
Phylogenetic Analysis of Alphacoronaviruses Based on 3c and M Gene Sequences Isolated from Cats with FIP in Romania

Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples
2.2. Coronavirus Antibodies
2.3. RNA Extraction from Effusions and Faecal Samples
2.4. RNA Extraction from Organ Samples
2.5. Amplification Using Real-Time RT-qPCR
2.6. Amplification Using RT-PCR
2.7. Sequencing
2.8. Phylogenetic Analysis
3. Results
3.1. Coronavirus Antibodies Titre
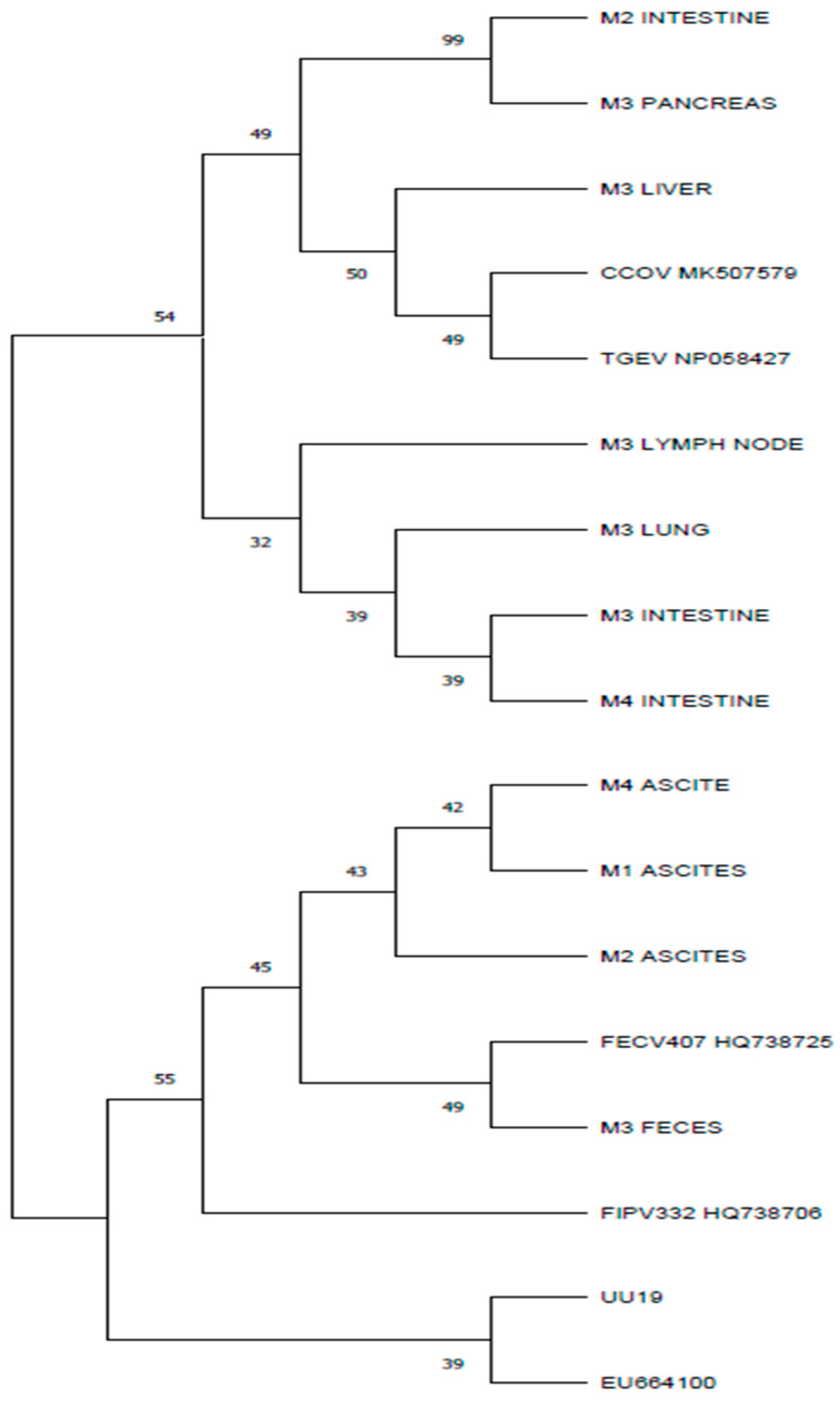
3.2. Amplification, Sequencing, and Phylogenetic Analysis
3.3. Results for the 3c Gene
3.4. Results for M Gene
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Cui, J.; Li, F.; Shi, Z.L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Woo, P.C.; Lau, S.K.; Huang, Y.; Yuen, K.Y. Coronavirus diversity, phylogeny and interspecies jumping. Exp. Biol. Med. 2009, 234, 1117–1127. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 588, E6. [Google Scholar] [CrossRef] [PubMed]
- Decaro, N.; Mari, V.; Campolo, M.; Lorusso, A.; Camero, M.; Elia, G.; Martella, V.; Cordioli, P.; Enjuanes, L.; Buonavoglia, C. Recombinant canine coronaviruses related to transmissible gastroenteritis virus of Swine are circulating in dogs. J. Virol. 2009, 83, 1532–1537. [Google Scholar] [CrossRef] [PubMed]
- Le Poder, S.; Pham-Hung d’Alexandryd’Orangiani, A.L.; Duarte, L.; Fournier, A.; Horhogea, C.; Pinhas, C.; Vabret, A.; Eloit, M. Infection of cats with atypical feline coronaviruses harbouring a truncated form of the canine type I non-structural ORF3 gene. Infect. Genet. Evol. 2013, 20, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Terada, Y.; Matsui, N.; Noguchi, K.; Kuwata, R.; Shimoda, H.; Soma, T.; Mochizuki, M.; Maeda, K. Emergence of Pathogenic Coronaviruses in Cats by Homologous Recombination between Feline and Canine Coronaviruses. PLoS ONE. 2014, 9, e106534. [Google Scholar] [CrossRef] [PubMed]
- Fehr, A.R.; Perlman, S. Coronaviruses: An overview of their replication and pathogenesis. Methods Mol. Biol. 2015, 1282, 1–23. [Google Scholar] [CrossRef]
- Borschensky, C.M.; Reinacher, M. Mutations in the 3c and 7b genes of feline coronavirus in spontaneously affected FIP cats. Res. Vet. Sci. 2014, 97, 333–340. [Google Scholar] [CrossRef]
- Myrrha, L.W.; Silva, F.M.F.; Vidigal, P.M.P.; Resende, M.; Bressan, G.C.; Fietto, J.L.R.; Santos, M.R.; Silva, L.M.N.; Assao, V.S.; Silva-JúNior, A.; et al. Feline coronavirus isolates from a part of Brazil: Insights into molecular epidemiology and phylogeny inferred from the 7b gene. J. Vet. Med. Sci. 2019, 81, 1455–1460. [Google Scholar] [CrossRef]
- Chang, H.W.; Egberink, H.F.; Halpin, R.; Spiro, D.J.; Rottier, P.J. Spike protein fusion peptide and feline coronavirus virulence. Emerg. Infect. Dis. 2012, 18, 1089–1095. [Google Scholar] [CrossRef]
- Chang, H.W.; de Groot, R.J.; Egberink, H.F.; Rottier, P.J. Feline infectious peritonitis: Insights into feline coronavirus pathobiogenesis and epidemiology based on genetic analysis of the viral 3c gene. J. Gen. Virol. 2010, 91 Pt 2, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Hora, A.S.; Tonietti, P.O.; Taniwaki, S.A.; Asano, K.M.; Maiorka, P.; Richtzenhain, L.J.; Brandão, P.E. Feline Coronavirus 3c Protein: A Candidate for a Virulence Marker? Biomed. Res. Int. 2016, 2016, 8560691. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.A.; Troyer, J.L.; Pecon-Slattery, J.; Roelke, M.E.; O’Brien, S.J. Genetics and pathogenesis of feline infectious peritonitis virus. Emerg. Infect. Dis. 2009, 15, 1445–1452. [Google Scholar] [CrossRef] [PubMed]
- Herrewegh, A.A.; Smeenk, I.; Horzinek, M.C.; Rottier, P.J.; de Groot, R.J. Feline coronavirus type II strains 79-1683 and 79–1146 originate from a double recombination between feline coronavirus type I and canine coronavirus. J. Virol. 1998, 72, 4508–4514. [Google Scholar] [CrossRef] [PubMed]
- Vennema, H.; Poland, A.; Foley, J.; Pedersen, N.C. Feline infectious peritonitis viruses arise by mutation from endemic feline enteric coronaviruses. Virology 1998, 243, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Herrewegh, A.A.; de Groot, R.J.; Cepica, A.; Egberink, H.F.; Horzinek, M.C.; Rottier, P.J. Detection of feline coronavirus RNA in feces, tissues, and body fluids of naturally infected cats by reverse transcriptase PCR. J. Clin. Microbiol. 1995, 33, 684–689. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.S.; Porter, E.; Matthews, D.; Kipar, A.; Tasker, S.; Helps, C.R.; Siddell, S.G. Genotyping coronaviruses associated with feline infectious peritonitis. J. Gen. Virol. 2015, 96 Pt 6, 1358–1368. [Google Scholar] [CrossRef] [PubMed]
- Oguma, K.; Ohno, M.; Yoshida, M.; Sentsui, H. Mutation of the S and 3c genes in genomes of feline coronaviruses. J. Vet. Med. Sci. 2018, 80, 1094–1100. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chang, H.W.; Egberink, H.F.; Rottier, P.J. Sequence analysis of feline coronaviruses and the circulating virulent/avirulent theory. Emerg. Infect. Dis. 2011, 17, 744. [Google Scholar] [CrossRef] [PubMed]
- Almazán, F.; González, J.M.; Pénzes, Z.; Izeta, A.; Calvo, E.; Plana-Durán, J.; Enjuanes, L. Engineering the largest RNA virus genome as an infectious bacterial artificial chromosome. Proc. Natl. Acad. Sci. USA 2000, 97, 5516-21. [Google Scholar] [CrossRef] [PubMed]
- Felten, S.; Klein-Richers, U.; Hofmann-Lehmann, R.; Bergmann, M.; Unterer, S.; Leutenegger, C.M.; Hartmann, K. Correlation of Feline Coronavirus Shedding in Feces with Coronavirus Antibody Titer. Pathogens 2020, 9, 598. [Google Scholar] [CrossRef]
- Pedersen, N.C.; Liu, H.; Dodd, K.A.; Pesavento, P.A. Significance of coronavirus mutants in feces and diseased tissues of cats suffering from feline infectious peritonitis. Viruses 2009, 1, 166–184. [Google Scholar] [CrossRef]
- Bank-Wolf, B.R.; Stallkamp, I.; Wiese, S.; Moritz, A.; Tekes, G.; Thiel, H.J. Mutations of 3c and spike protein genes correlate with the occurrence of feline infectious peritonitis. Vet. Microbiol. 2014, 173, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Hora, A.S.; Asano, K.M.; Guerra, J.M.; Mesquita, R.G.; Maiorka, P.; Richtzenhain, L.J.; Brandão, P.E. Intrahost diversity of feline coronavirus: A consensus between the circulating virulent/avirulent strains and the internal mutation hypotheses? Sci. World J. 2013, 27, 572325. [Google Scholar] [CrossRef]
- Dye, C.; Siddell, S.G. Genomic RNA sequence of feline coronavirus strain FCoV C1Je. J. Feline. Med. Surg. 2007, 9, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Addie, D.D.; Schaap, I.A.T.; Nicolson, L.; Jarrett, O. Persistence and transmission of natural type I feline coronavirus infection. J. Gen. Virol. 2003, 84 Pt 10, 2735–2744. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, N.C.; Liu, H.; Scarlett, J.; Leutenegger, C.M.; Golovko, L.; Kennedy, H.; Kamal, F.M. Feline infectious peritonitis: Role of the feline coronavirus 3c gene in intestinal tropism and pathogenicity based upon isolates from resident and adopted shelter cats. Virus Res. 2012, 165, 17–28. [Google Scholar] [CrossRef]
- Tekes, G.; Thiel, H.J. Feline Coronaviruses: Pathogenesis of Feline Infectious Peritonitis. Adv. Virus Res. 2016, 96, 193–218. [Google Scholar] [CrossRef]
- Thiel, V.; Thiel, H.J.; Tekes, G. Tackling feline infectious peritonitis via reverse genetics. Bioengineered 2014, 5, 396–400. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Coronavirus Strain | GenBank Accession No | Year of Collection | Host | Source |
|---|---|---|---|---|
| FIPV332 | HQ738706 | 2009 | cat | [19] |
| FECV407 | HQ738725 | 2010 | cat | [19] |
| TGEV_NP058427 | NP_058427 | Tissue culture adapted clone of the TGEV Purdue strain | pig | [20] |
| CCoV_MK507579 | MK507579 | 2018 | dog | Not available |
| EU664100 (FIPV) | EU664100 | 2004 | cat | [13] |
| UU19 (FECV-complete genome) | HQ392470 | 2007 | cat | [18] |
| Coronavirus Strain | GenBank Accession No | Year of Collection | Host | Source |
|---|---|---|---|---|
| Feline Coronavirus isolate 26 M | KP143512 | 2013 | cat | [17] |
| Feline coronavirus isolate 27C | KP143507 | 2013 | cat | [17] |
| Feline coronavirus isolate 28O | KP143508 | 2013 | cat | [17] |
| UU19 (FECV-complete genome) | HQ392470 | 2007 | cat | [18] |
| Canine coronavirus strain 119/08 | EU924791 | 2008 | dog | [4] |
| Cat | M1 | M2 | M3 | M4 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sample | Real-Time-RT-PCR | RT-PCR | Sequencing | real-Time-RT-PCR | RT-PCR | Sequencing | Real-Time-RT-PCR | RT-PCR | Sequencing | Real-Time--RT-PCR | RT-PCR | Sequencing |
| ascites | + | - | - | + | + | 471 pb | NA | NA | NA | + | + | 390 pb |
| feces | NA | NA | NA | NA | NA | NA | + | + | 727 pb | + | - | - |
| heart | NA | NA | NA | + | - | - | + | - | - | NA | NA | NA |
| kidney | + | - | - | + | - | - | + | - | - | + | - | - |
| large intestine | NA | NA | NA | NA | NA | NA | + | - | - | NA | - | - |
| liver | + | - | - | + | - | - | + | - | - | + | - | - |
| lung | + | - | - | + | - | - | + | - | - | + | NA | NA |
| lymph node | + | + | 430 pb | NA | NA | NA | + | + | 442 pb | + | + | - |
| pancreas | NA | NA | NA | NA | NA | NA | + | + | 405 pb | NA | NA | NA |
| small intestine | + | - | - | + | + | 432 pb | + | + | - | + | + | - |
| spleen | + | - | - | + | - | - | - | - | - | + | NA | NA |
| Cat | M1 | M2 | M3 | M4 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sample | Real-Time-RT-PCR | RT-PCR | Sequencing | Real-Time-RT-PCR | RT-PCR | Sequencing | Real-Time-RT-PCR | RT-PCR | Sequencing | Real-Time- RT-PCR | RT-PCR | Sequencing |
| ascites | + | + | 374 pb | + | + | 373 pb | NA | NA | NA | + | + | 368 pb |
| feces | NA | NA | NA | NA | NA | NA | + | + | 382 pb | + | NA | NA |
| heart | NA | NA | NA | + | NA | NA | + | NA | NA | NA | NA | NA |
| kidney | + | NA | NA | + | + | - | + | NA | NA | + | NA | NA |
| large intestine | NA | NA | NA | NA | NA | NA | + | + | 201 pb | + | NA | NA |
| liver | + | NA | NA | + | NA | NA | + | + | 104 pb | + | NA | NA |
| lung | + | NA | NA | + | NA | NA | + | + | 200 pb | + | NA | NA |
| lymph node | + | + | - | NA | NA | NA | + | + | 189 pb | NA | + | - |
| pancreas | NA | NA | NA | NA | NA | NA | + | + | 194 pb | + | NA | NA |
| small intestine | + | NA | NA | + | + | 181 pb | + | NA | NA | + | + | 263 pb |
| spleen | + | NA | NA | + | NA | NA | - | NA | NA | NA | NA | NA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Popovici, I.; Le Poder, S.; Rîmbu, C.-M.; Horhogea, C.-E. Phylogenetic Analysis of Alphacoronaviruses Based on 3c and M Gene Sequences Isolated from Cats with FIP in Romania. Microorganisms 2024, 12, 1557. https://doi.org/10.3390/microorganisms12081557
Popovici I, Le Poder S, Rîmbu C-M, Horhogea C-E. Phylogenetic Analysis of Alphacoronaviruses Based on 3c and M Gene Sequences Isolated from Cats with FIP in Romania. Microorganisms. 2024; 12(8):1557. https://doi.org/10.3390/microorganisms12081557
Chicago/Turabian StylePopovici, Ivona, Sophie Le Poder, Cristina-Mihaela Rîmbu, and Cristina-Elena Horhogea. 2024. "Phylogenetic Analysis of Alphacoronaviruses Based on 3c and M Gene Sequences Isolated from Cats with FIP in Romania" Microorganisms 12, no. 8: 1557. https://doi.org/10.3390/microorganisms12081557