
Sensitive and Accurate Quantification of Enterovirus-D68 (EV-D68) Viral Loads Using Droplet Digital PCR (ddPCR)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Primers and Probes
2.2. RNA Isolation and cDNA Preparation
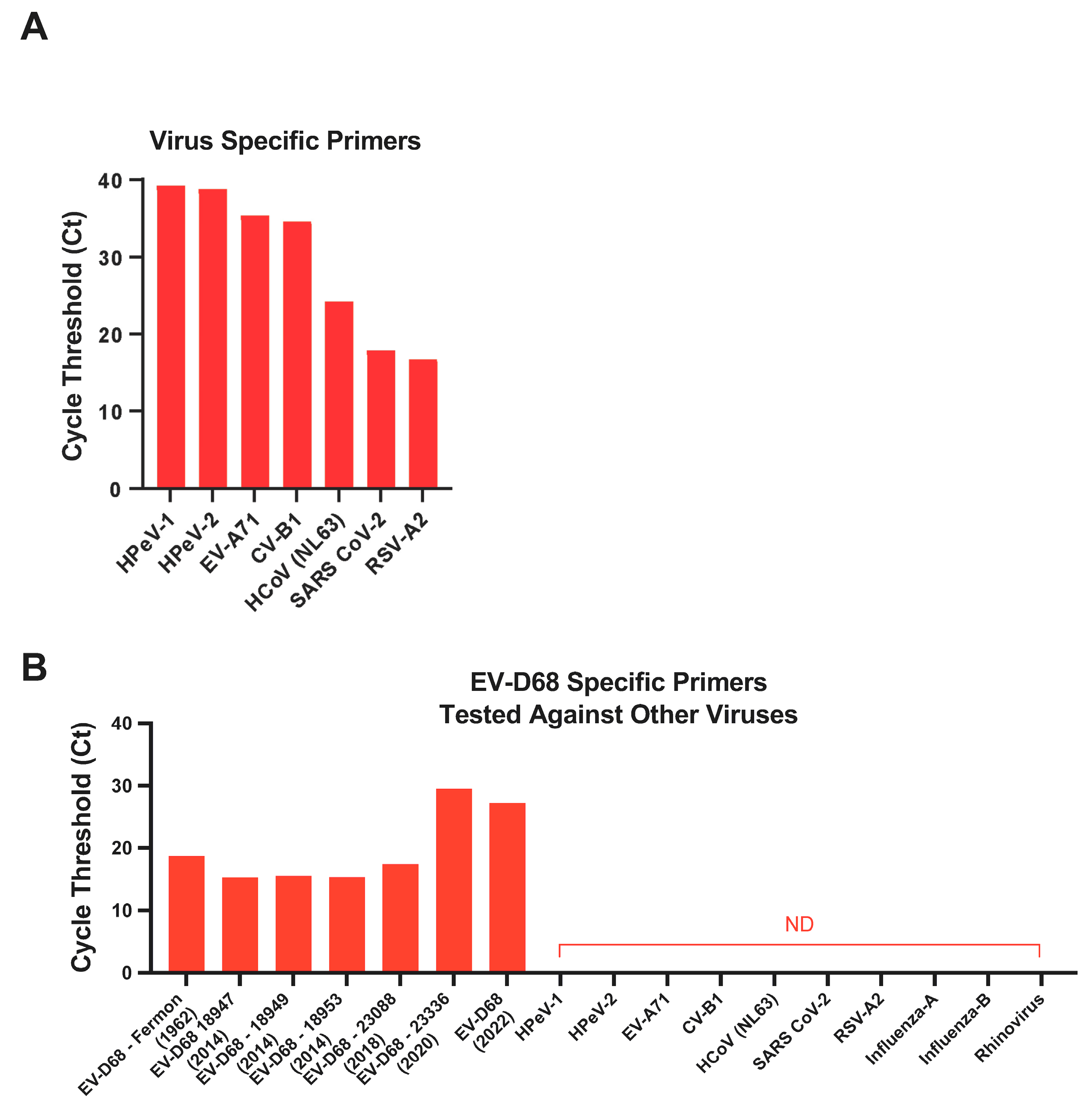
2.3. Specificity of EV-D68 Primers
2.4. Real-Time qPCR Assay
2.5. Droplet Digital PCR Assay
3. Results
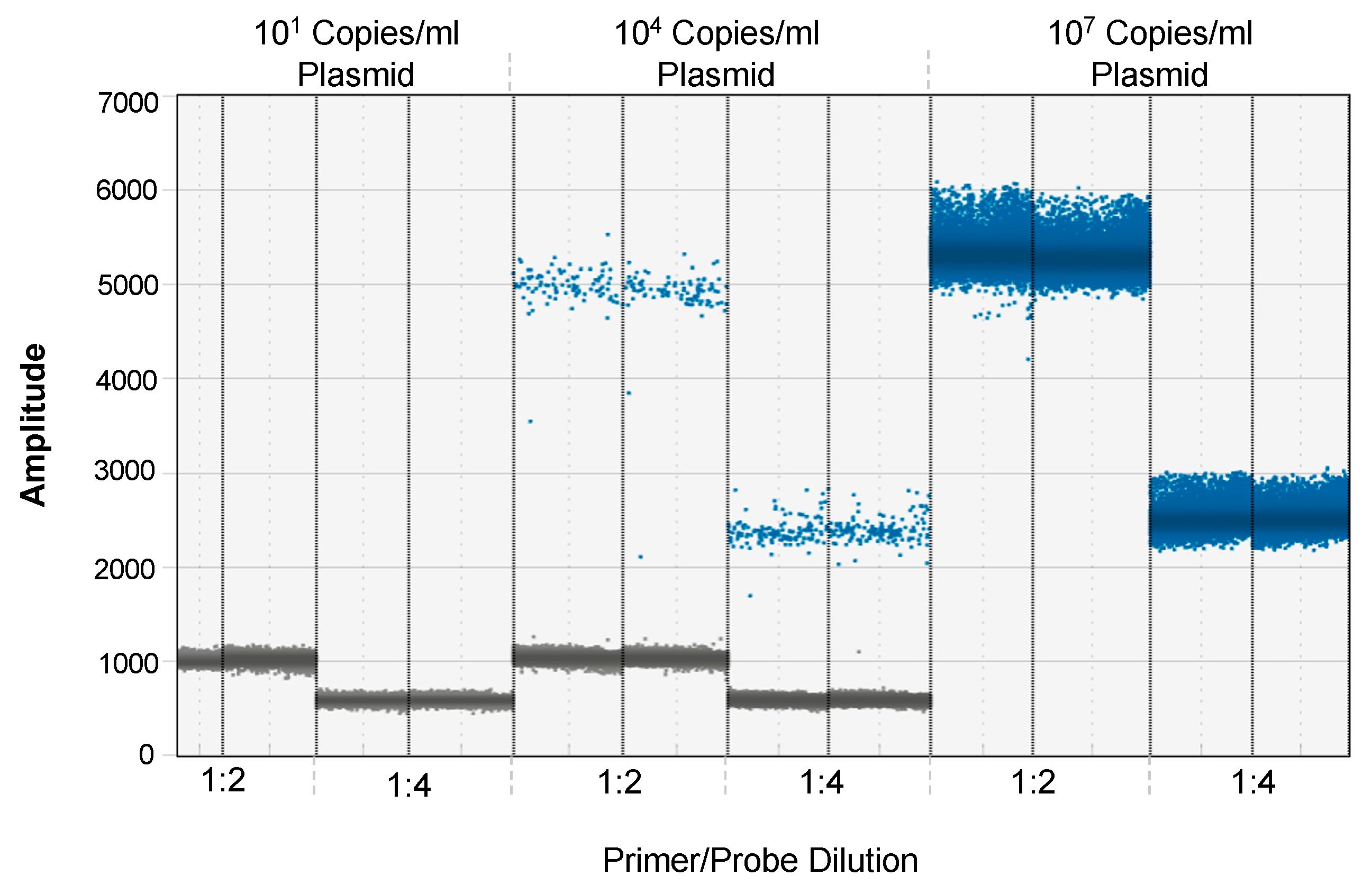
3.1. Optimization of EV-D68 Primer and Probe Concentration for ddPCR
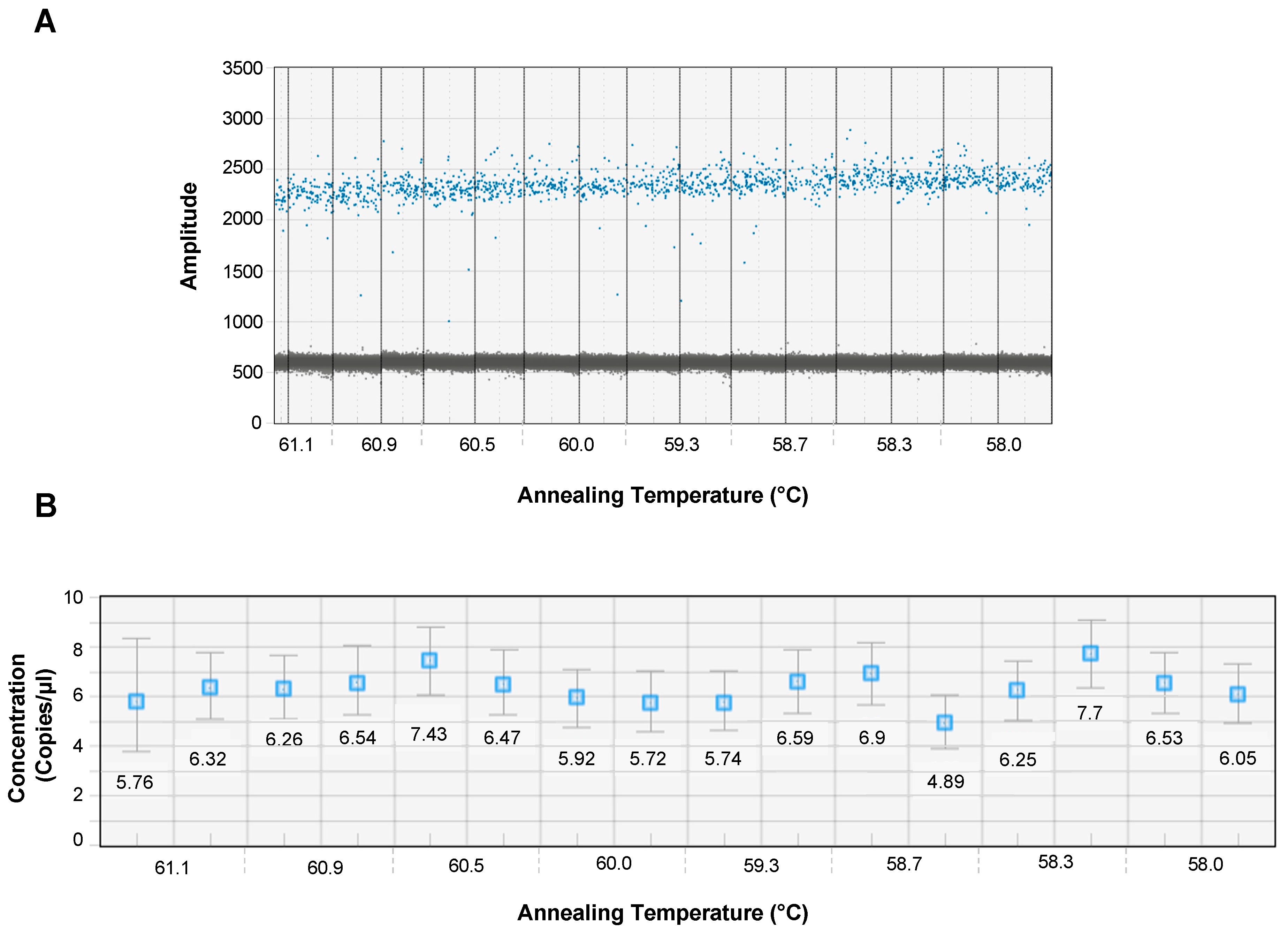
3.2. Optimal Annealing Temperature for ddPCR
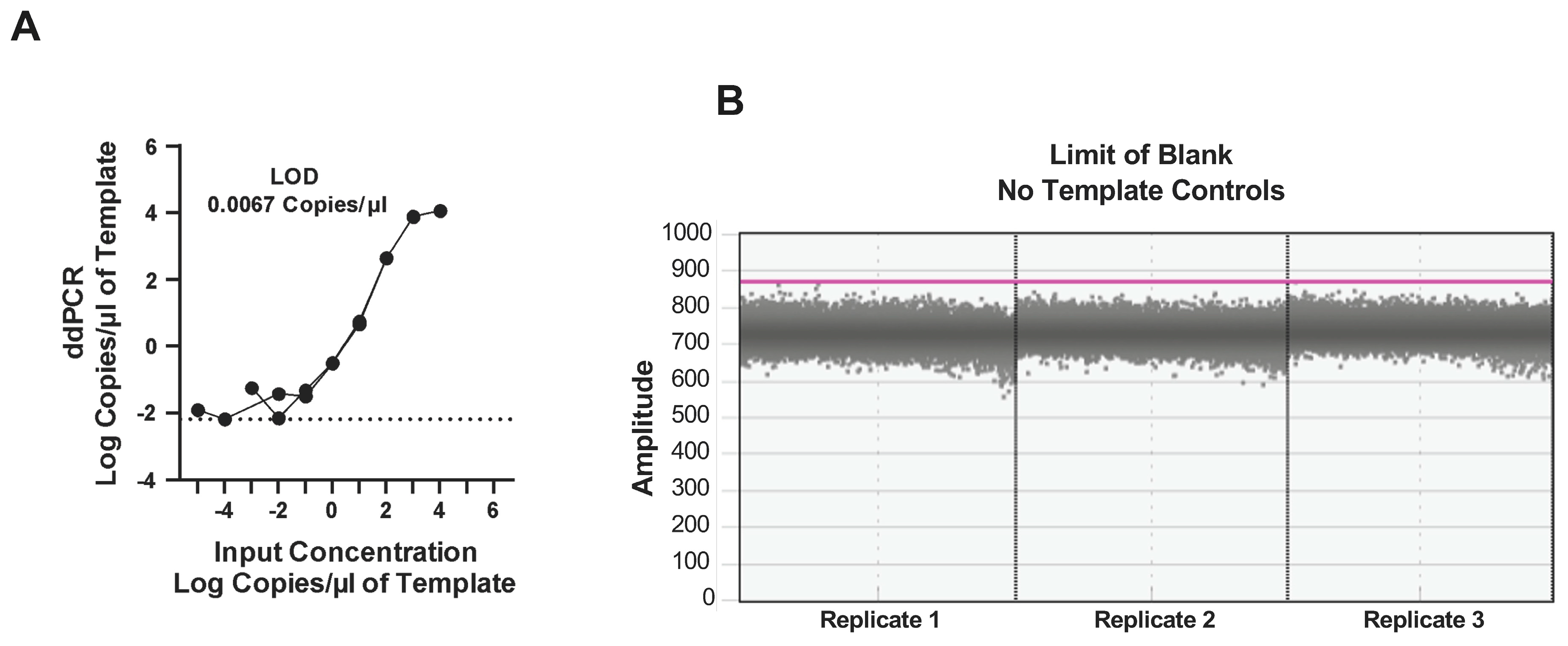
3.3. Dynamic Range of the ddPCR Assay
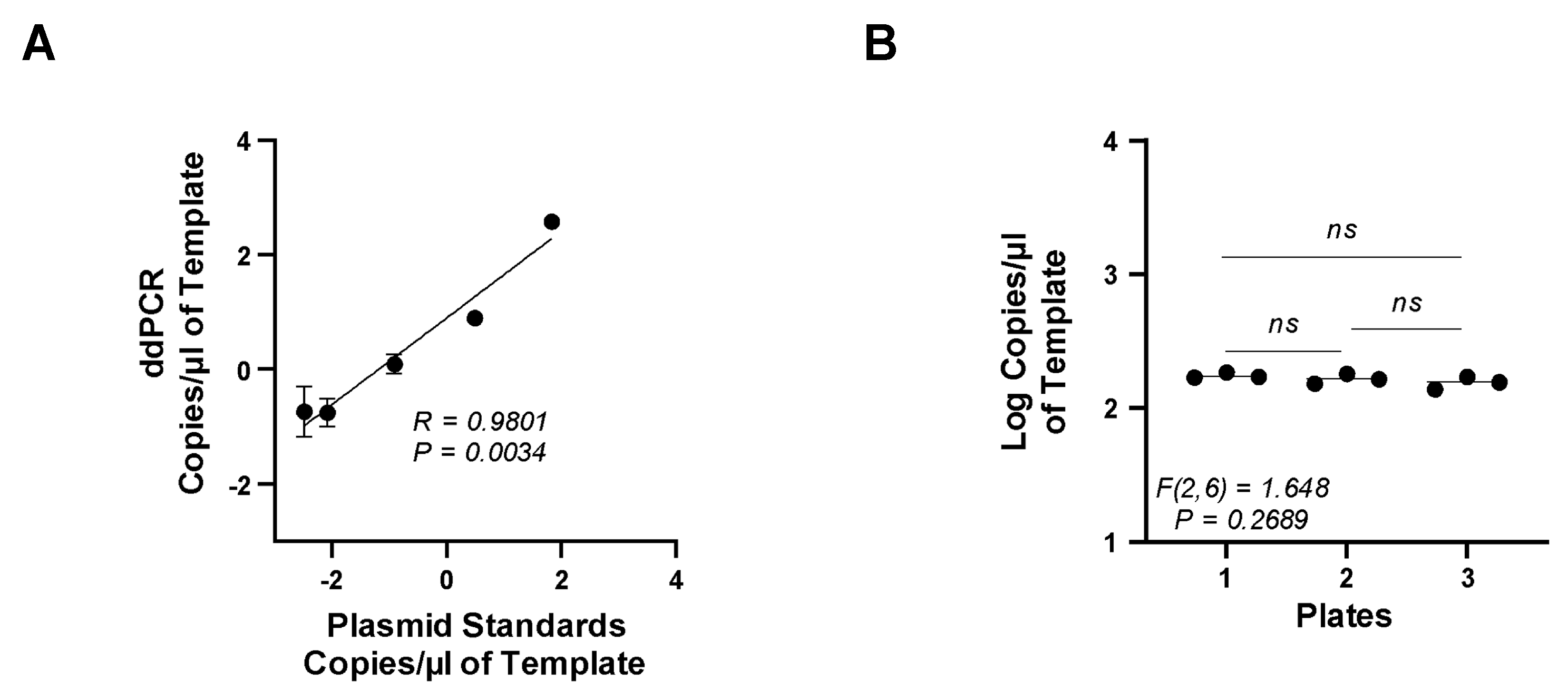
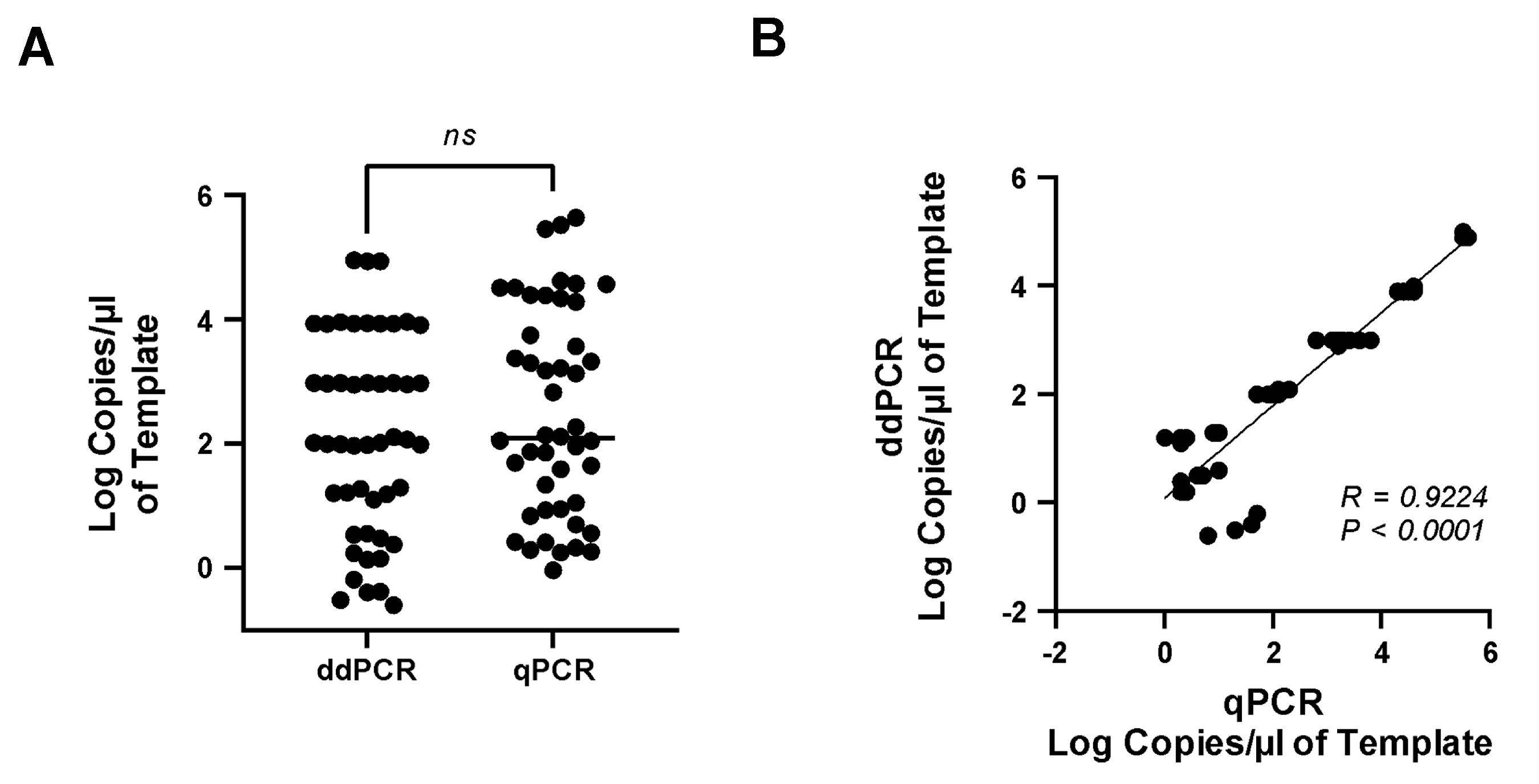
3.4. Accuracy and Precision of the ddPCR Assay
3.5. Specificity of EV-D68 Primers
3.6. Comparison of Real-Time qPCR and ddPCR Assays
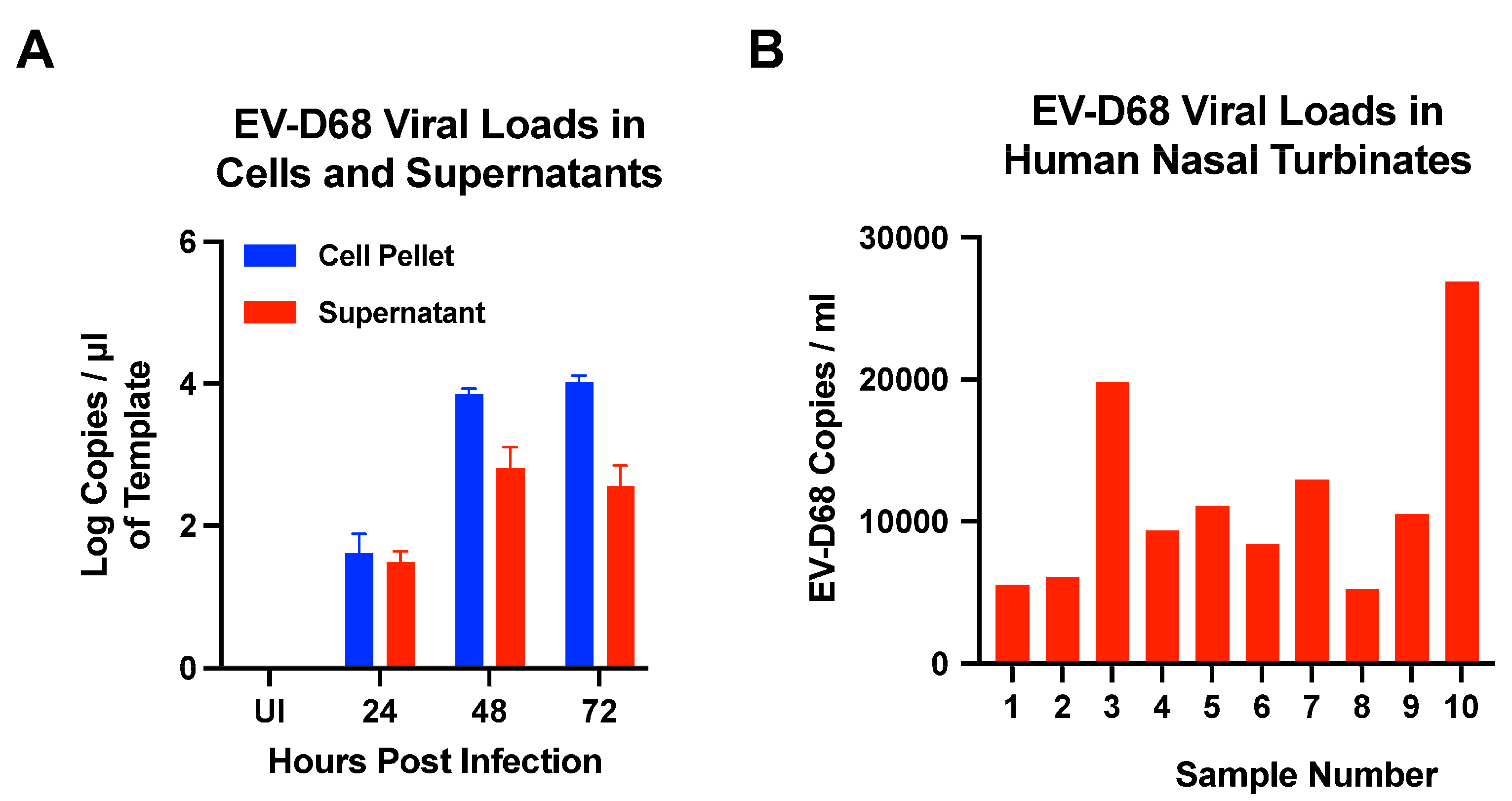
3.7. Quantification of EV-D68 Viral Loads in Cells, Supernatants, and Human Nasal Turbinates by ddPCR Assay
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Grizer, C.S.; Messacar, K.; Mattapallil, J.J. Enterovirus-D68—A reemerging non-polio enterovirus that causes severe respiratory and neurological disease in children. Front. Virol. 2024, 4, 1328457. [Google Scholar] [CrossRef]
- Messacar, K.; Schreiner, T.L.; Maloney, J.A.; Wallace, A.; Ludke, J.; Oberste, M.S.; Nix, W.A.; Robinson, C.C.; Glode, M.P.; Abzug, M.J.; et al. A cluster of acute flaccid paralysis and cranial nerve dysfunction temporally associated with an outbreak of enterovirus D68 in children in Colorado, USA. Lancet 2015, 385, 1662–1671. [Google Scholar] [CrossRef] [PubMed]
- Messacar, K.; Schreiner, T.L.; Van Haren, K.; Yang, M.; Glaser, C.A.; Tyler, K.L.; Dominguez, S.R. Acute flaccid myelitis: A clinical review of US cases 2012–2015. Ann. Neurol. 2016, 80, 326–338. [Google Scholar] [CrossRef] [PubMed]
- Holm-Hansen, C.C.; Midgley, S.E.; Fischer, T.K. Global emergence of enterovirus D68: A systematic review. Lancet Infect. Dis. 2016, 16, e64–e75. [Google Scholar] [CrossRef] [PubMed]
- Park, S.W.; Pons-Salort, M.; Messacar, K.; Cook, C.; Meyers, L.; Farrar, J.; Grenfell, B.T. Epidemiological dynamics of enterovirus D68 in the United States and implications for acute flaccid myelitis. Sci. Transl. Med. 2021, 13, eabd2400. [Google Scholar] [CrossRef] [PubMed]
- Kujawski, S.A.; Midgley, C.M.; Rha, B.; Lively, J.Y.; Nix, W.A.; Curns, A.T.; Payne, D.C.; Englund, J.A.; Boom, J.A.; Williams, J.V.; et al. Enterovirus D68-Associated Acute Respiratory Illness—New Vaccine Surveillance Network, United States, July–October, 2017 and 2018. MMWR Morb. Mortal Wkly Rep. 2019, 68, 277–280. [Google Scholar] [CrossRef] [PubMed]
- Mueller, Y.M.; Do, D.H.; Boyer, J.D.; Kader, M.; Mattapallil, J.J.; Lewis, M.G.; Weiner, D.B.; Katsikis, P.D. CD8+ Cell Depletion of SHIV89.6P-Infected Macaques Induces CD4+ T Cell Proliferation that Contributes to Increased Viral Loads1. J. Immunol. 2009, 183, 5006–5012. [Google Scholar] [CrossRef] [PubMed]
- McMahan, K.; Wegmann, F.; Aid, M.; Sciacca, M.; Liu, J.; Hachmann, N.P.; Miller, J.; Jacob-Dolan, C.; Powers, O.; Hope, D.; et al. Mucosal boosting enhances vaccine protection against SARS-CoV-2 in macaques. Nature 2024, 626, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.; Li, X.; Cai, X.; Li, W.; Li, J.; Yang, S.; Zhai, J.; Suolang, S.; Li, B. First Specific Detection of Mammalian Orthoreovirus from Goats Using TaqMan Real-Time RT-PCR Technology. Vet. Sci. 2024, 11, 141. [Google Scholar] [CrossRef]
- Madihi, S.; Laassili, C.; Boukaira, S.; Baha, W.; Khyatti, M.; Zyad, A.; Ben Mkaddem, S.; Benani, A. Development and validation of the first HBV qRT-PCR assay in the Mediterranean area targeting the X region. J. Virol. Methods 2024, 326, 114913. [Google Scholar] [CrossRef]
- Alhetheel, A.; Alsaeed, A.; Albarrag, A.; Hakami, A.; Binkhamis, K.; Somily, A.; Mujamammi, A.; Alswat, K. Evaluation of Quantitative Hepatitis C Virus Core Antigen Testing as a Tool for Screening and Confirmation of HCV Infection. Clin. Lab. 2023, 69, 1795–1801. [Google Scholar] [CrossRef] [PubMed]
- Rimal, S.; Shrestha, S.; Pandey, K.; Nguyen, T.V.; Bhandari, P.; Shah, Y.; Acharya, D.; Adhikari, N.; Rijal, K.R.; Ghimire, P.; et al. Co-Circulation of Dengue Virus Serotypes 1, 2, and 3 during the 2022 Dengue Outbreak in Nepal: A Cross-Sectional Study. Viruses 2023, 15, 507. [Google Scholar] [CrossRef] [PubMed]
- George, J.; Valiant, W.G.; Mattapallil, M.J.; Walker, M.; Huang, Y.S.; Vanlandingham, D.L.; Misamore, J.; Greenhouse, J.; Weiss, D.E.; Verthelyi, D.; et al. Prior Exposure to Zika Virus Significantly Enhances Peak Dengue-2 Viremia in Rhesus Macaques. Sci. Rep. 2017, 7, 10498. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.C.; Nadeau, K.; Abbasi, M.; Lachance, C.; Nguyen, M.; Fenrich, J. The Ultimate qPCR Experiment: Producing Publication Quality, Reproducible Data the First Time. Trends Biotechnol. 2019, 37, 761–774. [Google Scholar] [CrossRef] [PubMed]
- Hayden, R.T.; Gu, Z.; Ingersoll, J.; Abdul-Ali, D.; Shi, L.; Pounds, S.; Caliendo, A.M. Comparison of droplet digital PCR to real-time PCR for quantitative detection of cytomegalovirus. J. Clin. Microbiol. 2013, 51, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.H.; Chafets, D.M.; Busch, M.P.; Murphy, E.L. Quantitation of HTLV-I and II proviral load using real-time quantitative PCR with SYBR Green chemistry. J. Clin. Virol. 2004, 31, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, L.B.; Coleman, V.A.; Hindson, C.M.; Herrmann, J.; Hindson, B.J.; Bhat, S.; Emslie, K.R. Evaluation of a droplet digital polymerase chain reaction format for DNA copy number quantification. Anal. Chem. 2012, 84, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [PubMed]
- Koressaar, T.; Remm, M. Enhancements and modifications of primer design program Primer3. Bioinformatics 2007, 23, 1289–1291. [Google Scholar] [CrossRef]
- Koressaar, T.; Lepamets, M.; Kaplinski, L.; Raime, K.; Andreson, R.; Remm, M. Primer3_masker: Integrating masking of template sequence with primer design software. Bioinformatics 2018, 34, 1937–1938. [Google Scholar] [CrossRef]
- Moore, A.C.; Bixler, S.L.; Lewis, M.G.; Verthelyi, D.; Mattapallil, J.J. Mucosal and peripheral Lin- HLA-DR+ CD11c/123- CD13+ CD14- mononuclear cells are preferentially infected during acute simian immunodeficiency virus infection. J. Virol. 2012, 86, 1069–1078. [Google Scholar] [CrossRef] [PubMed]
- Kader, M.; Bixler, S.; Roederer, M.; Veazey, R.; Mattapallil, J.J. CD4 T cell subsets in the mucosa are CD28+Ki-67-HLA-DR-CD69+ but show differential infection based on alpha4beta7 receptor expression during acute SIV infection. J. Med. Primatol. 2009, 38 (Suppl. S1), 24–31. [Google Scholar] [CrossRef] [PubMed]
- Valiant, W.G.; Huang, Y.S.; Vanlandingham, D.L.; Higgs, S.; Lewis, M.G.; Mattapallil, J.J. Zika convalescent macaques display delayed induction of anamnestic cross-neutralizing antibody responses after dengue infection. Emerg. Microbes Infect. 2018, 7, 130. [Google Scholar] [CrossRef] [PubMed]
- Valiant, W.G.; Mattapallil, M.J.; Higgs, S.; Huang, Y.S.; Vanlandingham, D.L.; Lewis, M.G.; Mattapallil, J.J. Simultaneous Coinfection of Macaques with Zika and Dengue Viruses Does not Enhance Acute Plasma Viremia but Leads to Activation of Monocyte Subsets and Biphasic Release of Pro-inflammatory Cytokines. Sci. Rep. 2019, 9, 7877. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.M.; Hixon, A.M.; Oldfield, L.M.; Zhang, Y.; Novotny, M.; Wang, W.; Das, S.R.; Shabman, R.S.; Tyler, K.L.; Scheuermann, R.H. Contemporary Circulating Enterovirus D68 Strains Have Acquired the Capacity for Viral Entry and Replication in Human Neuronal Cells. mBio 2018, 9, e01954-18. [Google Scholar] [CrossRef] [PubMed]
- Selvaraju, S.B.; Nix, W.A.; Oberste, M.S.; Selvarangan, R. Optimization of a combined human parechovirus-enterovirus real-time reverse transcription-PCR assay and evaluation of a new parechovirus 3-specific assay for cerebrospinal fluid specimen testing. J. Clin. Microbiol. 2013, 51, 452–458. [Google Scholar] [CrossRef]
- Moes, E.; Vijgen, L.; Keyaerts, E.; Zlateva, K.; Li, S.; Maes, P.; Pyrc, K.; Berkhout, B.; van der Hoek, L.; Van Ranst, M. A novel pancoronavirus RT-PCR assay: Frequent detection of human coronavirus NL63 in children hospitalized with respiratory tract infections in Belgium. BMC Infect. Dis. 2005, 5, 6. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Wang, L.; Sakthivel, S.K.; Whitaker, B.; Murray, J.; Kamili, S.; Lynch, B.; Malapati, L.; Burke, S.A.; Harcourt, J.; et al. US CDC Real-Time Reverse Transcription PCR Panel for Detection of Severe Acute Respiratory Syndrome Coronavirus 2. Emerg. Infect. Dis. 2020, 26, 1654–1665. [Google Scholar] [CrossRef]
- Olivier, A.; Gallup, J.; de Macedo, M.M.; Varga, S.M.; Ackermann, M. Human respiratory syncytial virus A2 strain replicates and induces innate immune responses by respiratory epithelia of neonatal lambs. Int. J. Exp. Pathol. 2009, 90, 431–438. [Google Scholar] [CrossRef]
- Corona, A.K.; Saulsbery, H.M.; Corona Velazquez, A.F.; Jackson, W.T. Enteroviruses Remodel Autophagic Trafficking through Regulation of Host SNARE Proteins to Promote Virus Replication and Cell Exit. Cell Rep. 2018, 22, 3304–3314. [Google Scholar] [CrossRef]
- Yu, F.; Yan, L.; Wang, N.; Yang, S.; Wang, L.; Tang, Y.; Gao, G.; Wang, S.; Ma, C.; Xie, R.; et al. Quantitative Detection and Viral Load Analysis of SARS-CoV-2 in Infected Patients. Clin. Infect. Dis. 2020, 71, 793–798. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, X.; Dai, W.; Liu, Q.; Xiong, P.; Wang, S.; Geng, L.; Gong, S.; Huang, Z. A Mouse Model of Enterovirus D68 Infection for Assessment of the Efficacy of Inactivated Vaccine. Viruses 2018, 10, 58. [Google Scholar] [CrossRef] [PubMed]
- Freeman, W.M.; Walker, S.J.; Vrana, K.E. Quantitative RT-PCR: Pitfalls and potential. Biotechniques 1999, 26, 112–122, 124–125. [Google Scholar] [CrossRef]
- Bolufer, P.; Barragan, E.; Sanz, M.A.; Martin, G.; Bornstein, R.; Colomer, D.; Delgado, M.D.; Gonzalez, M.; Marugan, I.; Roman, J.; et al. Preliminary experience in external quality control of RT-PCR PML-RAR alpha detection in promyelocytic leukemia. Leukemia 1998, 12, 2024–2028. [Google Scholar] [CrossRef] [PubMed]
- Keilholz, U.; Willhauck, M.; Rimoldi, D.; Brasseur, F.; Dummer, W.; Rass, K.; de Vries, T.; Blaheta, J.; Voit, C.; Lethe, B.; et al. Reliability of reverse transcription-polymerase chain reaction (RT-PCR)-based assays for the detection of circulating tumour cells: A quality-assurance initiative of the EORTC Melanoma Cooperative Group. Eur. J. Cancer 1998, 34, 750–753. [Google Scholar] [CrossRef]
- Abachin, E.; Convers, S.; Falque, S.; Esson, R.; Mallet, L.; Nougarede, N. Comparison of reverse-transcriptase qPCR and droplet digital PCR for the quantification of dengue virus nucleic acid. Biologicals 2018, 52, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Loconsole, D.; Centrone, F.; Sallustio, A.; Casulli, D.; Zagaria, R.; Sacco, D.; Colella, V.; Albano, N.; Caselli, D.; Cardinale, F.; et al. Echovirus 11 lineage I and other enterovirus in hospitalized children with acute respiratory infection in Southern Italy (2022–2023). Int. J. Infect. Dis. 2024, 146, 107091. [Google Scholar] [CrossRef]
- Zhou, X.; Qian, K.; Zhu, C.; Yi, L.; Tu, J.; Yang, S.; Zhang, Y.; Zhang, Y.; Xia, W.; Ni, X.; et al. Surveillance, epidemiology, and impact of the coronavirus disease 2019 interventions on the incidence of enterovirus infections in Nanchang, China, 2010–2022. Front. Microbiol. 2023, 14, 1251683. [Google Scholar] [CrossRef]
- Lizasoain, A.; Mir, D.; Salvo, M.; Bortagaray, V.; Masachessi, G.; Farias, A.; Rodriguez-Osorio, N.; Nates, S.; Victoria, M.; Colina, R. First evidence of enterovirus A71 and echovirus 30 in Uruguay and genetic relationship with strains circulating in the South American region. PLoS ONE 2021, 16, e0255846. [Google Scholar] [CrossRef]
- Picone, S.; Mondi, V.; Di Palma, F.; Valli, M.B.; Rueca, M.; Bedetta, M.; Paolillo, P. Enterovirus and Paraechovirus Meningitis in Neonates: Which Is the Difference? Clin. Pediatr. 2024, 99228241235448. [Google Scholar] [CrossRef]
- Rose, E.B.; Wheatley, A.; Langley, G.; Gerber, S.; Haynes, A. Respiratory Syncytial Virus Seasonality—United States, 2014–2017. MMWR Morb. Mortal Wkly. Rep. 2018, 67, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, D.; Mambule, I.; Read, J.M.; Kiran, A.; Chilombe, M.; Bvumbwe, T.; Aston, S.; Menyere, M.; Masina, M.; Kamzati, M.; et al. Epidemiology of Human Seasonal Coronaviruses Among People with Mild and Severe Acute Respiratory Illness in Blantyre, Malawi, 2011–2017. J. Infect. Dis. 2024, jiad587. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grizer, C.S.; Li, Z.; Mattapallil, J.J. Sensitive and Accurate Quantification of Enterovirus-D68 (EV-D68) Viral Loads Using Droplet Digital PCR (ddPCR). Microorganisms 2024, 12, 1502. https://doi.org/10.3390/microorganisms12081502
Grizer CS, Li Z, Mattapallil JJ. Sensitive and Accurate Quantification of Enterovirus-D68 (EV-D68) Viral Loads Using Droplet Digital PCR (ddPCR). Microorganisms. 2024; 12(8):1502. https://doi.org/10.3390/microorganisms12081502
Chicago/Turabian StyleGrizer, Cassandra S., Zhaozhang Li, and Joseph J. Mattapallil. 2024. "Sensitive and Accurate Quantification of Enterovirus-D68 (EV-D68) Viral Loads Using Droplet Digital PCR (ddPCR)" Microorganisms 12, no. 8: 1502. https://doi.org/10.3390/microorganisms12081502