmission. Sustain. Cities Soc. 2020, 63, 102466. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. Likelihood ratio tests for detecting positive selection and application to primate lysozyme evolution. Mol. Biol. Evol. 1998, 15, 568–573. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wong, W.S.; Nielsen, R. Bayes empirical bayes inference of amino acid sites under positive selection. Mol. Biol. Evol. 2005, 22, 1107–1118. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Fang, B.; Liu, Y.; Cai, M.; Jun, J.; Ma, J.; Bu, D.; Wang, L.; Zhou, P.; Wang, H.; et al. Newly emerged porcine enteric alphacoronavirus in southern China: Identification, origin and evolutionary history analysis. Infect. Genet. Evol. 2018, 62, 179–187. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef]
- Allen, T.; Murray, K.A.; Zambrana-Torrelio, C.; Morse, S.S.; Rondinini, C.; Di Marco, M.; Breit, N.; Olival, K.J.; Daszak, P. Global hotspots and correlates of emerging zoonotic diseases. Nat. Commun. 2017, 8, 1124. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.; Normile, D. New SARS-like virus in China triggers alarm. Science 2020, 367, 234–235. [Google Scholar] [CrossRef] [PubMed]
- Han, B.A.; Schmidt, J.P.; Bowden, S.E.; Drake, J.M. Rodent reservoirs of future zoonotic diseases. Proc. Natl. Acad. Sci. USA 2015, 112, 7039–7044. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Zeng, L.P.; Yang, X.L.; Ge, X.Y.; Zhang, W.; Li, B.; Xie, J.Z.; Shen, X.R.; Zhang, Y.Z.; Wang, N.; et al. Discovery of a rich gene pool of bat SARS-related coronaviruses provides new insights into the origin of SARS coronavirus. PLoS Pathog. 2017, 13, e1006698. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.Y.; Yang, W.H.; Zhou, J.H.; Li, B.; Zhang, W.; Shi, Z.L.; Zhang, Y.Z. Detection of alpha- and betacoronaviruses in rodents from Yunnan, China. Virol. J. 2017, 14, 98. [Google Scholar] [CrossRef] [PubMed]
- Lohrasbi-Nejad, A. Detection of homologous recombination events in SARS-CoV-2. Biotechnol. Lett. 2022, 44, 399–414. [Google Scholar] [CrossRef]
- Li, F. Structure, Function, and Evolution of Coronavirus Spike Proteins. Annu. Rev. Virol. 2016, 3, 237–261. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples Type | Sampling Location | Sample Host | Samples | Positive Samples | Positive Rate (%) |
|---|---|---|---|---|---|
| Bat feces | MH | Hipposideros armiger | 66 | 0 | 0 |
| TC | Rhinolophus sinicus | 344 | 1 (TC-14) | 0.29 | |
| Rat intestine | GS | Apodemus ilex | 74 | 2 (GS-53, GS-56) | 2.7 |
| CoV | ORF | Start-End (Nucleotide Position) | Length (nt) | Length (aa) |
|---|---|---|---|---|
| TC-14 | 1ab | 304–20,489 | 20,186 | 6728 |
| S | 20,486–23,878 | 3393 | 1131 | |
| NS3 | 23,878–24,567 | 690 | 230 | |
| E | 24,548–24,775 | 228 | 76 | |
| M | 24,784–25,470 | 687 | 229 | |
| N | 25,482–26,609 | 1128 | 376 | |
| NS7a | 26,621–26,932 | 312 | 104 | |
| GS-56 | 1ab | 195–21,661 | 21,467 | 7155 |
| NS2a | 21,663–22,493 | 831 | 277 | |
| HE | 22,508–23,782 | 1275 | 425 | |
| S | 23,798–27,874 | 4077 | 1359 | |
| NS4 | 27,967–28,380 | 414 | 138 | |
| NS5 | 28,362–28,676 | 315 | 105 | |
| E | 28,669–28,917 | 249 | 83 | |
| M | 28,932–29,627 | 696 | 232 | |
| N2 | 29,620–30,312 | 693 | 231 | |
| N | 29,637–30,968 | 1332 | 444 |
| CoVs | Gene | ComparedModels | Positive Selected Sites (BEB Results, *: p > 95%, **: p > 99%) |
|---|---|---|---|
| α-CoV | S | M7–M8 | 80T, 86H, 114K, 133R *, 138G *, 281P, 311G, 312S *, 313T, 314L, 331V, 338K |
| ORF1ab | M7–M8 | 112N **, 161R *, 423S **, 450L, 533A, 632V, 1390S, 433T, 2292V *, 3128A | |
| β-CoV | S | M7–M8 | 68S, 364S |
| ORF1ab | M7–M8 | 269T, 1234L, 1475L |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Q.; Wang, D.-S.; Lian, Z.-H.; Fang, J.; Han, P.-Y.; Qiu, Y.; Zhao, J.-Y.; Zong, L.-D.; Zhang, Y.-Z.; Ge, X.-Y. Identification and Characterization of an Alphacoronavirus in Rhinolophus sinicus and a Betacoronavirus in Apodemus ilex in Yunnan, China. Microorganisms 2024, 12, 1490. https://doi.org/10.3390/microorganisms12071490
Liu Q, Wang D-S, Lian Z-H, Fang J, Han P-Y, Qiu Y, Zhao J-Y, Zong L-D, Zhang Y-Z, Ge X-Y. Identification and Characterization of an Alphacoronavirus in Rhinolophus sinicus and a Betacoronavirus in Apodemus ilex in Yunnan, China. Microorganisms. 2024; 12(7):1490. https://doi.org/10.3390/microorganisms12071490
Chicago/Turabian StyleLiu, Qian, Dan-Shu Wang, Zhong-Hao Lian, Jie Fang, Pei-Yu Han, Ye Qiu, Jun-Ying Zhao, Li-Dong Zong, Yun-Zhi Zhang, and Xing-Yi Ge. 2024. "Identification and Characterization of an Alphacoronavirus in Rhinolophus sinicus and a Betacoronavirus in Apodemus ilex in Yunnan, China" Microorganisms 12, no. 7: 1490. https://doi.org/10.3390/microorganisms12071490