Abstract
We previously reported the first-in-human assessment of three doses (2, 5, and 10 µg) of purified inactivated Zika virus vaccine (PIZV or TAK-426) in the Phase 1 ZIK-101 study (NCT03343626). Here, we report dose selection based on extended safety and immunogenicity data (6 months post-vaccination) and discuss considerations (e.g., immunological, historic, flavivirus immunological cross-reactions) for selecting a Zika virus (ZIKV) vaccine dose formulation. TAK-426 dose selection was conducted at the first interim analysis, and was based on cumulative safety data from both flavivirus-naïve (up to ≥28 days post-dose PD2) and flavivirus-primed participants (up to ≥28 days PD1), and on immunogenicity data from flavivirus-naïve participants only (at 28 days PD1 and 28 days PD2). The safety profile from TAK-426 recipients was compared to placebo recipients. Immunogenicity was assessed by geometric mean titer ratios of neutralizing anti-ZIKV antibodies and differences in seroconversion rates. There was no significant difference in safety between the three TAK-426 doses. The 10 μg dose provided the earliest and strongest immune response (with close to 100% seroconversion and higher antibody titers PD1 in flavivirus-naïve participants), and was well tolerated with acceptable safety profiles in both flavivirus-naïve and flavivirus-primed participants; this dose was selected for further development.
1. Introduction
Zika virus (ZIKV) is a single-stranded positive-sense RNA virus that is primarily a mosquito-borne flavivirus (FV). It was first isolated in 1947 from a rhesus monkey in the Zika Forest of Uganda [1,2]. The primary mode of ZIKV transmission is through the bite of infected female mosquitoes, prominently the Aedes aegypti and Aedes albopictus species that have spread globally [3]. Other recognized modes of transmission are mother-to-fetus during pregnancy, mother-to-child during the perinatal period, and sexual contact; few cases have been reported from blood component transfusion [3,4,5,6,7,8,9]. Exposure to ZIKV through laboratory work, solid organ transplants, and body fluids (e.g., tears, urine, breastmilk) are other potential mechanisms of disease transmission; however, in each of these cases, there is limited evidence [10,11,12,13,14,15]. The incubation period between exposure and the development of symptoms associated with ZIKV infection is approximately 3 to 11 days [16,17].
Available data strongly endorse links between ZIKV infection and congenital anomalies (congenital ZIKV syndrome) [18,19], as well as between ZIKV infection and Guillain–Barré syndrome (GBS) [19,20,21,22,23,24]. Congenital ZIKV syndrome is characterized by severe microcephaly, subcortical calcifications, macular scarring, focal pigmentary retinal mottling, congenital contractures, and early hypertonia [25]. GBS is characterized by a short time to onset following infection and a rapid evolution of the disease [26]. The pathogenesis of GBS in association with ZIKV infection is not understood, and the long-term clinical course is unknown.
Because of the low but sustained ZIKV transmission in endemic countries, as well as the risk of reemergence in areas with prior transmission, the development of a Zika vaccine is considered crucial to protect susceptible populations, including women of child-bearing potential living in and travelers visiting Zika-endemic areas [27,28,29,30]. Several platform technologies for ZIKV vaccine development have been employed: deoxyribose nucleic acid vaccines, messenger RNA vaccines, purified inactivated virus vaccines, and adenovirus-based vaccines. Takeda has developed an aluminum hydroxide-adjuvanted purified inactivated Zika virus vaccine (PIZV) candidate, also known as TAK-426, for the target indication of prevention of disease caused by ZIKV. In preclinical testing, TAK-426 was safe and well tolerated in New Zealand white rabbits (data on file), elicited robust immune responses, and protected mice and Indian rhesus macaques in a ZIKV challenge model [31,32].
An inactivated ZIKV vaccine cannot revert to a virulent form that is capable of causing and transmitting disease. Additionally, inactivated vaccines are not contraindicated during pregnancy. An inactivated ZIKV vaccine would be well suited and safer for the control of the current low and sustained transmission of ZIKV in endemic areas.
Immunogenicity data in two mice models (CD-1 and AG-129) and efficacy data in AG-129 mice demonstrated that two doses of TAK-426 formulated with an aluminum hydroxide adjuvant elicited robust immune responses [31]. Two doses of TAK-426, administered 28 days apart, have so far been shown to be well tolerated in over 100 participants with an acceptable safety profile in both FV-naïve and FV-primed healthy adults aged 18–49 years. TAK-426 induced a dose-dependent humoral immune response in both FV-naïve and FV-primed participants [33]. TAK-426 also induced neutralizing antibody (nAb) magnitudes and kinetics that appear to be comparable to those elicited by ZIKV natural infection [34]. Previously, we described the selection of the TAK-426 vaccine dose formulation during Phase 1 studies based on characterization of the immunogenicity dose–response in FV-naïve adults only and characterization of the safety profile in FV-naïve and FV-primed adults who received at least one vaccine dose [33]. We now present and discuss the dose selection with the extended safety follow-up and the TAK-426-induced immunogenicity data 6 months after vaccination with low (2 μg) and medium (5 μg) doses, as well as with a high (10 μg) dose that was selected for further clinical development. We also discuss how historic considerations of other inactivated virus vaccines and flavivirus immunological cross-reactions ought to be considered when selecting a ZIKV vaccine regimen and dose.
2. Materials and Methods
This Phase 1, randomized, observer-blind, placebo-controlled ZIK-101 study was the first trial of TAK-426 in humans. The dose-selection study assessing the safety and immunogenicity of TAK-426 enrolled FV-naïve and FV-primed healthy adults aged 18 to 49 years from the United States and Puerto Rico [33,34]. Due to known cross-reactivity between FVs, FV-naïve and FV-primed participants were sequentially enrolled. The ZIK-101 protocol was approved by the local ethics committee or institutional review board of each study center, registered on ClinicalTrials.gov (NCT03343626), and implemented in accordance with International Council for Harmonisation, Good Clinical Practice guidelines, the Declaration of Helsinki, and applicable local regulatory requirements.
A total of 271 participants were enrolled and randomized (1:1:1:1) into four groups: placebo (saline solution), and 2, 5, or 10 µg of TAK-426. Each dosing group comprised more than 60 participants, with at least 30 participants in the FV-naïve cohort and 30 in the FV-primed cohort. A total of 125 participants were enrolled in the FV-naïve cohort and 146 participants in the FV-primed cohort. All participants were followed for a minimum of 6 months post-dose 2 (PD2) for the assessment of safety and persistence of the vaccine-induced immune response. Although the placebo group and the selected dose group (10 µg) were followed for up to 24 months PD2 for the assessment of the persistence of immunity and long-term safety, only the safety and immunogenicity data 6 months after vaccination were used to discuss dose selection. An independent Data Monitoring Committee had the safety oversight of this study.
Flavivirus antibody status at the time of screening of participants was performed utilizing a Multiplex Luminex® IgG enzyme-linked immunosorbent assay (Luminex Corp, Austin, TX, USA). The assay included multiple relevant flavivirus antigens, which were conjugated to Luminex beads (Table 1).

Table 1.
Antigens included in the initial Multiplex Luminex® assay for detection of flaviviral antibodies (IgG) in serum or plasma samples for the Phase 1 study.
The vaccination regimen consisted of two doses of TAK-426 administered intramuscularly 28 days apart. The primary objectives of this Phase 1 study were as follows: (1) to describe the safety of two doses of TAK-426 with three different antigen dose levels (2, 5, or 10 μg) in FV-naïve and FV-primed healthy adults through 28 days PD2; and (2) to select a single dose level from three different antigen concentrations (2, 5, or 10 µg) of TAK-426 for further clinical development.
The primary rationale for selection of the three vaccine dose levels was as follows: (1) the low-dose level (2 µg) was selected for purposes of potentially identifying an immunological threshold, and was based on the limit of the analytical methods for the drug product available at the time; (2) the mid-dose level (5 µg) was selected to provide a mid-point on the dose–response curve. In addition, this dose level was based on a Phase 1 study of the Walter Reed Army Institute of Research Zika vaccine candidate (ZPIV) [35], which was originally based on initial studies in mice and non-human primates; and (3) the high-dose level (10 µg) was selected to follow an approximate 2-fold range and to further define the potential dose–response curve in both FV-naïve and FV-primed participants.
2.1. Safety Assessments
Safety assessments for the ZIK-101 study have been previously described [33,34]. Following earlier assessments of solicited local and systemic AEs (7 days after each dose) and unsolicited AEs (28 days after each dose) [33], serious AEs (SAEs) and new medical conditions (including neurological and neuroinflammatory disorders) were reported through the 2-year follow-up [34].
2.2. Acceptability Assessment of the Safety Profile
The acceptability of the safety profile was based on the comparison to the placebo (saline) group. In addition, marketed inactivated aluminum hydroxide-adjuvanted vaccines (the most relevant being FV vaccines) were also used for comparison, particularly for the injection site reactions (local reactogenicity) [36,37,38,39]. The acceptability of the safety profile (PD1 and PD2) was determined not only based on the frequency of the relevant safety parameters (solicited events, unsolicited events, and safety laboratory parameters), but also on other parameters that characterized the AE(s), such as nature, latency, duration, outcome, potential impact on public health or the healthcare system (e.g., ER visits, hospitalizations), safety laboratory parameters, and change from baseline. Any increase in reactogenicity PD2 also impacted the assessment of the safety profile of the vaccine. In case any related SAE and/or any SAE leading to withdrawal of the investigational medicinal product occurred, the acceptability of the safety profile of a vaccine dose relied on the detailed analyses of such (S)AEs. If it appeared that such events were segregated to one vaccine dose level with unclear causality to the study vaccine, then there may not be an acceptable safety profile at that dose level.
2.3. Immunogenicity Assessments
To evaluate post-vaccination immune responses, serum samples for all participants were obtained at baseline (day 1) and at 1, 2, and 8 months. Anti-ZIKV nAb levels were measured using a qualified plaque reduction neutralization test (PRNT) at Q2 Solutions (San Juan Capistrano, CA, USA) The Zika PRNT test had a lower limit of detection of 1:10 dilution and a lower limit of quantitation of 26 (reciprocal dilution) [33,34]. ZIK-101 serum ZIKV NAb levels were also measured using a fit-for-purpose ZIKV reporter virus particle (RVP) assay, developed and performed in the Takeda Laboratory in Cambridge, Massachusetts, with a lower limit of quantitation of 105, which was used as the threshold to define seropositivity or seronegativity.
The assessment of TAK-426 immunogenicity data from FV-naïve and FV-primed participants occurred at 28 days PD1, 28 days PD2, and 6 months PD2.
2.4. Dose Selection (Phase 1 Interim Analysis)
As previously described [33], dose selection was based on the magnitude of the immune response in FV-naïve participants, as measured by the ratios of GMTs of anti-ZIKV nAbs and differences in the seroconversion rate between the dosing groups, as well as safety outcome measures of FV-naïve and FV-primed participants. The interim analyses were performed by a separate set of unblinded statisticians and programmers at a Clinical Research Organization, who had access to individual treatment assignments. All personnel involved in the conduct of the trial, including those at Takeda, the Clinical Research Organization, and the trial sites, remained blinded to the individual participant treatment assignment. The study team had access to the group level unblinded results only. The preliminary TAK-426 efficacy (extrapolated from preclinical anti-ZIKV nAbs associated with protection against a ZIKV challenge in non-human primates [rhesus macaques]) was also taken into consideration as supplemental information in dose selection [40].
The final dose-selection decision was made following the pre-specified statistical analysis plan and its consistency with one of the four different qualitative scenarios described in Table 2.
Table 2.
Decision-making scenarios for dose selection.
2.5. Statistical Analysis
The sample size was not determined by formal statistical power calculations. However, stochastic simulations (with 1 million simulation runs) suggested that 60 participants per group were adequate to select one of the three tested doses by the ratios of geometric mean titers (GMTs) between the dosing groups [33]. With 60 participants in each group, the probability of observing a common adverse event (AE) (10% true rate) was over 80%. Safety assessments were performed on all randomized participants who received ≥1 dose of vaccine or placebo (safety set). Safety endpoints are summarized descriptively with frequency and percentage for categorical data. The number and percentage of participants with at least 1 solicited local and systemic AE are reported. For participants with more than 1 episode of the same event, the maximum severity is used for tabulations. Unsolicited AEs, up to 28 days after each injection, are coded using MedDRA and summarized by SOC and PT by trial arm. Safety summaries are provided overall and by severity (solicited AEs). Immunogenicity assessments were based on the per-protocol set, comprising all participants with no major protocol violations who received ≥1 dose of the investigational vaccine or placebo and provided valid baseline serology and ≥1 postvaccination time point. Anti-ZIKV antibody levels were compared between the dosing groups using GMTs ratios and seroconversion rate (SCR) differences. The point estimates of GMT ratios ≥2 were considered meaningful to differentiate the immunogenicity in terms of GMTs between different dose levels. Seroconversion was defined as seronegative participants at baseline who became seropositive after vaccination or seropositive participants at baseline with ≥4-fold increases in GMTs. For the calculation of GMTs, seronegative samples were assigned a titer of 5 (half of the limit of detection), and the 95% confidence intervals (CIs) were calculated using the exact Clopper–Pearson method for each dosing group and time point. ANOVA analysis of immunogenicity as measured by the pairwise ratios of GMT of anti-ZIKV nAbs were also performed. The distribution of neutralization titers of each dose level was also evaluated using reverse cumulative distribution curves (RCDCs), and comparisons were made based on individuals reaching no less than 70% of high anti-ZIKV nAb titers. We used SAS, version 9.2, for statistical analyses.
3. Results
FV-naïve cohort (n = 125) PD1 and PD2 safety data as well as FV-primed cohort (n = 146) PD1 data only were used to select the TAK-426 dose for further vaccine development. This early selection based on PD1 data allowed for the initiation of manufacturing Phase 2 clinical trial materials. Demographic characteristics and disposition of participants of both cohorts were reported previously [33]. Notably, no TAK-426-related SAEs were reported in any FV cohort.
3.1. FV-Naïve Cohort
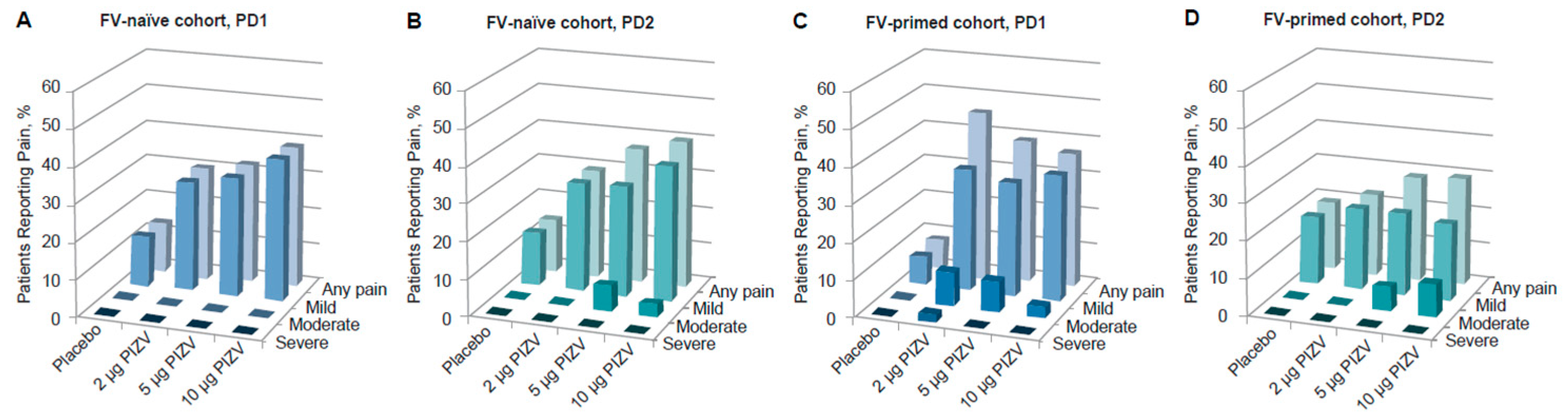
Solicited local reactions and systemic AEs occurring during the 7 days after each dose were previously reported [33]. Overall, the reporting rates of the total solicited AEs (local and systemic after both doses) in the TAK-426 groups were comparable to those reported in the placebo group. While solicited local AE rates were higher among those vaccinated with TAK-426 as compared to the placebo group, systemic AE rates were higher among placebo recipients (as compared to TAK-426 vaccinees). Pain at the injection site was the most frequent solicited AE, with no significant difference between doses, but slightly higher pain incidence rates were reported with increasing TAK-426 dose levels after the first and second doses. TAK-426 10 µg induced pain reactions were not severe (Grade 3, as defined by the FDA Toxicity Grading Scale) (Figure 1). The only Grade 3 (severe) solicited AE, fever, was reported in the placebo group PD2 (Table 3). Unsolicited AEs were higher among placebo recipients, with the lowest rate reported by those receiving the 10 µg TAK-426 dose (19% vs. 42% in the placebo group).
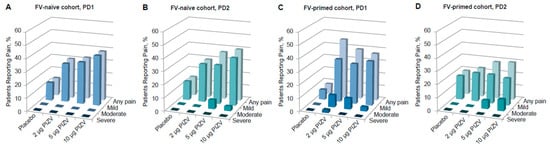
Figure 1.
Incidence of solicited local pain in the FV-naïve cohort PD1 (A) and PD2 (B), and in the FV-primed cohort PD1 (C) and PD2 (D). FV: flavivirus, PIZV: purified inactivated Zika virus vaccine, PD: post-dose.
Table 3.
Total number of Grade 3 (severe) solicited and unsolicited AEs by cohort (FV-naïve or FV-primed) and TAK-426 dose.
3.2. FV-Primed Cohort
Solicited local reactions and systemic AEs occurring during the 7 days PD1 of TAK-426 were reported previously [33]. In summary, the reporting rates of the total solicited AEs (local and systemic after both doses) in the TAK-426 groups were higher than those reported in the placebo group. PD1, the reporting rates of solicited local AEs were higher in the TAK-426 groups than in the placebo group (45%, 42%, and 37% in the 2 µg, 5 µg, and 10 µg TAK-426 groups, respectively, vs. 14% in the placebo group). A similar reporting pattern was observed PD2, albeit with lower rates, as compared to PD1 in all TAK-426 groups (22%, 31%, and 32% in the 2 µg, 5 µg, and 10 µg TAK-426 groups, respectively, vs. 18% in the placebo group). Systemic AE rates were comparable or lower than placebo PD1 (24%, 36%, and 37% in the 2 µg, 5 µg, and 10 µg TAK-426 groups, respectively, vs. 33% in the placebo group) but higher than or comparable to placebo PD2 (19%, 31%, and 21% in the 2 µg, 5 µg, and 10 µg TAK-426 groups, respectively, vs. 21% in the placebo group). Like the FV-naïve cohort, previously FV-exposed individuals reported fewer unsolicited AEs in the TAK-426 groups (27%, 30%, and 22%, respectively) as compared to the placebo (31%), with the lowest rates reported by those receiving the 10 µg TAK-426 dose (22%).
Pain at the injection site was the most frequent solicited AE reported in both FV-naïve and FV-primed cohorts. Most of these events were reports of mild to moderate pain at the injection site, with onset on day 1 and with a mean duration of less than 2 days. There was one pain case only, in a FV-primed participant, described as severe following the first dose of TAK-426 2 µg (see Table 3). As shown in Figure 1, among FV-naïve participants, the reported rates of pain after any dose were slightly higher in those receiving the TAK-426 10 µg dose as compared to 2 µg and 5 µg; the opposite was reported among FV-primed individuals following the first dose, whereby the lowest rate was reported in those receiving the highest dose.
A total of 22 Grade 3 (severe) solicited and unsolicited AEs were reported, with most (20 out of 22) reported by FV-primed participants. Most Grade 3 AEs were reported among those receiving TAK-426 5 µg (n = 10), followed by the 2 µg (n = 6), 10 µg (n = 4), and placebo (n = 2) groups. Details on the Grade 3 AEs by dose are provided in Table 3. No deaths and no Grade 4 AEs were reported in the FV-naïve or the FV-primed cohorts. There were no safety laboratory changes of concern at 7 days post-vaccination in any TAK-426 group in either cohort. Most blood chemistry and hematology values remained within normal limits or were not above Grade 1. No participants in the FV-naïve cohort and two participants in the FV-primed cohort had Grade 4 prothrombin time (>1.25 upper limit of normal). Of the two participants with Grade 4 prothrombin time (PT), one participant received TAK-426 (5 µg) and the other participant received placebo. PTs of both participants were within normal ranges on day 57.
In summary, among participants previously exposed to FVs, there were some apparent differences in the safety profile between doses, favoring TAK-426 10 µg; the lower the TAK-426 dose, the higher the rates of solicited AEs PD2 and unsolicited AEs after any dose. Similarly, the lower the dose, the higher the number of Grade 3 (severe) solicited AEs, which occurred more often in FV-primed than in FV-naïve participants.
3.3. TAK-426-Induced Immune Responses
TAK-426 was immunogenic in both FV-naïve and FV-primed adults aged 18 to 49 years. TAK-426 2 µg and 5 µg recipients were followed up to 6 months PD2, and TAK-426 10 µg recipients up to 24 months PD2, as previously reported [34]. Briefly, TAK-426 induced a dose-dependent humoral immune response in both FV-naïve and FV-primed participants. TAK-426-induced ZIKV nAbs persisted for up to at least 24 months among FV-naïve individuals and up to at least 12 months in FV-primed individuals. Anti-ZIKV nAb titers declined after 6 months in both cohorts. While FV-naïve ZIKV nAbs reached a steady state from 12 to 24 months PD2, FV-primed ZIKV nAbs continued dropping to levels similar to those in the placebo group (95% CIs overlapped). FV-naïve seropositivity rates (SPRs) and SCRs were high (>94%) and remained elevated up to 24 months PD2. FV-primed SPRs remained at 100% up to 12 months and declined to 76% by 24 months PD2.
3.4. FV-Naïve TAK-426-Induced nAbs Comparison (2 µg, 5 µg, and 10 µg)
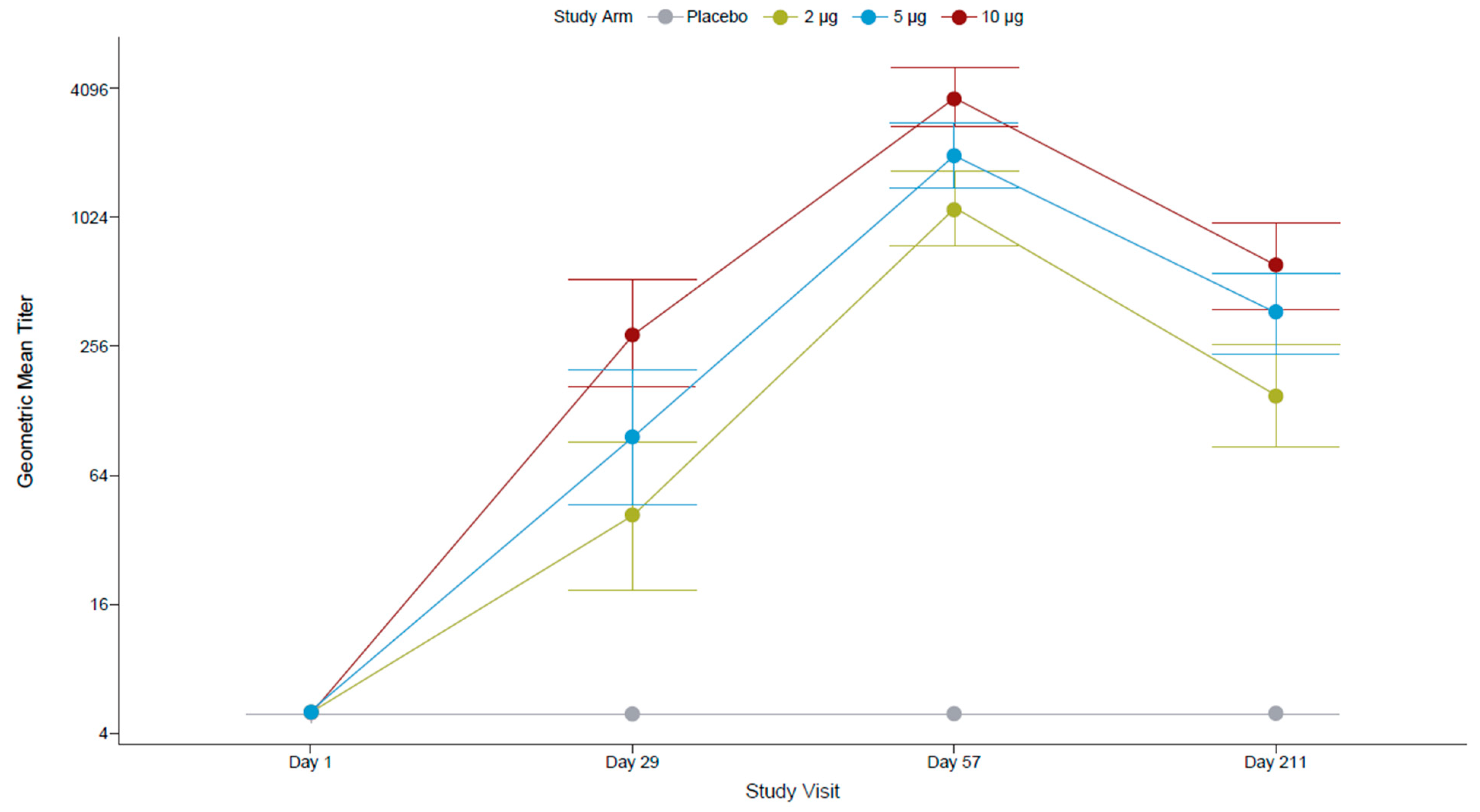
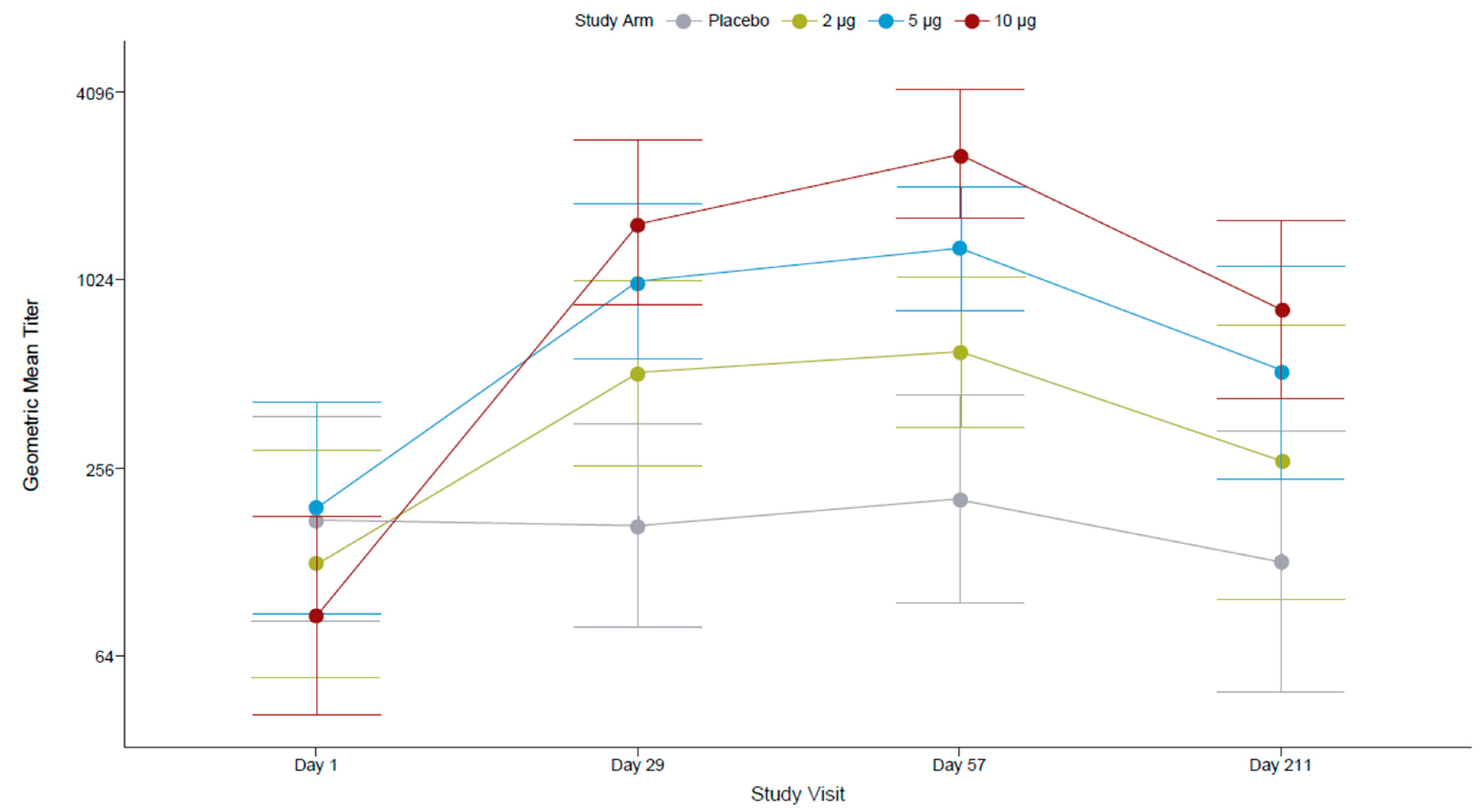
All FV-naïve participants at screening were seronegative for antibodies against FVs (IgG ELISA assessing 12 FV antigens) and for anti-ZIKV nAbs (by PRNT). After the first dose, most participants (96.4%) who had received the 10 µg TAK-426 dose had seroconverted (titer ≥ 10), compared to 82.1% in the 5 µg TAK-426 group and 72.0% in the 2 µg TAK-426 group. The second dose increased the GMTs by over 10 times in all TAK-426 groups (Figure 2). The placebo group remained seronegative throughout the study.
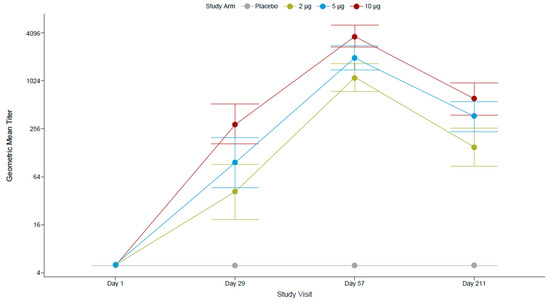
Figure 2.
TAK-426-induced nAbs in the FV-naïve cohort by TAK-426 dose (2 µg, 5 µg, and 10 µg). GMTs (PRNT) and 95% CIs are reported at baseline, PD1 (day 29), 28 days PD2 (day 57), and 6 months PD2 (day 211) for the per-protocol set. FV: flavivirus, GMT: geometric mean titer, PD: post-dose, PRNT: plaque reduction neutralization test.
Pairwise comparison of PRNT GMTs (Table 4) showed no statistically significant differences between TAK-426 groups PD1, whereas at 1 month PD2, the GMT of the 10 µg TAK-426 group was significantly higher than the GMT of the 2 µg TAK-426 group (3.27 times higher; p < 0.001) and the GMT of the 5 µg TAK-426 group (1.85 times higher; p = 0.012); the GMT of the 5 µg TAK-426 group was significantly higher than the GMT of the 2 µg TAK-426 group (1.76 times higher; p = 0.027). The 95% CIs around the GMTs did not overlap between the 2 µg and 10 µg TAK-426 groups at either time point. Comparable pairwise results were observed with the reporter virus particle (RVP) assay.
Table 4.
GMT ratios (by PRNT and RVP) in the FV-naïve cohort (per-protocol set).
Anti-ZIKV nAb titers declined after 6 months (a reduction of 87%, 82%, and 84% in the 2 µg, 5 µg, and 10 µg TAK-426 groups, respectively), though SCRs remained at 100% in all groups. Similar to GMT ratios at 1 month PD2, at 6 months, the GMT of the 10 µg TAK-426 group was significantly higher than the GMT of the 2 µg TAK-426 group (GMT ratio: 3.9; 95% CI: 2.2–7.1; p = 0.0004) and 2-fold higher than the GMT of the 5 µg TAK-426 group (GMT ratio: 1.9; 95% CI: 1.1–3.4; p = 0.1160).
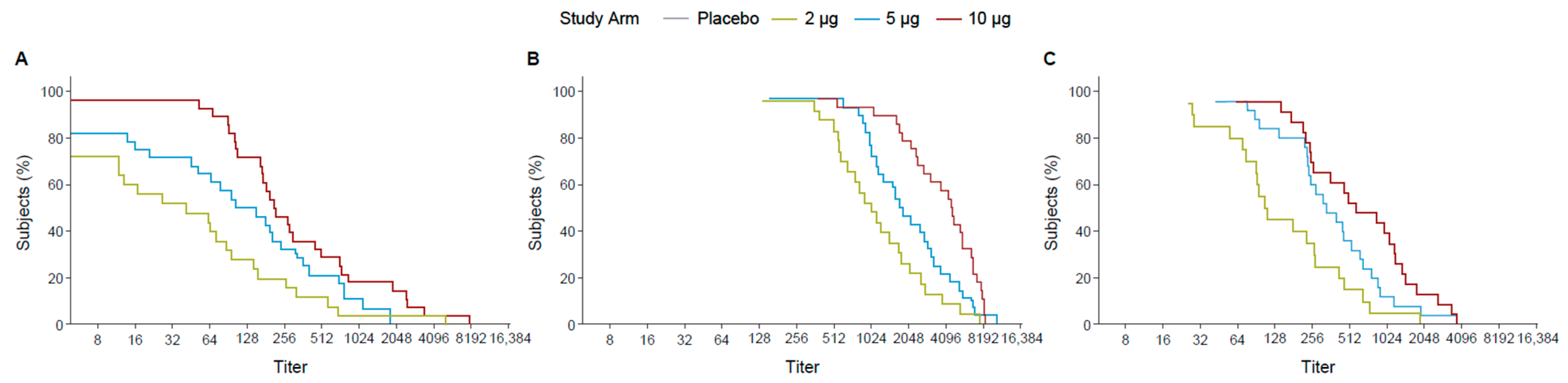
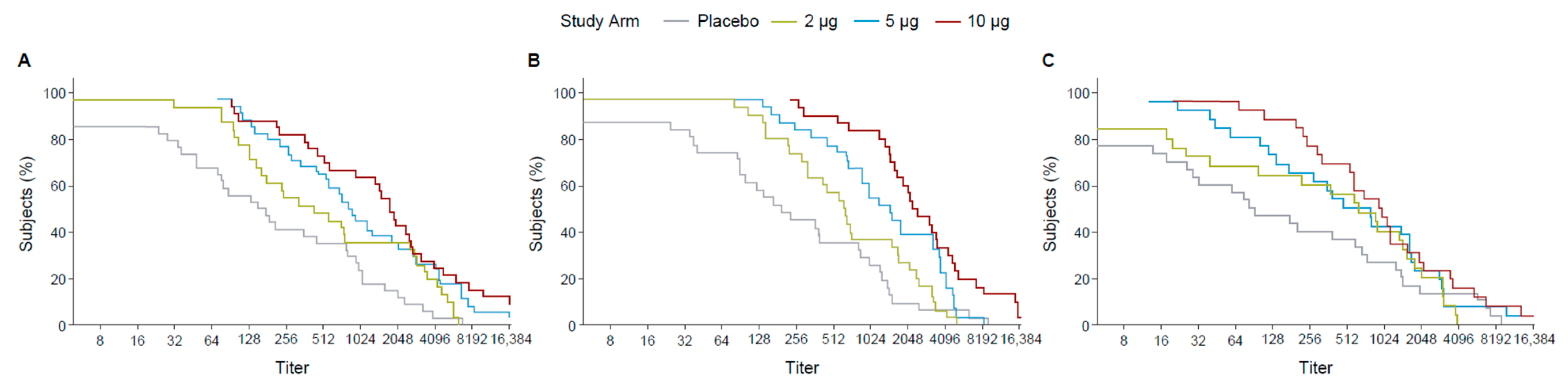
Following the first dose, the 10 µg TAK-426 group had 70% of participants with titers ≥ 128 while the 5 µg TAK-426 and the 2 µg TAK-426 groups had 70% of the participants with titers ≥ 64 and ≥16, respectively. The difference was greater at 2 months PD2, where 70% of the participants had titers ≥ 2048, ≥1024, and ≥512 in the 10, 5, and 2 µg groups, respectively. After 6 months following the second dose, the 10 µg and the 5 µg TAK-426 groups had similar antibody levels with 70% of the participants having titers ≥ 256, while titers in the 2 µg group dropped to ≥64 (Figure 3).
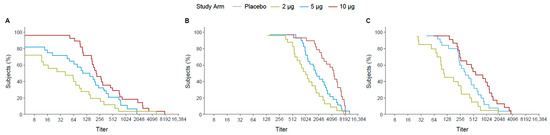
Figure 3.
RCDCs of FV-naïve ZIKV nAbs 28 days PD1 (day 29) (A), 28 days PD2 (day 57) (B), and 6 months PD2 (day 211) (C). FV: flavivirus, PD: post-dose, PRNT: plaque reduction neutralization test, RCDC: reverse cumulative distribution curve.
3.5. FV-Primed TAK-426-Induced nAbs Comparison (2 µg, 5 µg, and 10 µg)
All FV-primed participants at screening were seropositive for antibodies against FVs (IgG ELISA assessing 12 FV antigens), and most (81.3%) had anti-ZIKV nAbs (PRNT). After the first dose, 100% of participants who received the 5 µg and the 10 µg TAK-426 dose, and 97% who received the 2 µg dose had nAbs against ZIKV. The highest seroconversion rates of PD1 and PD2 were reported for the 10 µg TAK-426 group (70% and 77%, respectively), as compared to the 5 µg (50% for PD1 and 55% for PD2) and 2 µg (39% for PD1 and 47% for PD2) groups. A limited number of participants in the placebo group seroconverted during the follow-up (3% PD1 and 6% PD2).
TAK-426 10 µg dose 1 (day 29) elicited the highest (21-fold) increase from baseline ZIKV nAb titers as compared to the 2 µg (4-fold) and the 5 µg (5-fold) groups. The magnitude of the increase in GMTs PD2 was <2-fold (1.6 for 10 µg, 1.3 for 5 µg, 1.2 for 2 µg) as compared to the increase in GMTs PD1 (≥4-fold). While the 95% CIs around the GMTs overlapped between all TAK-426 dosing groups at 28 days PD1, they did not overlap between the 2 µg and 10 µg TAK-426 groups at 28 days PD2 (Figure 4).
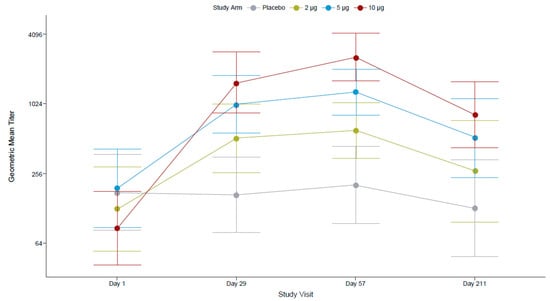
Figure 4.
TAK-426-induced nAbs in the FV-primed cohort by TAK-426 dose (2 µg, 5 µg, and 10 µg). GMTs (PRNT) and 95% CIs are reported at baseline, PD1 (day 29), 28 days PD2 (day 57), and 6 months PD2 (day 211) for the per-protocol set. FV: flavivirus, GMT: geometric mean titer, PD: post-dose.
In the 10 µg TAK-426 group, GMTs decreased at 6 months PD2 (866.26, 95% CI: 445.59–1684.08) as compared to 28 days PD2 (2590.54, 95% CI: 1649.18–4069.22). However, they remained 11-fold higher than at baseline (73.46, 95% CI: 35.59–151.62). The fold increase from baseline to 6 months PD2 was 2 for the 2 µg group and 3 for 5 µg group; no fold increase was observed in the placebo group. The decline in anti-ZIKV nAb titers at 6 months PD2 was 57%, 61%, and 67% in the 2 µg, 5 µg, and 10 µg TAK-426 groups, respectively.
The GMTs overlapped between the 5 µg and 10 µg TAK-426 groups, suggesting they were comparable at either time point up to 6 months PD2 (GMT ratios ≤ 2). The GMT ratios between the 2 µg and 10 µg TAK-426 groups were 3.1 (95% CI: 1.3–7.1; p = 0.01) at 28 days PD1, 3.7 (95% CI: 2.0–6.8; p < 0.0001) at 28 days PD2, and 2.2 (95% CI: 1.1, 4.8; p < 0.05) at 6 months PD2.
The RCDCs indicated that across all dose groups at three time points (1 month PD1, 1 month PD2, and 6 months PD2), 70% of the participants achieved titers ≥ 256. The curves overlapped for titers ≥ 512 (Figure 5).
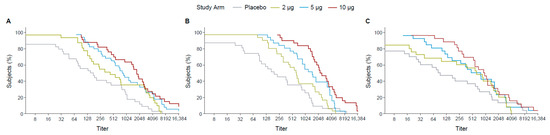
Figure 5.
RCDCs of FV-primed ZIKV nAbs at 28 days PD1 (day 29) (A), 28 days PD2 (day 57) (B), and 6 months PD2 (day 211) (C). FV: flavivirus, PD: post-dose, PRNT: plaque reduction neutralization test, RCDC: reverse cumulative distribution curve.
4. Discussion
Phase 1 safety and immunogenicity data supported the selection of two doses of TAK-426 10 µg administered 28 days apart for further clinical development. The dose selection was based mainly on data from FV-naïve participants and was reached following execution of a pre-specified interim analysis plan that generated data meeting pre-defined safety and immunogenicity criteria consistent with decision-making scenario 1 described in Table 2. The key decision driver was TAK-426-induced humoral immune response differentiation, leading to the selection of the highest antigen content dose (10 µg). The Phase 1 dose-selection decision was supported by safety and immunogenicity data, as well as by predicted efficacy data [40], up to 6 months PD2 in the FV-naïve cohort.
One limitation of our early dose selection is that most of the FV-primed safety and immunogenicity data were not included (as per protocol). These FV-primed data may fit best with the second decision-making scenario (Table 2) in which the major decision driver would be safety, because, as compared to FV-naïve participants, FV-primed adults reported more severe solicited systemic AEs than the placebo group. Among FV-primed participants, there were more Grade 3 (severe) solicited systemic events PD2 (10 events) than after the first dose (four events). Most FV-primed adults were residents in Puerto Rico and therefore most likely had been exposed to dengue virus (DENV) prior to, during, and/or after trial enrollment. DENV cross-reactive immune responses might have contributed to such apparent AE increases. FV-primed data seemed to indicate that higher doses of the TAK-426 antigen led to less severe AEs, supporting the early selection of the 10 µg antigen content.
The FV-primed data also suggested that a single TAK-426 5 µg or 10 µg dose would suffice since they both induced comparable ZIKV nAb titers 28 days PD1, in which case a second dose would not be required. Indeed, a single 5 µg dose of another Zika-inactivated vaccine (ZPIV), albeit less immunogenic than TAK-426, was sufficient to elicit cross-reactive neutralizing antibodies against ZIKV and DENV in a participant with prior DENV exposure [41]. TAK-426 5 µg could induce fewer severe AE cases as compared to a single dose of 2 µg (Table 3 and scenario 4 in Table 2). A single TAK-426 5 µg dose, administered to FV-primed individuals (i.e., participants with prior DENV natural infection), would have a similar safety profile as a single TAK-426 10 µg dose with some manufacturing advantages and less cost. However, the benefit of a booster dose to ensure long-term protection, if not achieved through natural exposure, should also be explored.
Injection-site pain was the most frequently reported solicited AE during the ZIK-101 study. Participants reported pain more frequently than those who received other licensed FV vaccines such as YF-VAX® (<5% in uncontrolled clinical trials) [38], TICOVAC (13.2% in adults 16-65 years old) [39], and IXIARO (>25% in adults > 18 years-old) [37]. Pain was reported less frequently in the ZIK-101 study compared to another trial of a Zika-inactivated candidate vaccine (ZPIV) [42].
Two precedents challenge our ZIKV dose-selection approach assumption that the human dose–response to an inactivated ZIKV vaccine is saturating. Firstly, past studies of virus-inactivated vaccines have shown that low-antigen content candidate vaccines developed for mycoplasma pneumoniae [43] and for other infectious diseases (e.g., Japanese encephalitis, Rocky Mountain spotted fever, typhus, and lymphocytic choriomeningitis) [44] led to vaccine-associated enhancement of the target disease (homotypic vaccine-associated enhanced disease [VAED]) in humans and animal models. This increase in homotypic VAED rates was also reported for high-antigen content candidate vaccines developed to prevent trachoma [45], respiratory syncytial virus [46,47], and measles virus [48]. So far, very limited published data support the occurrence of homotypic ZIKV immune enhancement [49,50]. In contrast, heterotypic FV-immune enhancement has been reported in humans for DENV [51,52,53], ZIKV [54], and West Nile virus [55]. Heterotypic VAED has also been observed with an inactivated dengue vaccine candidate followed by a live-attenuated booster [56] and a licensed attenuated dengue vaccine [57]. Secondly, past dengue studies have demonstrated that a range of FV antibody concentration (dose-dependent in vaccinees) is a key factor predicting dengue disease enhancement [53]. Dengue disease enhancement has been studied in humans [58], non-human primates [59], in vitro experiments [60,61], and molecular simulations [62]. It remains unknown if TAK-426 could induce homotypic or heterotopic VAED. These two precedents indicate that a peaked dose–response curve model may best fit the TAK-426-induced immune responses, as seen in HIV [63], influenza [64], and malaria [65] vaccines. If so, a ZIKV vaccine candidate selection should not be based purely on high antibody responses but also on the likelihood of causing VAED. Although the ZIK-101 study was not designed or powered to determine safety differences across dosages, the higher total number of severe solicited AEs observed in the 2 µg dose group (3 AEs) compared to the 5 µg and 10 µg dose groups (1 AE in each dose group) of FV-primed participants PD1 could be an early indicator of a potential low-dose VAED.
In the case of Zika, two recent FV cross-reaction events are also to be factored in when developing and selecting the regimen and the dose for any ZIKV vaccine: the observed increase in the risk of severe dengue disease after natural ZIKV infection [54], and the lower Zika risk in dengue-seropositive recipients of a dengue vaccine [66]. When planning future TAK-426 development steps, FV immunological cross-reactions (in particular those that enhance or protect against DENV or ZIKV) are to be leveraged.
Zika antigen content (dose) is therefore a critical PIZV modifiable vaccine attribute likely to predict a better safety profile (i.e., less likely to induce homotypic and/or heterotopic VAED). The tendency to select a vaccine candidate based purely on high antibody responses may have been due to the association in early studies of low-antigen content with enhanced disease; for instance, when it was noted in a publication for the polio vaccine that “with a low antigen killed vaccine you stand the danger of actually doing more harm than good” [44]. Caution should be exercised now that fractional doses of licensed vaccines are recommended and being used [67]. The other critical safety-related modifiable vaccine attribute is the actual inactivation manufacturing process. Such inactivation processes should preserve the epitopes necessary to induce an optimal (e.g., neither the lowest nor the highest) immune response. With the current worldwide use of inactivated vaccines to protect millions of people against several infectious diseases, and with the accumulated TAK-426 data and learnings from the ZIK-101 study, we are cautiously optimistic and more confident on the next steps toward the development of an inactivated vaccine against ZIKV.
Author Contributions
Conceptualization, C.J.A., F.N., E.K., K.J.M., P.K. and K.H.; Methodology, F.N. and C.J.A.; Formal Analysis, F.N. and C.J.A.; Writing—Original Draft Preparation, C.J.A. and E.K.; Writing—Review and Editing, C.J.A., F.N., E.K., K.J.M., P.K. and K.H. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by Federal funds from the US Department of Health and Human Services (HHS); Administration for Strategic Preparedness and Response (ASPR); Biomedical Advanced Research and Development Authority (BARDA), under Contract No. HHSO100201600015C). Any opinions, findings, and conclusions expressed in this material are those of the authors and do not necessarily reflect the views of US HHS, ASPR, or BARDA. Medical Writing and Editorial support were provided by Excel Scientific Solutions, and funded by Takeda. Funding to pay the Open Access publication charges for this article was provided by Takeda Vaccines.
Data Availability Statement
The datasets from the clinical study NCT03343626, will be made available within 3 months from initial request to researchers who provide a methodologically sound proposal. The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request. The data will be provided after its de-identification, in compliance with applicable privacy laws, data protection, and requirements for consent and anonymization.
Acknowledgments
This study was sponsored by Takeda. We wish to thank all the volunteers who participated in this study and the study staff at the different centers. The following members of the Takeda team made notable contributions to the manufacture of the Zika vaccine candidate, the study design, and interpretation of data: Gary Dubin, Joseph Santangelo, Htay-Htay Han, Filippo Pacciarini, Vincent Mwangi, Harold Sofield, Laurence De Moerlooze, Elke Walter, Hansi Dean, Holli Giebler, Jamie Gifford, Sushma Kommareddy, Jill Livengood, Mark Lyons, Jackie Marks, Masafumi Misaki, Nao Ogasawara, Hetal Patel, Tatsuki Satou, Asae Shintani.
Conflicts of Interest
C.J.A., F.N., E.K., K.J.M., P.K. and K.H. are employees of Takeda and hold stock/stock options in Takeda.
References
- Dick, G.W. Zika virus (II). Pathogenicity and physical properties. Trans. R. Soc. Trop. Med. Hyg. 1952, 46, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Dick, G.W.; Kitchen, S.F.; Haddow, A.J. Zika virus. I. Isolations and serological specificity. Trans. R. Soc. Trop. Med. Hyg. 1952, 46, 509–520. [Google Scholar] [CrossRef] [PubMed]
- European Centre for Disease Prevention and Control. Rapid Risk Assessment. Zika Virus Disease Epidemic, Tenth Update. 4 April 2017. Available online: https://ecdc.europa.eu/sites/portal/files/media/en/publications/Publications/21-03-2017-RRA%20UPDATE%209-Zika%20virus-Americas%2C%20Caribbean%2C%20Oceania%2C%20Asia.pdf (accessed on 24 January 2023).
- Hamer, D.H.; Wilson, M.E.; Jean, J.; Chen, L.H. Epidemiology, prevention, and potential future treatments of sexually transmitted Zika virus infection. Curr. Infect. Dis. Rep. 2017, 19, 16. [Google Scholar] [CrossRef] [PubMed]
- Basu, R.; Tumban, E. Zika virus on a spreading spree: What we now know that was unknown in the 1950’s. Virol. J. 2016, 13, 165. [Google Scholar] [CrossRef] [PubMed]
- Paixao, E.S.; Barreto, F.; Teixeira Mda, G.; Costa Mda, C.; Rodrigues, L.C. History, epidemiology, and clinical manifestations of Zika: A systematic review. Am. J. Public Health 2016, 106, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Plourde, A.R.; Bloch, E.M. A literature review of Zika virus. Emerg. Infect. Dis. 2016, 22, 1185–1192. [Google Scholar] [CrossRef] [PubMed]
- Moreira, J.; Peixoto, T.M.; Siqueira, A.M.; Lamas, C.C. Sexually acquired Zika virus: A systematic review. Clin. Microbiol. Infect. 2017, 23, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Magnus, M.M.; Esposito, D.L.A.; Costa, V.A.D.; Melo, P.S.; Costa-Lima, C.; Fonseca, B.; Addas-Carvalho, M. Risk of Zika virus transmission by blood donations in Brazil. Hematol. Transfus. Cell Ther. 2018, 40, 250–254. [Google Scholar] [CrossRef] [PubMed]
- Gregory, C.J.; Oduyebo, T.; Brault, A.C.; Brooks, J.T.; Chung, K.W.; Hills, S.; Kuehnert, M.J.; Mead, P.; Meaney-Delman, D.; Rabe, I.; et al. Modes of transmission of Zika virus. J. Infect. Dis. 2017, 216, S875–S883. [Google Scholar] [CrossRef]
- Tan, J.J.L.; Balne, P.K.; Leo, Y.S.; Tong, L.; Ng, L.F.P.; Agrawal, R. Persistence of Zika virus in conjunctival fluid of convalescence patients. Sci. Rep. 2017, 7, 11194. [Google Scholar] [CrossRef]
- Baud, D.; Gubler, D.J.; Schaub, B.; Lanteri, M.C.; Musso, D. An update on Zika virus infection. Lancet 2017, 390, 2099–2109. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Zika Transmission. Available online: https://www.cdc.gov/zika/prevention/transmission-methods.html (accessed on 24 January 2023).
- Hemachudha, P.; Wacharapluesadee, S.; Buathong, R.; Petcharat, S.; Bunprakob, S.; Ruchiseesarod, C.; Roeksomtawin, P.; Hemachudha, T. Lack of transmission of Zika virus infection to breastfed infant. Clin. Med. Insights Case Rep. 2019, 12, 1179547619835179. [Google Scholar] [CrossRef] [PubMed]
- Sampieri, C.L.; Montero, H. Breastfeeding in the time of Zika: A systematic literature review. Peer J. 2019, 7, e6452. [Google Scholar] [CrossRef] [PubMed]
- Zanluca, C.; Dos Santos, C.N. Zika virus—An overview. Microbes Infect. 2016, 18, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Musso, D.; Bossin, H.; Mallet, H.P.; Besnard, M.; Broult, J.; Baudouin, L.; Levi, J.E.; Sabino, E.C.; Ghawche, F.; Lanteri, M.C.; et al. Zika virus in French Polynesia 2013-14: Anatomy of a completed outbreak. Lancet Infect. Dis. 2018, 18, e172–e182. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Congenital Zika syndrome & Other Birth Defects. Available online: https://www.cdc.gov/pregnancy/zika/testing-follow-up/zika-syndrome-birth-defects.html (accessed on 24 January 2023).
- Pan American Health Organization. Zika—Epidemiological Update. 27 April 2017. Available online: https://www.paho.org/en/documents/27-april-2017-zika-epidemiological-update-0 (accessed on 5 March 2024).
- Oehler, E.; Watrin, L.; Larre, P.; Leparc-Goffart, I.; Lastere, S.; Valour, F.; Baudouin, L.; Mallet, H.; Musso, D.; Ghawche, F. Zika virus infection complicated by Guillain-Barre syndrome--case report, French Polynesia, December 2013. Eurosurveillance 2014, 19, 20720. [Google Scholar] [CrossRef] [PubMed]
- Araujo, L.M.; Ferreira, M.L.; Nascimento, O.J. Guillain-Barre syndrome associated with the Zika virus outbreak in Brazil. Arq. Neuropsiquiatr. 2016, 74, 253–255. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Situation Report: Zika Virus, Microcephaly, Guillain-Barré Syndrome. Available online: http://apps.who.int/iris/bitstream/handle/10665/251905/zikasitrep8Dec2016-eng.pdf?sequence=1 (accessed on 5 March 2024).
- Dirlikov, E.; Major, C.G.; Mayshack, M.; Medina, N.; Matos, D.; Ryff, K.R.; Torres-Aponte, J.; Alkis, R.; Munoz-Jordan, J.; Colon-Sanchez, C.; et al. Guillain-Barre Syndrome During Ongoing Zika Virus Transmission—Puerto Rico, January 1–July 31, 2016. MMWR Morb. Mortal. Wkly. Rep. 2016, 65, 910–914. [Google Scholar] [CrossRef]
- Parra, B.; Lizarazo, J.; Jimenez-Arango, J.A.; Zea-Vera, A.F.; Gonzalez-Manrique, G.; Vargas, J.; Angarita, J.A.; Zuniga, G.; Lopez-Gonzalez, R.; Beltran, C.L.; et al. Guillain-Barre syndrome associated with Zika virus infection in Colombia. N. Engl. J. Med. 2016, 375, 1513–1523. [Google Scholar] [CrossRef]
- Moore, C.A.; Staples, J.E.; Dobyns, W.B.; Pessoa, A.; Ventura, C.V.; Fonseca, E.B.; Ribeiro, E.M.; Ventura, L.O.; Neto, N.N.; Arena, J.F.; et al. Characterizing the pattern of anomalies in congenital Zika syndrome for pediatric clinicians. JAMA Pediatr. 2017, 171, 288–295. [Google Scholar] [CrossRef]
- Cao-Lormeau, V.M.; Blake, A.; Mons, S.; Lastere, S.; Roche, C.; Vanhomwegen, J.; Dub, T.; Baudouin, L.; Teissier, A.; Larre, P.; et al. Guillain-Barre syndrome outbreak associated with Zika virus infection in French Polynesia: A case-control study. Lancet 2016, 387, 1531–1539. [Google Scholar] [CrossRef] [PubMed]
- Vannice, K.S.; Giersing, B.K.; Kaslow, D.C.; Griffiths, E.; Meyer, H.; Barrett, A.; Durbin, A.P.; Wood, D.; Hombach, J. Meeting Report: WHO consultation on considerations for regulatory expectations of Zika virus vaccines for use during an emergency. Vaccine 2019, 37, 7443–7450. [Google Scholar] [CrossRef] [PubMed]
- Seers, T.; Rothe, C.; Hamer, D.H.; Denny, S.; Spindler, R.; Schwartz, E.; Johnston, V. Zika virus infection in European travellers returning from Thailand in 2022: A GeoSentinel case series. Trop. Med. Int. Health 2023, 28, 576–579. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Information for Travellers Visiting Countries with Zika Virus Transmission. Available online: https://www.who.int/publications/m/item/information-for-travellers-visiting-countries-with-zika-virus-transmission (accessed on 5 March 2024).
- World Health Organization. Zika Epidemiology Update. Available online: https://www.who.int/publications/m/item/zika-epidemiology-update---february-2022 (accessed on 5 March 2024).
- Baldwin, W.R.; Livengood, J.A.; Giebler, H.A.; Stovall, J.L.; Boroughs, K.L.; Sonnberg, S.; Bohning, K.J.; Dietrich, E.A.; Ong, Y.T.; Danh, H.K.; et al. Purified inactivated Zika vaccine candidates afford protection against lethal challenge in mice. Sci. Rep. 2018, 8, 16509. [Google Scholar] [CrossRef] [PubMed]
- Young, G.; Bohning, K.J.; Zahralban-Steele, M.; Hather, G.; Tadepalli, S.; Mickey, K.; Godin, C.S.; Sanisetty, S.; Sonnberg, S.; Patel, H.K.; et al. Complete protection in macaques conferred by purified inactivated Zika vaccine: Defining a correlate of protection. Sci. Rep. 2020, 10, 3488. [Google Scholar] [CrossRef] [PubMed]
- Han, H.H.; Diaz, C.; Acosta, C.J.; Liu, M.; Borkowski, A. Safety and immunogenicity of a purified inactivated Zika virus vaccine candidate in healthy adults: An observer-blind, randomised, phase 1 trial. Lancet Infect. Dis. 2021, 21, 1282–1292. [Google Scholar] [CrossRef]
- Acosta, C.J.; Diaz, C.; Nordio, F.; Han, H.H.; Moss, K.J.; Bohning, K.; Kumar, P.; Liu, M.; Patel, H.; Pacciarini, F.; et al. Persistence of immunogenicity of a purified inactivated Zika virus vaccine candidate in healthy adults: 2 years of follow-up compared with natural infection. J. Infect. Dis. 2023, 227, 1303–1312. [Google Scholar] [CrossRef] [PubMed]
- Modjarrad, K.; Lin, L.; George, S.L.; Stephenson, K.E.; Eckels, K.H.; De La Barrera, R.A.; Jarman, R.G.; Sondergaard, E.; Tennant, J.; Ansel, J.L.; et al. Preliminary aggregate safety and immunogenicity results from three trials of a purified inactivated Zika virus vaccine candidate: Phase 1, randomised, double-blind, placebo-controlled clinical trials. Lancet 2018, 391, 563–571. [Google Scholar] [CrossRef] [PubMed]
- US Food and Drug Administration. Vaccines Licensed for Use in the United States. Available online: https://www.fda.gov/vaccines-blood-biologics/vaccines/vaccines-licensed-use-united-states (accessed on 18 March 2024).
- US Food and Drug Administration. Package Insert and Patient Information—IXIARO®. Available online: https://www.fda.gov/media/75777/download?attachment (accessed on 18 March 2024).
- US Food and Drug Administration. Package Insert—YF-VAX®. Available online: https://www.fda.gov/media/76015/download?attachment (accessed on 18 March 2024).
- US Food and Drug Administration. Package Insert—TICOVAC®. Available online: https://www.fda.gov/media/151502/download?attachment (accessed on 18 March 2024).
- Acosta, C.J.; Nordio, F.; Boltz, D.A.; Baldwin, W.R.; Hather, G.; Kpamegan, E. Predicting efficacy of a purified inactivated Zika virus vaccine (PIZV) in humans using an immunological correlate of protection in non-human primates. Microorganisms 2024, 12, 1177. [Google Scholar] [CrossRef]
- Dussupt, V.; Sankhala, R.S.; Gromowski, G.D.; Donofrio, G.; De La Barrera, R.A.; Larocca, R.A.; Zaky, W.; Mendez-Rivera, L.; Choe, M.; Davidson, E.; et al. Potent Zika and dengue cross-neutralizing antibodies induced by Zika vaccination in a dengue-experienced donor. Nat. Med. 2020, 26, 228–235. [Google Scholar] [CrossRef]
- Stephenson, K.E.; Tan, C.S.; Walsh, S.R.; Hale, A.; Ansel, J.L.; Kanjilal, D.G.; Jaegle, K.; Peter, L.; Borducchi, E.N.; Nkolola, J.P.; et al. Safety and immunogenicity of a Zika purified inactivated virus vaccine given via standard, accelerated, or shortened schedules: A single-centre, double-blind, sequential-group, randomised, placebo-controlled, phase 1 trial. Lancet Infect. Dis. 2020, 20, 1061–1070. [Google Scholar] [CrossRef] [PubMed]
- Chanock, R.M.; Smith, C.B.; Friedewald, W.T.; Gutekunst, R.; Steinberg, P.; Fuld, S.; Jensen, K.E.; Senterfit, L.B.; Prescott, B. Mycoplasma pneumoniae infection—Prospects for live and inactivated vaccines. In Proceedings of the First International Conference on Vaccines Against Viral and Rickettsial Diseases of Man, Washington, DC, USA, 7–11 November 1966; Pan American Health Organization: Washington, DC, USA, 1967; p. 132. [Google Scholar]
- Cox, H.R.; Greenberg, B.G.; Kleinman, H.; Meier, P. The present status of polio vaccines. Ill. Med. J. 1960, 118, 160–168. [Google Scholar]
- Grayston, J.T.; Woolridge, R.L.; Wang, S. Trachoma vaccine studies on Taiwan. Ann. N. Y. Acad. Sci. 1962, 98, 352–367. [Google Scholar] [CrossRef] [PubMed]
- Kapikian, A.Z.; Mitchell, R.H.; Chanock, R.M.; Shvedoff, R.A.; Stewart, C.E. An epidemiologic study of altered clinical reactivity to respiratory syncytial (RS) virus infection in children previously vaccinated with an inactivated RS virus vaccine. Am. J. Epidemiol. 1969, 89, 405–421. [Google Scholar] [CrossRef]
- Kim, H.W.; Canchola, J.G.; Brandt, C.D.; Pyles, G.; Chanock, R.M.; Jensen, K.; Parrott, R.H. Respiratory syncytial virus disease in infants despite prior administration of antigenic inactivated vaccine. Am. J. Epidemiol. 1969, 89, 422–434. [Google Scholar] [CrossRef] [PubMed]
- Holt, E.A.; Moulton, L.H.; Siberry, G.K.; Halsey, N.A. Differential mortality by measles vaccine titer and sex. J. Infect. Dis. 1993, 168, 1087–1096. [Google Scholar] [CrossRef] [PubMed]
- Marques, E.T.A.; Drexler, J.F. Complex scenario of homotypic and heterotypic Zika virus immune enhancement. mBio 2019, 10, e01849-19. [Google Scholar] [CrossRef] [PubMed]
- Shim, B.S.; Kwon, Y.C.; Ricciardi, M.J.; Stone, M.; Otsuka, Y.; Berri, F.; Kwal, J.M.; Magnani, D.M.; Jackson, C.B.; Richard, A.S.; et al. Zika virus-immune plasmas from symptomatic and asymptomatic individuals enhance Zika pathogenesis in adult and pregnant mice. mBio 2019, 10, e00758-19. [Google Scholar] [CrossRef] [PubMed]
- Kliks, S.C.; Nimmanitya, S.; Nisalak, A.; Burke, D.S. Evidence that maternal dengue antibodies are important in the development of dengue hemorrhagic fever in infants. Am. J. Trop. Med. Hyg. 1988, 38, 411–419. [Google Scholar] [CrossRef]
- Chau, T.N.; Hieu, N.T.; Anders, K.L.; Wolbers, M.; Lien, L.B.; Hieu, L.T.; Hien, T.T.; Hung, N.T.; Farrar, J.; Whitehead, S.; et al. Dengue virus infections and maternal antibody decay in a prospective birth cohort study of Vietnamese infants. J. Infect. Dis. 2009, 200, 1893–1900. [Google Scholar] [CrossRef]
- Katzelnick, L.C.; Gresh, L.; Halloran, M.E.; Mercado, J.C.; Kuan, G.; Gordon, A.; Balmaseda, A.; Harris, E. Antibody-dependent enhancement of severe dengue disease in humans. Science 2017, 358, 929–932. [Google Scholar] [CrossRef]
- Katzelnick, L.C.; Narvaez, C.; Arguello, S.; Lopez Mercado, B.; Collado, D.; Ampie, O.; Elizondo, D.; Miranda, T.; Bustos Carillo, F.; Mercado, J.C.; et al. Zika virus infection enhances future risk of severe dengue disease. Science 2020, 369, 1123–1128. [Google Scholar] [CrossRef]
- Bardina, S.V.; Bunduc, P.; Tripathi, S.; Duehr, J.; Frere, J.J.; Brown, J.A.; Nachbagauer, R.; Foster, G.A.; Krysztof, D.; Tortorella, D.; et al. Enhancement of Zika virus pathogenesis by preexisting antiflavivirus immunity. Science 2017, 356, 175–180. [Google Scholar] [CrossRef]
- Lyke, K.E.; Chua, J.V.; Koren, M.; Friberg, H.; Gromowski, G.D.; Rapaka, R.R.; Waickman, A.T.; Joshi, S.; Strauss, K.; McCracken, M.K.; et al. Efficacy and immunogenicity following dengue virus-1 human challenge after a tetravalent prime-boost dengue vaccine regimen: An open-label, phase 1 trial. Lancet Infect. Dis. 2024, 4, S1473-3099(24)00100-2. [Google Scholar] [CrossRef]
- Sridhar, S.; Luedtke, A.; Langevin, E.; Zhu, M.; Bonaparte, M.; Machabert, T.; Savarino, S.; Zambrano, B.; Moureau, A.; Khromava, A.; et al. Effect of dengue serostatus on dengue vaccine safety and efficacy. N. Engl. J. Med. 2018, 379, 327–340. [Google Scholar] [CrossRef]
- Simmons, C.P.; Chau, T.N.; Thuy, T.T.; Tuan, N.M.; Hoang, D.M.; Thien, N.T.; Lien, L.B.; Quy, N.T.; Hieu, N.T.; Hien, T.T.; et al. Maternal antibody and viral factors in the pathogenesis of dengue virus in infants. J. Infect. Dis. 2007, 196, 416–424. [Google Scholar] [CrossRef] [PubMed]
- Halstead, S.B. In vivo enhancement of dengue virus infection in rhesus monkeys by passively transferred antibody. J. Infect. Dis. 1979, 140, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Halstead, S.B.; O’Rourke, E.J. Antibody-enhanced dengue virus infection in primate leukocytes. Nature 1977, 265, 739–741. [Google Scholar] [CrossRef]
- Kliks, S.C.; Nisalak, A.; Brandt, W.E.; Wahl, L.; Burke, D.S. Antibody-dependent enhancement of dengue virus growth in human monocytes as a risk factor for dengue hemorrhagic fever. Am. J. Trop. Med. Hyg. 1989, 40, 444–451. [Google Scholar] [CrossRef]
- Ripoll, D.R.; Wallqvist, A.; Chaudhury, S. Molecular simulations reveal the role of antibody fine specificity and viral maturation state on antibody-dependent enhancement of infection in dengue virus. Front. Cell. Infect. Microbiol. 2019, 9, 200. [Google Scholar] [CrossRef]
- Evans, T.G.; McElrath, M.J.; Matthews, T.; Montefiori, D.; Weinhold, K.; Wolff, M.; Keefer, M.C.; Kallas, E.G.; Corey, L.; Gorse, G.J.; et al. QS-21 promotes an adjuvant effect allowing for reduced antigen dose during HIV-1 envelope subunit immunization in humans. Vaccine 2001, 19, 2080–2091. [Google Scholar] [CrossRef] [PubMed]
- Nassim, C.; Christensen, S.; Henry, D.; Holmes, S.; Hohenboken, M.; Kanesa-Thasan, N. Identification of antigen and adjuvant doses resulting in optimal immunogenicity and antibody persistence up to 1 year after immunization with a pandemic A/H1N1 influenza vaccine in children 3 to <9 years of age. Pediatr. Infect. Dis. J. 2012, 31, e59–e65. [Google Scholar] [CrossRef] [PubMed]
- Regules, J.A.; Cicatelli, S.B.; Bennett, J.W.; Paolino, K.M.; Twomey, P.S.; Moon, J.E.; Kathcart, A.K.; Hauns, K.D.; Komisar, J.L.; Qabar, A.N.; et al. Fractional third and fourth dose of RTS, S/AS01 malaria candidate vaccine: A phase 2a controlled human malaria parasite infection and immunogenicity study. J. Infect. Dis. 2016, 214, 762–771. [Google Scholar] [CrossRef] [PubMed]
- Zambrano, B.; Noriega, F.; Dayan, G.H.; Rivera, D.M.; Arredondo, J.L.; Reynales, H.; Luz, K.; Deseda, C.; Bonaparte, M.I.; Langevin, E.; et al. Zika and dengue interactions in the context of a large dengue vaccine clinical trial in Latin America. Am. J. Trop. Med. Hyg. 2021, 104, 136–144. [Google Scholar] [CrossRef]
- Meeting of the Strategic Advisory Group of Experts on immunization, October 2016—Conclusions and recommendations. Wkly. Epidemiol. Rec. 2016, 91, 581–582.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).