
Development of a Robust Saccharomyces cerevisiae Strain for Efficient Co-Fermentation of Mixed Sugars and Enhanced Inhibitor Tolerance through Protoplast Fusion
 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Microbial Strains and Media

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strains and Plasmids | Description | Source/Reference |
|---|---|---|
| Strains | ||
| CEN.PK102-5B | Haploid S. cerevisiae strain MATa; URA3-52, HIS3Δ1, LEU2-3, 112 | Laboratory reserved [41] |
| LF1 | Recombinant glucose/xylose co-fermenting strain derived from BSIF (pho13::XI, 3δ::XI, gre3::PPP, XK, AE-PCS, N360F, AE) | Laboratory preserved [11] |
| BLN26 | RC212 derivative; (δ::BGL& KanMX, ADH2::IBX, δ::IBX) | Laboratory preserved [12] |
| BLH01 | Protoplast fusion strain from LF1 and BLN26 | This work |
| BLH02 | Protoplast fusion strain from LF1 and BLN26 | This work |
| BLH172 | Protoplast fusion strain with stable inheritance of traits | This work |
| BLH508 | BLH172 derivative; adh2Δ::BGL | This work |
| BLH510 | BLH508 derivative; adh2Δ::BGL, δ::IBX | This work |
| Plasmids | ||
| YEp-CH | YEp24 derivative; GAL1p-Cre-CYC1t, TEF1p-hygB-TEF1t | Laboratory preserved [11] |
| pUG6 | E. coli plasmid with segment loxP-KanMX4-loxP | Laboratory preserved [42] |
| pJXIHIBX | pJXIH-PC with β-xylosidase gene xyl3A from P. oxalicum; signal peptide of Kluyveromyces INU | Laboratory preserved [19] |
| pUCδBK | pUC19-based yeast integration plasmid containing the δ-sequence-targeting recombinant arms, an expression cassette of BGL, and the selectable marker loxP-KanMX4-loxP | Laboratory preserved [12] |
2.2. Construction of Plasmids and Integrating Fragments
2.3. The Preparation, Inactivation, and Fusion of Yeast Protoplasts
2.3.1. Protoplasts Preparation
2.3.2. Protoplasts Inactivation
2.3.3. Protoplasts Fusion
2.4. The Screening of Yeast Fusants Derived from Protoplasts Fusion
2.5. Ploidy Determination of S. cerevisiae
2.6. Oxygen-Limited Growth and Fermentation
2.7. Determination of Intracellular Trehalose Content
2.8. Determination of the Ratio of Reduced Glutathione (GSH) to Oxidized Glutathione (GSSG)
2.9. Assays of β-Glucosidase, β-Xylosidase, and Xylose Isomerase Activity
2.10. Analysis of Hydrolysis and Fermentation Products
3. Results and Discussion
3.1. Optimization of Protoplast Fusion Operating Conditions
3.1.1. Optimization of Protoplast Preparation and Regeneration Conditions for LF1 and BLN26
3.1.2. Optimization of Inactivation Conditions for Protoplasts of LF1 and BLN26
3.1.3. Optimization of Fusion Conditions for Protoplasts of LF1 and BLN26
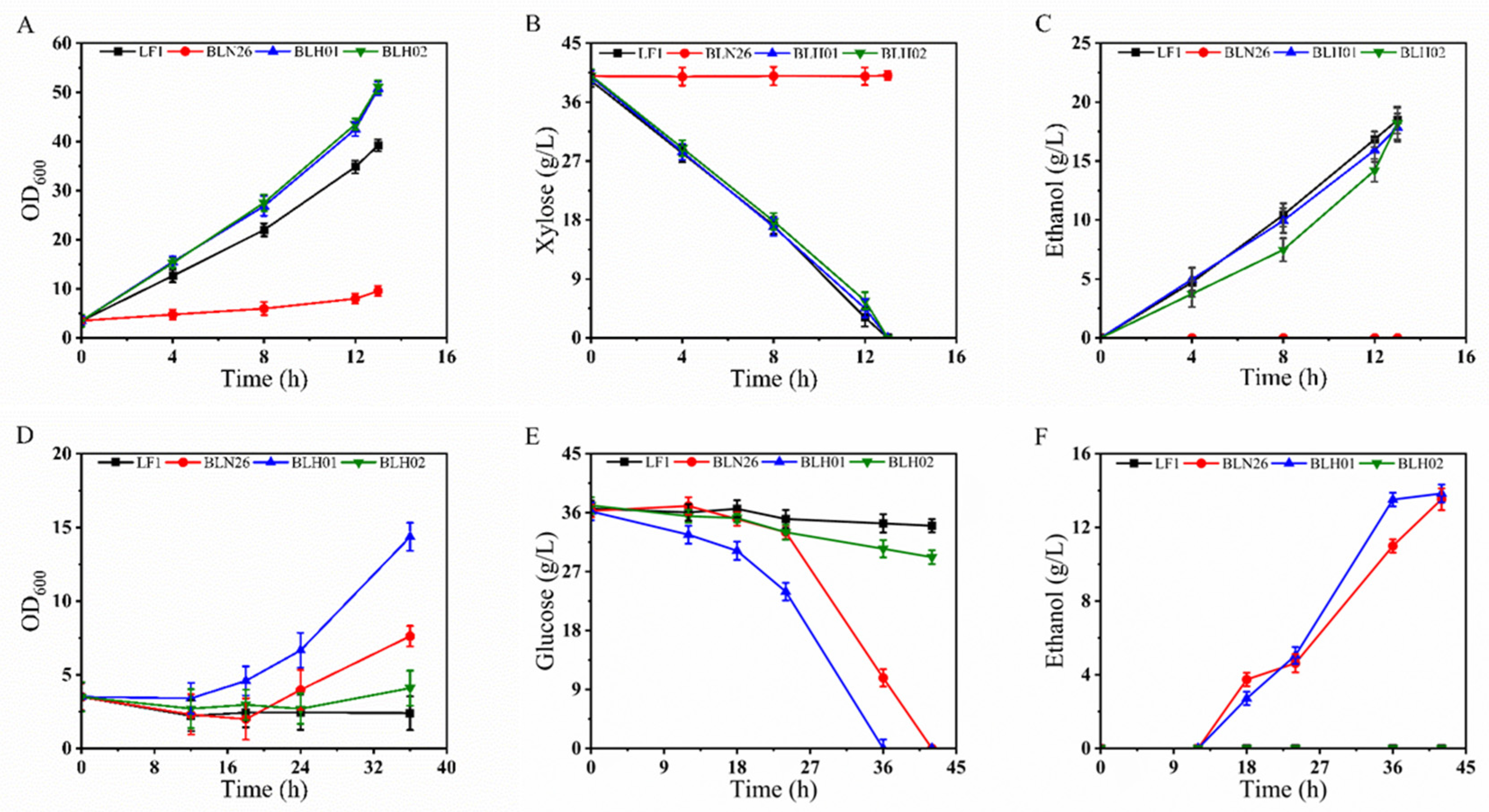
3.2. Screening and Oxygen-Limited Fermentation of Fusants Derived from LF1 and BLN26
3.2.1. Primary Screening of Fusants
3.2.2. Secondary Screening of Fusants
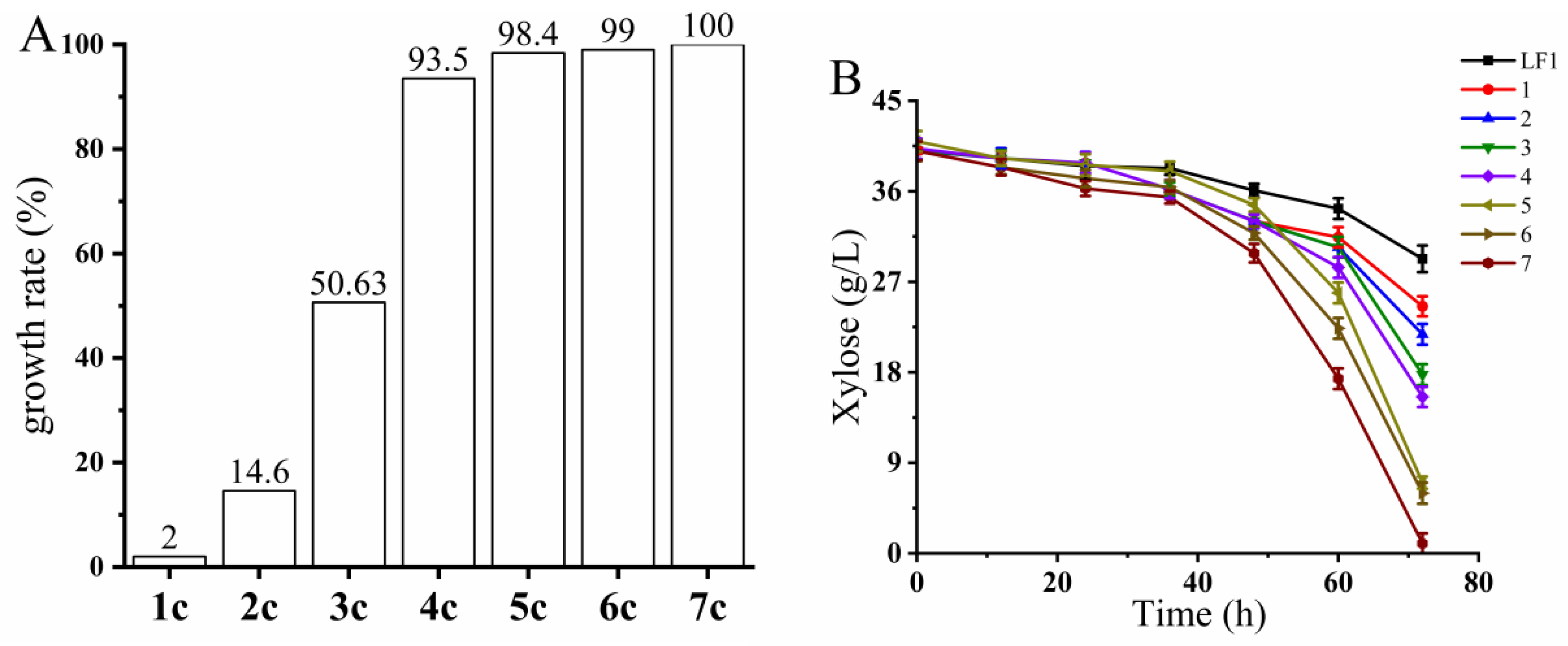
3.2.3. Ploidy Analysis of the Fusant Strain BLH01
3.2.4. The Genetic Stability Assessment of Fusant BLH01
3.3. Selection and Oxygen-Limited Fermentation of Stable Fusants
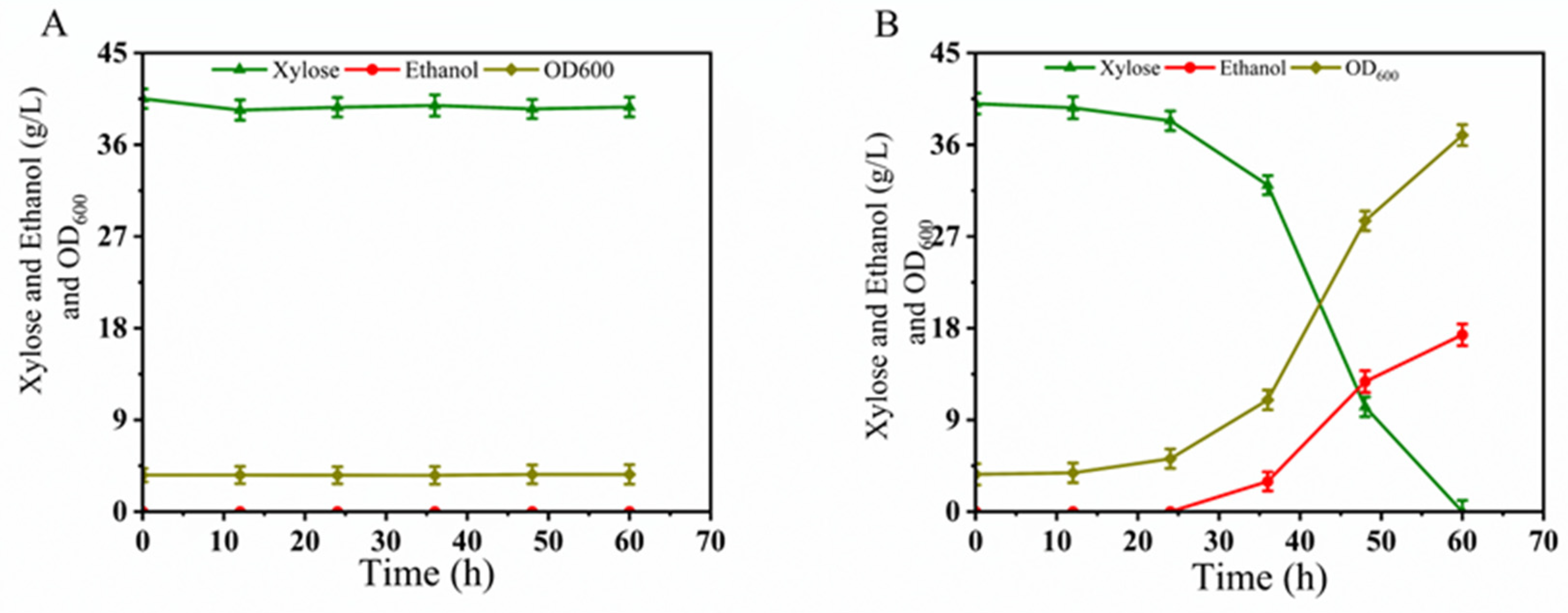
3.3.1. Selection of Stable Fusant
3.3.2. Oxygen-Limited Fermentation Performance of BLH172 in YPX40 and YPD40I2×
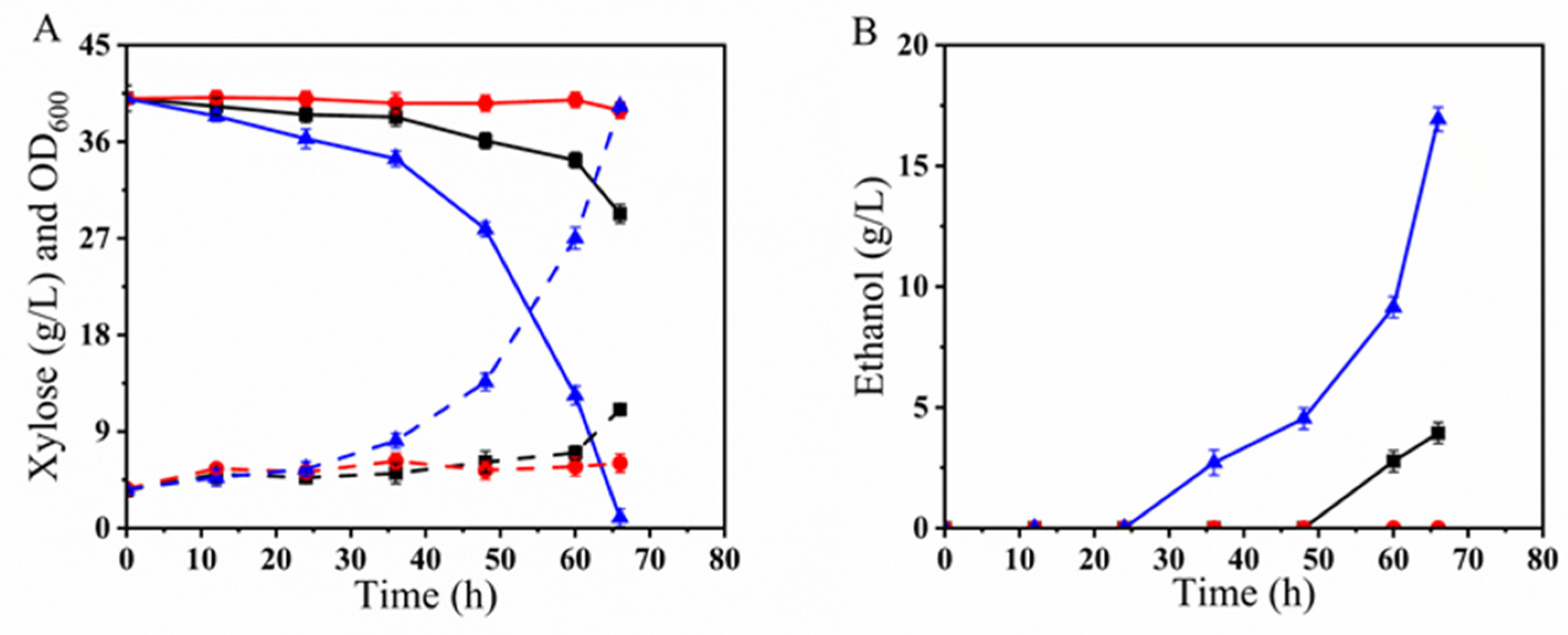
3.3.3. Oxygen-Limited Fermentation Performance of BLH172 in YPX40I1.5×
3.4. Construction of BLH510 with β-Glucosidase and β-Xylosidase Expression Activity
3.4.1. Integration of the β-Glucosidase (BGL) Gene at the ADH2 Locus
3.4.2. Integration of the β-Xylosidase Gene at the δ-Sequence Locus
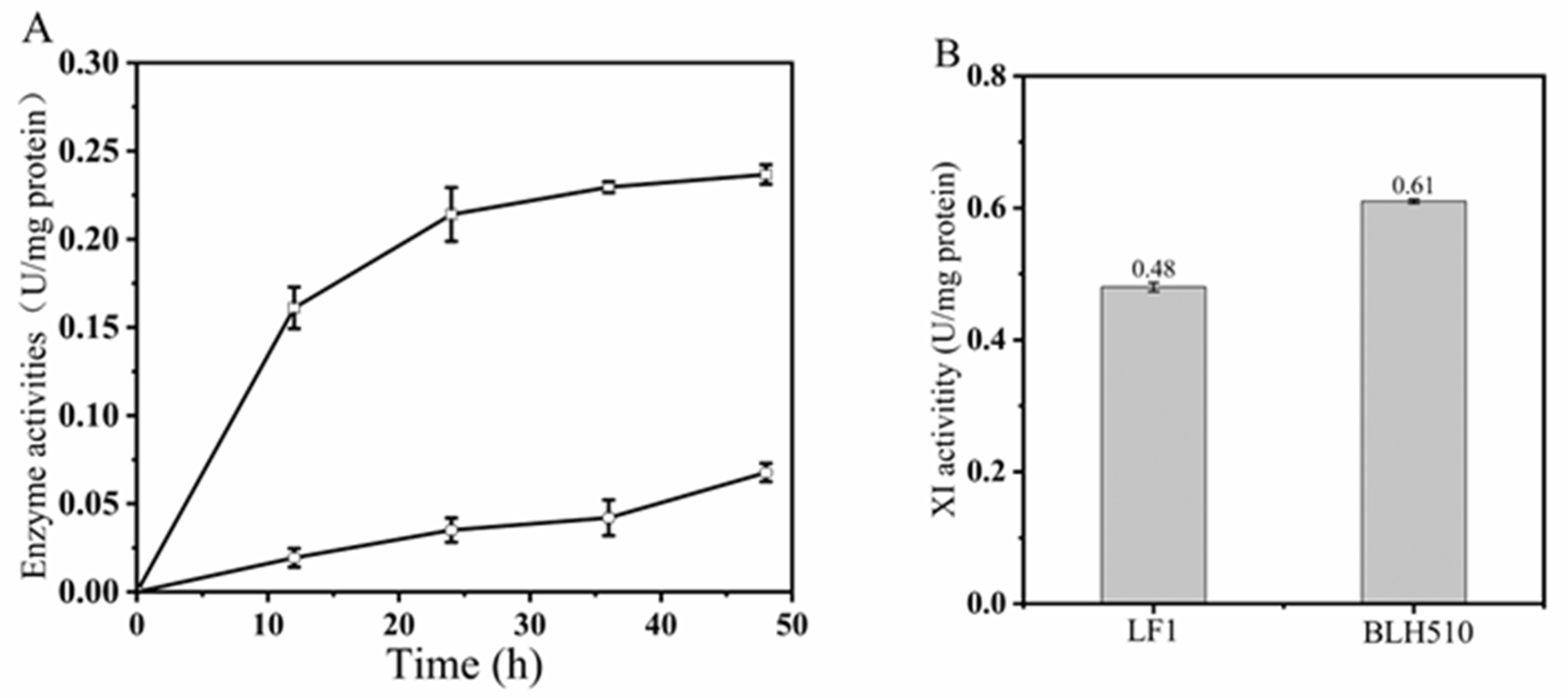
3.4.3. The Extracellular β-Glucosidase and β-Xylosidase Activities of BLH510
3.5. Oxygen-Limited Fermentation of BLH510
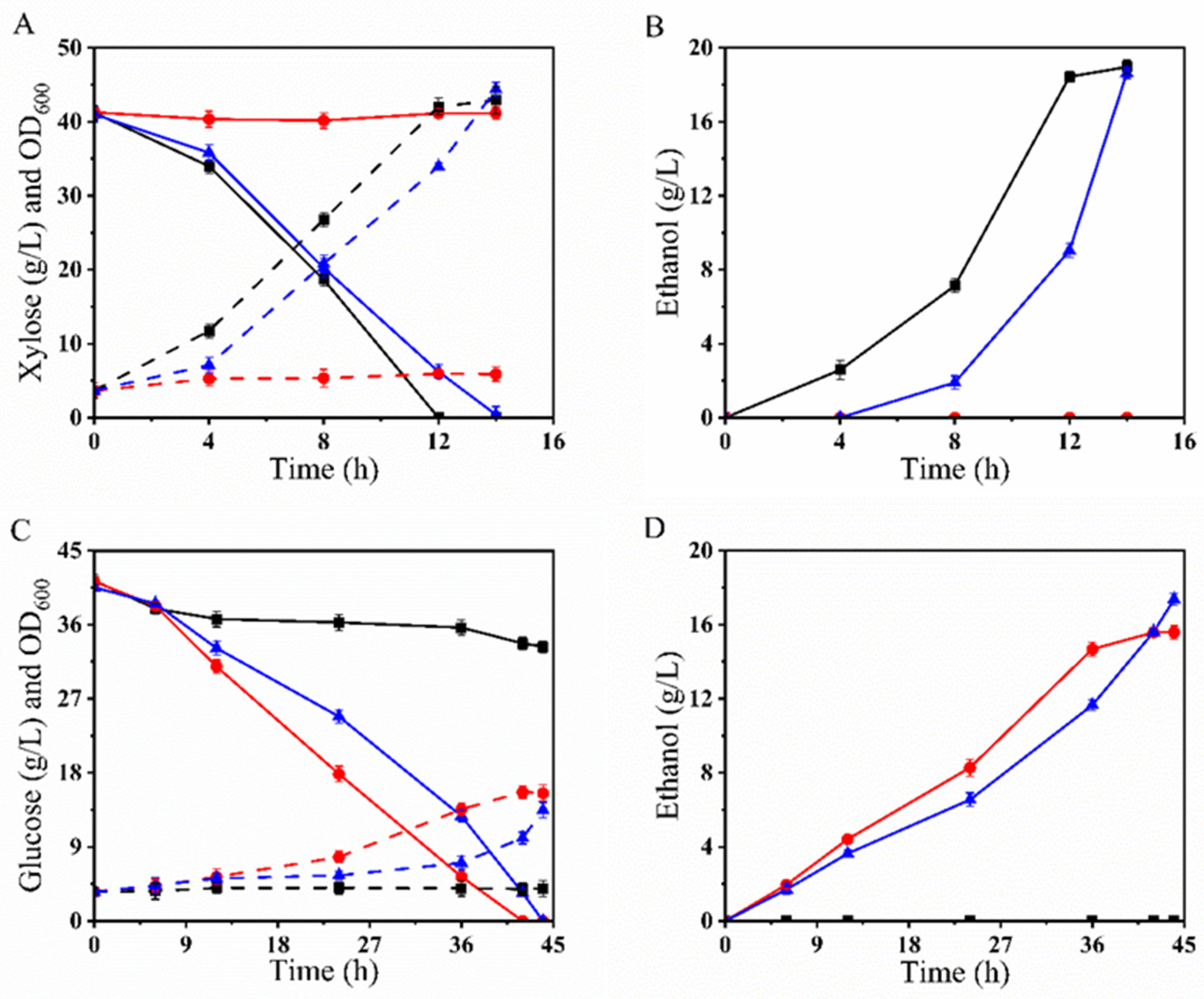
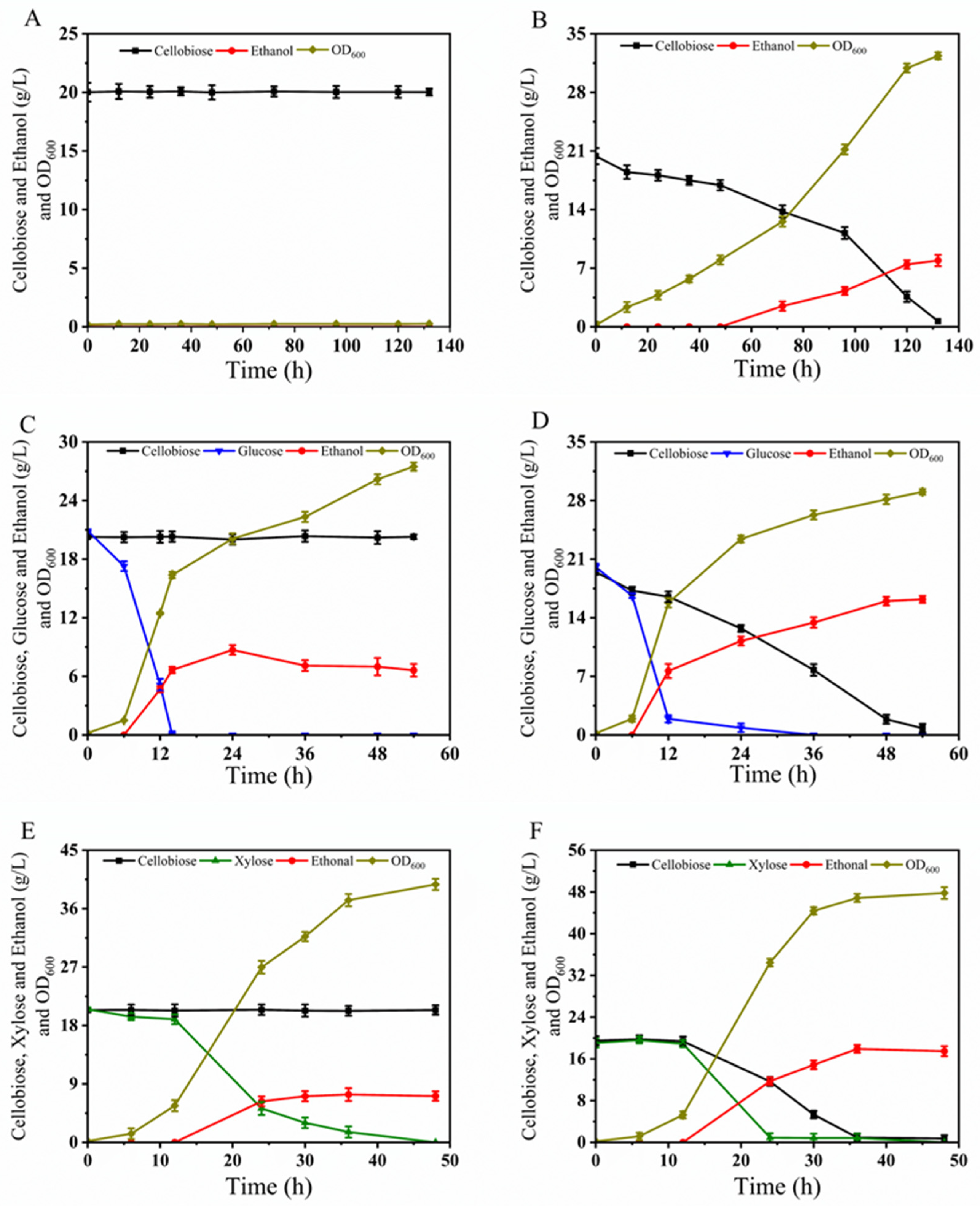
3.5.1. Fermentation of BLH510 with Cellobiose and the Mixture of Glucose–Cellobiose or Xylose–Cellobiose
3.5.2. Fermentation of BLH510 with the Mixture of Glucose–Xylose–Cellobiose
3.5.3. Fermentation of BLH510 with the Mixture of Glucose–Xylose–Cellobiose-XOSpre
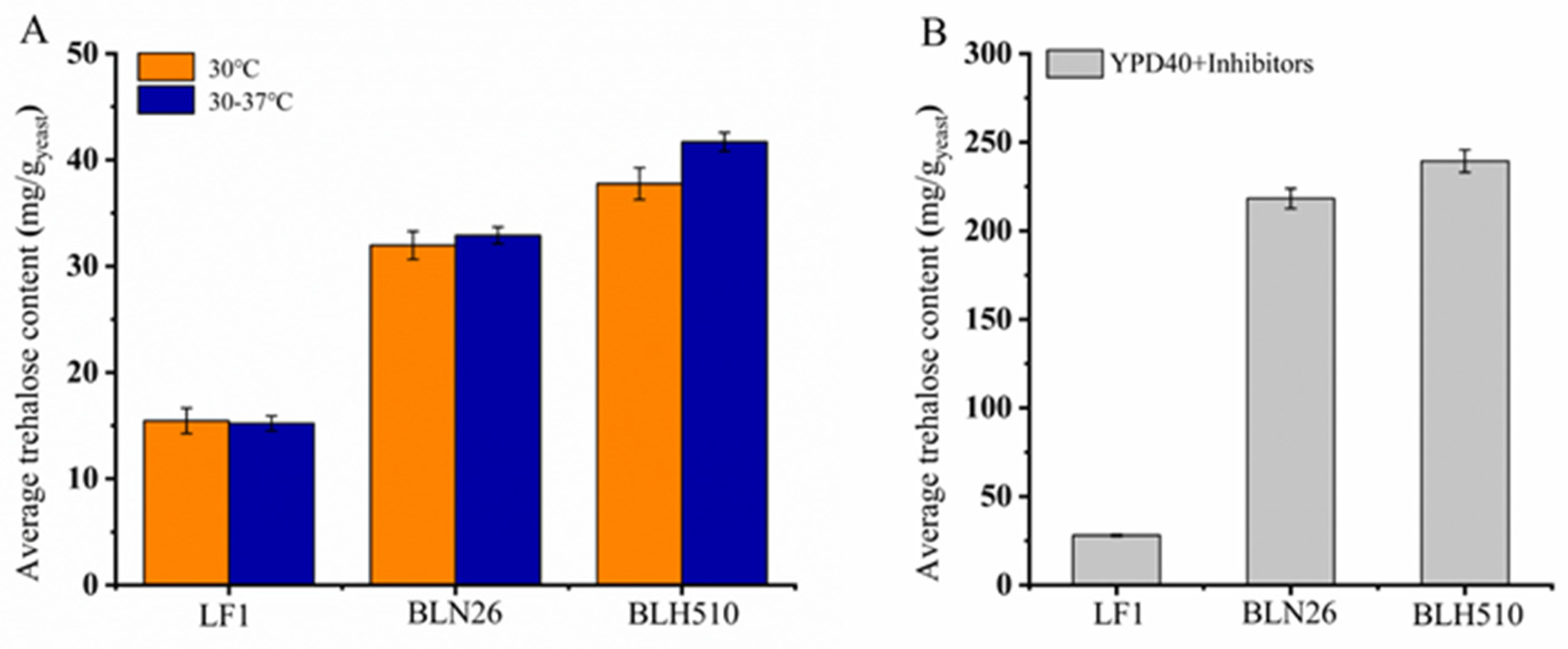
3.6. Determination of Intracellular Trehalose Content in S. cerevisiae Strains
3.7. Determination of the Ratio of Reduced and Oxidized Glutathione (GSH/GSSG)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Broda, M.; Yelle, D.J.; Serwańska, K. Bioethanol Production from Lignocellulosic Biomass—Challenges and Solutions. Molecules 2022, 27, 8717. [Google Scholar] [CrossRef] [PubMed]
- Raj, T.; Chandrasekhar, K.; Kumar, A.N.; Banu, J.R.; Yoon, J.-J.; Bhatia, S.K.; Yang, Y.-H.; Varjani, S.; Kim, S.-H. Recent Advances in Commercial Biorefineries for Lignocellulosic Ethanol Production: Current Status, Challenges and Future Perspectives. Bioresour. Technol. 2022, 344, 126292. [Google Scholar] [CrossRef]
- Liu, C.-G.; Xiao, Y.; Xia, X.-X.; Zhao, X.-Q.; Peng, L.; Srinophakun, P.; Bai, F.-W. Cellulosic Ethanol Production: Progress, Challenges and Strategies for Solutions. Biotechnol. Adv. 2019, 37, 491–504. [Google Scholar] [CrossRef]
- Carriquiry, M.A.; Du, X.; Timilsina, G.R. Second Generation Biofuels: Economics and Policies. Energy Policy 2011, 39, 4222–4234. [Google Scholar] [CrossRef]
- Alvira, P.; Tomás-Pejó, E.; Ballesteros, M.; Negro, M. Pretreatment Technologies for an Efficient Bioethanol Production Process Based on Enzymatic Hydrolysis: A Review. Bioresour. Technol. 2010, 101, 4851–4861. [Google Scholar] [CrossRef]
- Su, T.; Zhao, D.; Khodadadi, M.; Len, C. Lignocellulosic Biomass for Bioethanol: Recent Advances, Technology Trends, and Barriers to Industrial Development. Curr. Opin. Green Sustain. Chem. 2020, 24, 56–60. [Google Scholar] [CrossRef]
- Menegol, D.; Fontana, R.C.; Dillon, A.J.P.; Camassola, M. Second-Generation Ethanol Production from Elephant Grass at High Total Solids. Bioresour. Technol. 2016, 211, 280–290. [Google Scholar] [CrossRef]
- Biodegradation: Natural and Synthetic Materials|SpringerLink. Available online: https://link.springer.com/book/10.1007/978-1-4471-3470-1 (accessed on 7 July 2024).
- Ronan, P.; William Yeung, C.; Schellenberg, J.; Sparling, R.; Wolfaardt, G.M.; Hausner, M. A Versatile and Robust Aerotolerant Microbial Community Capable of Cellulosic Ethanol Production. Bioresour. Technol. 2013, 129, 156–163. [Google Scholar] [CrossRef]
- Demeke, M.M.; Dietz, H.; Li, Y.; Foulquié-Moreno, M.R.; Mutturi, S.; Deprez, S.; Den Abt, T.; Bonini, B.M.; Liden, G.; Dumortier, F.; et al. Development of a D-Xylose Fermenting and Inhibitor Tolerant Industrial Saccharomyces cerevisiae Strain with High Performance in Lignocellulose Hydrolysates Using Metabolic and Evolutionary Engineering. Biotechnol. Biofuels 2013, 6, 89. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Shen, Y.; Wu, M.; Hou, J.; Jiao, C.; Li, Z.; Liu, X.; Bao, X. Engineering a Wild-Type Diploid Saccharomyces cerevisiae Strain for Second-Generation Bioethanol Production. Bioresour. Bioprocess. 2016, 3, 51. [Google Scholar] [CrossRef]
- Yan, N.; Luan, T.; Yin, M.; Niu, Y.; Wu, L.; Yang, S.; Li, Z.; Li, H.; Zhao, J.; Bao, X. Co-Fermentation of Glucose–Xylose–Cellobiose–XOS Mixtures Using a Synthetic Consortium of Recombinant Saccharomyces cerevisiae Strains. Fermentation 2023, 9, 775. [Google Scholar] [CrossRef]
- Ko, J.K.; Um, Y.; Woo, H.M.; Kim, K.H.; Lee, S.-M. Ethanol Production from Lignocellulosic Hydrolysates Using Engineered Saccharomyces cerevisiae Harboring Xylose Isomerase-Based Pathway. Bioresour. Technol. 2016, 209, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Hahn-Hägerdal, B.; Karhumaa, K.; Fonseca, C.; Spencer-Martins, I.; Gorwa-Grauslund, M.F. Towards Industrial Pentose-Fermenting Yeast Strains. Appl. Microbiol. Biotechnol. 2007, 74, 937–953. [Google Scholar] [CrossRef] [PubMed]
- Palmqvist, E.; Hahn-Hägerdal, B. Fermentation of Lignocellulosic Hydrolysates. II: Inhibitors and Mechanisms of Inhibition. Bioresour. Technol. 2000, 74, 25–33. [Google Scholar] [CrossRef]
- Nagar, S.; Gupta, V.K.; Kumar, D.; Kumar, L.; Kuhad, R.C. Production and Optimization of Cellulase-Free, Alkali-Stable Xylanase by Bacillus Pumilus SV-85S in Submerged Fermentation. J. Ind. Microbiol. Biotechnol. 2010, 37, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Hou, J.; Shen, Y.; Xu, L.; Yang, H.; Fang, X.; Bao, X. High β-Glucosidase Secretion in Saccharomyces cerevisiae Improves the Efficiency of Cellulase Hydrolysis and Ethanol Production in Simultaneous Saccharification and Fermentation. J. Microbiol. Biotechnol. 2013, 23, 1577–1585. [Google Scholar] [CrossRef] [PubMed]
- Mert, M.J.; la Grange, D.C.; Rose, S.H.; van Zyl, W.H. Engineering of Saccharomyces cerevisiae to Utilize Xylan as a Sole Carbohydrate Source by Co-Expression of an Endoxylanase, Xylosidase and a Bacterial Xylose Isomerase. J. Ind. Microbiol. Biotechnol. 2016, 43, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Wu, L.; Shen, Y.; Zhao, J.; Zhang, J.; Yi, Y.; Li, H.; Bao, X. Coexpression of β-Xylosidase and Xylose Isomerase in Saccharomyces cerevisiae Improves the Efficiency of Saccharification and Fermentation from Xylo-Oligosaccharides. Cellulose 2019, 26, 7923–7937. [Google Scholar] [CrossRef]
- Gao, M.; Ploessl, D.; Shao, Z. Enhancing the Co-Utilization of Biomass-Derived Mixed Sugars by Yeasts. Front. Microbiol. 2019, 9, 3264. [Google Scholar] [CrossRef]
- Wei, S.; Bai, P.; Liu, Y.; Yang, M.; Ma, J.; Hou, J.; Liu, W.; Bao, X.; Shen, Y. A Thi2p Regulatory Network Controls the Post-Glucose Effect of Xylose Utilization in Saccharomyces cerevisiae. Front. Microbiol. 2019, 10, 1649. [Google Scholar] [CrossRef]
- Tang, R.; Ye, P.; Alper, H.S.; Liu, Z.; Zhao, X.; Bai, F. Identification and Characterization of Novel Xylose Isomerases from a Bos Taurus Fecal Metagenome. Appl. Microbiol. Biotechnol. 2019, 103, 9465–9477. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Huang, S.; Geng, A. Recombinant Diploid Saccharomyces cerevisiae Strain Development for Rapid Glucose and Xylose Co. Fermentation 2018, 4, 59. [Google Scholar] [CrossRef]
- Ho, N.W.; Chen, Z.; Brainard, A.P. Genetically Engineered Saccharomyces Yeast Capable of Effective Cofermentation of Glucose and Xylose. Appl. Environ. Microbiol. 1998, 64, 1852–1859. [Google Scholar] [CrossRef]
- Wei, F.; Li, M.; Wang, M.; Li, H.; Li, Z.; Qin, W.; Wei, T.; Zhao, J.; Bao, X. A C6/C5 Co-Fermenting Saccharomyces cerevisiae Strain with the Alleviation of Antagonism between Xylose Utilization and Robustness. GCB Bioenergy 2021, 13, 83–97. [Google Scholar] [CrossRef]
- Chen, Y.; Stabryla, L.; Wei, N. Improved Acetic Acid Resistance in Saccharomyces cerevisiae by Overexpression of the WHI2 Gene Identified through Inverse Metabolic Engineering. Appl. Environ. Microbiol. 2016, 82, 2156–2166. [Google Scholar] [CrossRef] [PubMed]
- Oh, E.J.; Wei, N.; Kwak, S.; Kim, H.; Jin, Y.-S. Overexpression of RCK1 Improves Acetic Acid Tolerance in Saccharomyces cerevisiae. J. Biotechnol. 2019, 292, 1–4. [Google Scholar] [CrossRef]
- Zhang, M.-M.; Xiong, L.; Tang, Y.-J.; Mehmood, M.A.; Zhao, Z.K.; Bai, F.-W.; Zhao, X.-Q. Enhanced Acetic Acid Stress Tolerance and Ethanol Production in Saccharomyces cerevisiae by Modulating Expression of the de Novo Purine Biosynthesis Genes. Biotechnol. Biofuels 2019, 12, 116. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.; van Rensburg, E.; Görgens, J.F. Simultaneously Improving Xylose Fermentation and Tolerance to Lignocellulosic Inhibitors through Evolutionary Engineering of Recombinant Saccharomyces cerevisiae harbouring Xylose Isomerase. BMC Biotechnol. 2014, 14, 41. [Google Scholar] [CrossRef]
- Mans, R.; Daran, J.-M.G.; Pronk, J.T. Under Pressure: Evolutionary Engineering of Yeast Strains for Improved Performance in Fuels and Chemicals Production. Curr. Opin. Biotechnol. 2018, 50, 47–56. [Google Scholar] [CrossRef]
- Demeke, M.M.; Dumortier, F.; Li, Y.; Broeckx, T.; Foulquié-Moreno, M.R.; Thevelein, J.M. Combining Inhibitor Tolerance and D-Xylose Fermentation in Industrial Saccharomyces cerevisiae for Efficient Lignocellulose-Based Bioethanol Production. Biotechnol. Biofuels 2013, 6, 120. [Google Scholar] [CrossRef]
- Koppram, R.; Albers, E.; Olsson, L. Evolutionary Engineering Strategies to Enhance Tolerance of Xylose Utilizing Recombinant Yeast to Inhibitors Derived from Spruce Biomass. Biotechnol. Biofuels 2012, 5, 32. [Google Scholar] [CrossRef] [PubMed]
- Kumari, J.A.; Panda, T. Intergeneric Hybridization of Trichoderma reesei QM9414 and Saccharomyces cerevisiae NCIM 3288 by Protoplast Fusion. Enzym. Microb. Technol. 1994, 16, 870–882. [Google Scholar] [CrossRef]
- Pasha, C.; Kuhad, R.C.; Rao, L.V. Strain Improvement of Thermotolerant Saccharomyces cerevisiae VS3 Strain for Better Utilization of Lignocellulosic Substrates. J. Appl. Microbiol. 2007, 103, 1480–1489. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, R.; Louie, S.; Gavrilovic, V.; Perry, K.; Stemmer, W.P.C.; Ryan, C.M.; del Cardayré, S. Genome Shuffling of Lactobacillus for Improved Acid Tolerance. Nat. Biotechnol. 2002, 20, 707–712. [Google Scholar] [CrossRef]
- Shitut, S.; Bergman, G.Ö.; Kros, A.; Rozen, D.E.; Claessen, D. Use of Permanent Wall-Deficient Cells as a System for the Discovery of New-to-Nature Metabolites. Microorganisms 2020, 8, 1897. [Google Scholar] [CrossRef] [PubMed]
- Dai, M.; Copley, S.D. Genome Shuffling Improves Degradation of the Anthropogenic Pesticide Pentachlorophenol by Sphingobium chlorophenolicum ATCC 39723. Appl. Environ. Microbiol. 2004, 70, 2391–2397. [Google Scholar] [CrossRef] [PubMed]
- Xin, Y.; Yang, M.; Yin, H.; Yang, J. Improvement of Ethanol Tolerance by Inactive Protoplast Fusion in Saccharomyces cerevisiae. BioMed Res. Int. 2020, 2020, 1979318. [Google Scholar] [CrossRef] [PubMed]
- Verma, N.; Bansal, M.C.; Kumar, V. Protoplast Fusion Technology and Its Biotechnological Applications. Chem. Eng. Trans. 2008, 14, 113–120. [Google Scholar]
- Wimalasena, T.T.; Greetham, D.; Marvin, M.E.; Liti, G.; Chandelia, Y.; Hart, A.; Louis, E.J.; Phister, T.G.; Tucker, G.A.; Smart, K.A. Phenotypic Characterisation of Saccharomyces spp. Yeast for Tolerance to Stresses Encountered during Fermentation of Lignocellulosic Residues to Produce Bioethanol. Microb. Cell Factories 2014, 13, 47. [Google Scholar] [CrossRef]
- Entian, K.-D.; Kötter, P. 23 Yeast Mutant and Plasmid Collections. In Methods in Microbiology; Brown, A.J.P., Tuite, M., Eds.; Yeast Gene Analysis; Academic Press: Cambridge, MA, USA, 1998; Volume 26, pp. 431–449. [Google Scholar]
- Güldener, U.; Heck, S.; Fielder, T.; Beinhauer, J.; Hegemann, J.H. A New Efficient Gene Disruption Cassette for Repeated Use in Budding Yeast. Nucleic Acids Res. 1996, 24, 2519–2524. [Google Scholar] [CrossRef]
- Li, H.; Wu, M.; Xu, L.; Hou, J.; Guo, T.; Bao, X.; Shen, Y. Evaluation of Industrial Saccharomyces cerevisiae Strains as the Chassis Cell for Second-Generation Bioethanol Production. Microb. Biotechnol. 2015, 8, 266–274. [Google Scholar] [CrossRef]
- Santos, A.; Alonso, A.; Belda, I.; Marquina, D. Cell Cycle Arrest and Apoptosis, Two Alternative Mechanisms for PMKT2 Killer Activity. Fungal Genet. Biol. 2013, 50, 44–54. [Google Scholar] [CrossRef]
- Sánchez, Ó.J.; Cardona, C.A. Trends in Biotechnological Production of Fuel Ethanol from Different Feedstocks. Bioresour. Technol. 2008, 99, 5270–5295. [Google Scholar] [CrossRef]
- Shen, Y.; Chen, X.; Peng, B.; Chen, L.; Hou, J.; Bao, X. An Efficient Xylose-Fermenting Recombinant Saccharomyces cerevisiae Strain Obtained through Adaptive Evolution and Its Global Transcription Profile. Appl. Microbiol. Biotechnol. 2012, 96, 1079–1091. [Google Scholar] [CrossRef]
- Kuyper, M.; Harhangi, H.R.; Stave, A.K.; Winkler, A.A.; Jetten, M.S.M.; de Laat, W.T.A.M.; den Ridder, J.J.J.; Op den Camp, H.J.M.; van Dijken, J.P.; Pronk, J.T. High-Level Functional Expression of a Fungal Xylose Isomerase: The Key to Efficient Ethanolic Fermentation of Xylose by Saccharomyces cerevisiae? FEMS Yeast Res. 2003, 4, 69–78. [Google Scholar] [CrossRef]
- Shi, H.; Zhang, Y.; Li, X.; Huang, Y.; Wang, L.; Wang, Y.; Ding, H.; Wang, F. A Novel Highly Thermostable Xylanase Stimulated by Ca2+ from Thermotoga thermarum: Cloning, Expression and Characterization. Biotechnol. Biofuels 2013, 6, 26. [Google Scholar] [CrossRef]
- Feng, D.; Li, L.; Yang, F.; Tan, W.; Zhao, G.; Zou, H.; Xian, M.; Zhang, Y. Separation of Ionic Liquid [Mmim][DMP] and Glucose from Enzymatic Hydrolysis Mixture of Cellulose Using Alumina Column Chromatography. Appl. Microbiol. Biotechnol. 2011, 91, 399–405. [Google Scholar] [CrossRef]
- Salazar-Cerezo, S.; de Vries, R.P.; Garrigues, S. Strategies for the Development of Industrial Fungal Producing Strains. J. Fungi 2023, 9, 834. [Google Scholar] [CrossRef]
- Peberdy, J.F.; Davis, B. Factors Influencing Protoplast Isolation; Peberdy, J.F., Ferenczy, L., Eds.; CRC Press: Boca Raton, FL, USA, 2020; pp. 45–71. [Google Scholar]
- Lyu, Y.; Wu, P.; Zhou, J.; Yu, Y.; Lu, H. Protoplast Transformation of Kluyveromyces marxianus. Biotechnol. J. 2021, 16, e2100122. [Google Scholar] [CrossRef]
- Carvalho, V.S.D.; Gómez-Delgado, L.; Curto, M.Á.; Moreno, M.B.; Pérez, P.; Ribas, J.C.; Cortés, J.C.G. Analysis and Application of a Suite of Recombinant Endo-β(1,3)-D-Glucanases for Studying Fungal Cell Walls. Microb. Cell Fact. 2021, 20, 126. [Google Scholar] [CrossRef]
- Ning, Y.; Hu, B.; Yu, H.; Liu, X.; Jiao, B.; Lu, X. Optimization of Protoplast Preparation and Establishment of Genetic Transformation System of an Arctic-Derived Fungus Eutypella sp. Front. Microbiol. 2022, 13, 769008. [Google Scholar] [CrossRef] [PubMed]
- Kavanagh, K.; Whittaker, P.A. Application of Protoplast Fusion to the Nonconventional Yeast. Enzym. Microb. Technol. 1996, 18, 45–51. [Google Scholar] [CrossRef]
- Li, J.; Liu, L.; Xu, L.; Wang, S.; Zhang, N.; Sun, C.; Yan, M. Interspecific Hybridization between Ganoderma lingzhi and G. resinaceum by PEG-Induced Double-Inactivated Protoplast Fusion. Horticulturae 2023, 9, 1129. [Google Scholar] [CrossRef]
- Rastogi, R.P.; Richa; Kumar, A.; Tyagi, M.B.; Sinha, R.P. Molecular Mechanisms of Ultraviolet Radiation-Induced DNA Damage and Repair. J. Nucleic Acids 2010, 2010, 592980. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.S.; Anjum, N.A.; Gill, R.; Jha, M.; Tuteja, N. DNA Damage and Repair in Plants under Ultraviolet and Ionizing Radiations. Sci. World J. 2015, 2015, 250158. [Google Scholar] [CrossRef]
- Zhao, K.; Ping, W.; Zhang, L.; Liu, J.; Lin, Y.; Jin, T.; Zhou, D. Screening and Breeding of High Taxol Producing Fungi by Genome Shuffling. Sci. China Ser. C-Life Sci. 2008, 51, 222–231. [Google Scholar] [CrossRef]
- Kavanagh, K.; Walsh, M.; Whittaker, P.A. Enhanced Intraspecific Protoplast Fusion in Yeast. FEMS Microbiol. Lett. 1991, 81, 283–286. [Google Scholar] [CrossRef]
- Reeves, E.; Kavanagh, K.; Whittaker, P.A. Multiple Transformation of Saccharomyces cerevisiae by Protoplast Fusion. FEMS Microbiol. Lett. 1992, 99, 193–197. [Google Scholar] [CrossRef]
- Curran, B.P.; Bugeja, V.C. Protoplast Fusion in Saccharomyces cerevisiae. Methods Mol. Biol. 1996, 53, 45–49. [Google Scholar] [CrossRef]
- Kawai, S.; Hashimoto, W.; Murata, K. Transformation of Saccharomyces cerevisiae and Other Fungi. Bioeng. Bugs 2010, 1, 395–403. [Google Scholar] [CrossRef]
- Gerstein, A.C.; Lim, H.; Berman, J.; Hickman, M.A. Ploidy Tug-of-War: Evolutionary and Genetic Environments Influence the Rate of Ploidy Drive in a Human Fungal Pathogen. Evolution 2017, 71, 1025–1038. [Google Scholar] [CrossRef]
- Andreychuk, Y.; Zhuk, A.; Tarakhovskaya, E.; Inge-Vechtomov, S.; Stepchenkova, E. Rate of Spontaneous Polyploidization in Haploid Yeast Saccharomyces cerevisiae. Biol. Commun. 2022, 67, 88–96. [Google Scholar] [CrossRef]
- Fang, A.Q.; Li, S.L.; Chen, Y.W.; Li, P. Study on the Protoplast Fusion between a Thermotolerant Yeast and Saccharomyces cerevisiae. Chin. J. Biotechnol. 1990, 6, 207–213. [Google Scholar] [PubMed]
- Sarachek, A.; Weber, D.A. Segregant-Defective Heterokaryons of Candida albicans. Curr. Genet. 1986, 10, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Maráz, A.; Šubík, J. Transmission and Recombination of Mitochondrial Genes in Saccharomyces cerevisiae after Protoplast Fusion. Mol. Gen. Genet. 1981, 181, 131–133. [Google Scholar] [CrossRef]
- Ezeji, U.; Kalu, I.; Chinakwe, E. Production of High-Ethanol-Yielding Saccharomyces cerevisiae of Palm Wine Origin by Protoplast Fusion. Life Sci. 2008, 5, 64–68. [Google Scholar]
- Suzuki, T.; Yamada, M.; Sakaguchi, S. Occurrence of Chromosome Rearrangements during the Fusion Process in the Imperfect Yeast Candida albicans. Microbiology 1994, 140 Pt 12, 3319–3328. [Google Scholar] [CrossRef] [PubMed]
- Lacefield, S.; Magendantz, M.; Solomon, F. Consequences of Defective Tubulin Folding on Heterodimer Levels, Mitosis and Spindle Morphology in Saccharomyces cerevisiae. Genetics 2006, 173, 635–646. [Google Scholar] [CrossRef]
- Yu, W.; Yuan, R.; Liu, M.; Liu, K.; Ding, X.; Hou, Y. Effects of rpl1001 Gene Deletion on Cell Division of Fission Yeast and Its Molecular Mechanism. Curr. Issues Mol. Biol. 2024, 46, 2576–2597. [Google Scholar] [CrossRef]
- Kara, M.; Öztaş, E.; Boran, T.; Sevim, Ç.; Keskin, S.E.; Veskoukis, A.S.; Kuzmin, S.V.; Tsatsakis, A.M. The Sesquiterpenoid Valerenic Acid Protects Neuronal Cells from the Detrimental Effects of the Fungicide Benomyl on Apoptosis and DNA Oxidation. Hum. Exp. Toxicol. 2022, 41, 9603271221101038. [Google Scholar] [CrossRef]
- Wiebe, M.G.; Rintala, E.; Tamminen, A.; Simolin, H.; Salusjärvi, L.; Toivari, M.; Kokkonen, J.T.; Kiuru, J.; Ketola, R.A.; Jouhten, P.; et al. Central Carbon Metabolism of Saccharomyces cerevisiae in Anaerobic, Oxygen-Limited and Fully Aerobic Steady-State Conditions and Following a Shift to Anaerobic Conditions. FEMS Yeast Res. 2008, 8, 140–154. [Google Scholar] [CrossRef] [PubMed]
- Ye, W.; Zhang, W.; Liu, T.; Tan, G.; Li, H.; Huang, Z. Improvement of Ethanol Production in Saccharomyces cerevisiae by High-Efficient Disruption of the ADH2 Gene Using a Novel Recombinant TALEN Vector. Front. Microbiol. 2016, 7, 1067. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.N.; Jiang, X.Z.; Tian, B.Y.; Shu, Z.Y.; Huang, J.Z. Overexpression of Alcohol Dehydrogenase I in Saccharomyces cerevisiae. Microbiol. China 1992, 35, 0261–0266. [Google Scholar]
- Claes, A.; Deparis, Q.; Foulquié-Moreno, M.R.; Thevelein, J.M. Simultaneous Secretion of Seven Lignocellulolytic Enzymes by an Industrial Second-Generation Yeast Strain Enables Efficient Ethanol Production from Multiple Polymeric Substrates. Metab. Eng. 2020, 59, 131–141. [Google Scholar] [CrossRef]
- Schebor, C.; Galvagno, M.; del Pilar Buera, M.; Chirife, J. Glass Transition Temperatures and Fermentative Activity of Heat-Treated Commercial Active Dry Yeasts. Biotechnol. Prog. 2000, 16, 163–168. [Google Scholar] [CrossRef]



| Name | Primes Sequence (5′→3′) | Description |
|---|---|---|
| BGL-F | AGATCTACGTATGGACTTTCTTCTTCAG | BGL |
| BGL-R | AAGGGTTGTCGACCTGCAGCTATTCCTTCAATTCAGCAACAAAATTTGAA | |
| loxP-F | GCTGCAGGTCGACAACCCTTAAT | KanMX |
| loxP-R | GGCCACTAGTGGATCTGATATCACC | |
| XYL-F | GGAAGTACCTTCAAAGAATGGGGTC | IBX |
| XYL-R | GGGTTGTCGACCTGCAGCCTTCGAGCGTCCCAAAACCTTC | |
| U1-ADH2-F | GAAGGTGCCGGTGTCGTT | UP1 adh2 |
| U1-ADH2-R | GCAAATACGTAGATCTGCGTCAGCGGTAGCGTATt | |
| D-ADH2-F | CCACTAGTGGCCGGCTACCAACGGCGGT | DOWN adh2 |
| D-ADH2-R | CAGCTCTGTTCCCCACGTAAGA | |
| U2-ADH2-F | CAATCTTGTGTGCTGGTATCACCG | UP2 adh2 |
| U2-ADH2-R | GACCATACGTAGATCTTTCACCACCGAGCGAGGTAAA | |
| UP1-δ-F | TATGTATTTTTAATCGTCCTTGTATGGAAGTATCAAAGG | δ 1 |
| UP1-δ-R | GACCCCATTCTTTGAAGGTACTTCCCAACAACCTCTTGCTATCAAACTTACTTTTG | |
| D-δ-F | GGTGATATCAGATCCACTAGTGGCCGGCTTCATTGTAACATGTAAGTGAACATC | δ 2 |
| D-δ-R | CAGGCTTATAATGTCAGTATGCATTGTTACG | |
| UP2-δ-F | AACGTTAAAAGAGTAGCTCAGACAGTG | δ 3 |
| UP2-δ-R | GACCCCATTCTTTGAAGGTACTTCCGAATATGGATCTTAATATAATCGTATAAGAGGGGC |
| Zymolyase Concentration (U/g Wet Cell Weight) | Protoplast Preparation Rate | Protoplast Regeneration Rate | ||
|---|---|---|---|---|
| LF1 | BLN26 | LF1 | BLN26 | |
| 5 | 99.99% | 99.99% | Not detected | Not detected |
| 3 | 99.99% | 99.99% | 0.09% | 1.219% |
| 2 | 99.95% | 99.85% | 12.50% | 22.07% |
| 1 | 99.98% | 99.76% | 42.60% | 54.25% |
| 0.5 | 91.97% | 90.5% | 40.21% | 45.82% |
| Heat treatment (min) | 2 | 5 | 10 | 15 | 20 |
| lethality rate of LF1 (%) | 15.8 ± 0.01 | 90.1 ± 0.02 | 98.5 ± 0.00 | 100 ± 0.00 | 100 ± 0.00 |
| UV treatment (min) | 1 | 2 | 5 | 7 | 10 |
| lethality rate of BLN26 (%) | 56.3 ± 0.02 | 87.5 ± 0.01 | 95.4 ± 0.02 | 100 ± 0.00 | 100 ± 0.00 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, J.; Zhao, Y.; Wu, L.; Yan, N.; Yang, S.; Xu, L.; He, D.; Li, H.; Bao, X. Development of a Robust Saccharomyces cerevisiae Strain for Efficient Co-Fermentation of Mixed Sugars and Enhanced Inhibitor Tolerance through Protoplast Fusion. Microorganisms 2024, 12, 1526. https://doi.org/10.3390/microorganisms12081526
Zhao J, Zhao Y, Wu L, Yan N, Yang S, Xu L, He D, Li H, Bao X. Development of a Robust Saccharomyces cerevisiae Strain for Efficient Co-Fermentation of Mixed Sugars and Enhanced Inhibitor Tolerance through Protoplast Fusion. Microorganisms. 2024; 12(8):1526. https://doi.org/10.3390/microorganisms12081526
Chicago/Turabian StyleZhao, Jianzhi, Yuping Zhao, Longhao Wu, Ning Yan, Shuo Yang, Lili Xu, Deyun He, Hongxing Li, and Xiaoming Bao. 2024. "Development of a Robust Saccharomyces cerevisiae Strain for Efficient Co-Fermentation of Mixed Sugars and Enhanced Inhibitor Tolerance through Protoplast Fusion" Microorganisms 12, no. 8: 1526. https://doi.org/10.3390/microorganisms12081526