Abstract
A novel Gram-stain-negative, facultatively anaerobic, and mixotrophic bacterium, designated as strain LZ166T, was isolated from the bathypelagic seawater in the western Pacific Ocean. The cells were short rod-shaped, oxidase- and catalase-positive, and motile by means of lateral flagella. The growth of strain LZ166T was observed at 10–45 °C (optimum 34–37 °C), at pH 5–10 (optimum 6–8), and in the presence of 0–5% NaCl (optimum 1–3%). A phylogenetic analysis based on the 16S rRNA gene showed that strain LZ166T shared the highest similarity (98.58%) with Aquibium oceanicum B7T and formed a distinct branch within the Aquibium genus. The genomic characterization, including average nucleotide identity (ANI, 90.73–76.79%), average amino identity (AAI, 88.50–79.03%), and digital DNA–DNA hybridization (dDDH, 36.1–22.2%) values between LZ166T and other species within the Aquibium genus, further substantiated its novelty. The genome of strain LZ166T was 6,119,659 bp in size with a 64.7 mol% DNA G+C content. The predominant fatty acid was summed feature 8 (C18:1ω7c and/or C18:1ω6c). The major polar lipids identified were diphosphatidylglycerol (DPG), phosphatidylethanolamine (PE), glycolipid (GL), and phosphatidylglycerol (PG), with ubiquinone-10 (Q-10) as the predominant respiratory quinone. The genomic annotation indicated the presence of genes for a diverse metabolic profile, including pathways for carbon fixation via the Calvin–Benson–Bassham cycle and inorganic sulfur oxidation. Based on the polyphasic taxonomic results, strain LZ166T represented a novel species of the genus Aquibium, for which the name Aquibium pacificus sp. nov. is proposed, with the type strain LZ166T (=MCCC M28807T = KACC 23148T = KCTC 82889T).
1. Introduction
The genus Aquibium, derived from its isolation from aquatic environments, was first proposed by Kim et al. and classified within the family Phyllobacteriaceae [1]. At the time of writing, only three species within the genus Aquibium have been validly published according to the List of Prokaryotic names with Standing in Nomenclature (LPSN, http://lpsn.dsmz.de/search?word=Aquibium accessed on 20 June 2024) [2]. The type species is Aquibium microcysteis, with the other two species, Aquibium carbonis and Aquibium oceanicum, having been reclassified from the genus Mesorhizobium [3]. While most of the species of Mesorhizobium were isolated from the rhizospheres of leguminous plants, the Aquibium species originated from aquatic environments such as coal bed water [4], deep-sea water [5], and cultures of Microcystis aeruginos [1]. The genomic analysis suggested that Aquibium species might be ancestral to those in Mesorhizobium, with some Mesorhizobium members acquiring nitrogen-fixing genes over the course of evolution [6]. Species within Aquibium consistently exhibit characteristics such as being Gram-straining-negative, aerobic, and catalase- and oxidase-positive. The genomic DNA G+C content, predominant fatty acid, and respiratory quinone for these species are 65.1–67.9 mol%, summed feature 8 (C18:1ω7c and/or C18:1ω6c), and ubiquinone-10 (Q-10), respectively [1].
Marine microorganisms play a crucial role in global carbon cycling and ecological interactions. Mixotrophy, a key trophic strategy involving the concurrent use of both autotrophic and heterotrophic nutrition, significantly influences these cycles [7]. This metabolic flexibility, as observed in ubiquitous species like Prochlorococcus [8], SAR324 [9], and Arcobacteraceae [10], enables adaptation to the fluctuating marine environment [11]. While some progress has been achieved, gaps still exist in the aspect of the mixotrophic microorganisms, their metabolic mechanisms, and their ecological roles.
In this study, a novel strain, designated as LZ166T, was isolated from bathypelagic seawater in the western Pacific Ocean. A comprehensive investigation of the morphology, physiology, chemotaxonomy, and phylogeny was conducted to elucidate its taxonomy status and consequently denominate it as Aquibium pacificus sp. nov. Furthermore, genomic analyses were performed to delineate the mixotrophic lifestyle supported by the flexible metabolic functions of carbon, nitrogen, and sulfur in this novel strain.
2. Materials and Methods
2.1. Isolation and Culture
Strain LZ166T was isolated from the bathypelagic seawater of the western Pacific Ocean at station CTD-11 (1000 m depth, 16°13′12″ N, 130°22′12″ E) during the DY60 cruise in January 2021. Seawater samples were collected and filtered through a 0.2 μm polycarbonate (PC) membrane (Merck, Rahway, NJ, USA) on board. The membrane was then immediately transferred to Axygen screw cap tubes containing 30% (v/v) glycerol, and subsequently stored at −80 °C for preservation prior to laboratory analysis. Once in the laboratory, microbes attached to the membrane were washed off and serially diluted with sterile artificial seawater and spread onto Marine Agar (MA, BD Difco, Sparks, MD, USA) plates. Individual colonies were isolated and transferred to fresh MA plates to obtain pure cultures. Strain LZ166T was successfully isolated and cultivated following these procedures. For long-term preservation, cultures were mixed with 30% glycerol and stored at −80 °C.
Closely related type strains, including A. microcysteis NIBR3T and A. oceanicum B7T, were purchased from the Korean Agricultural Culture Collection (KACC) and the Marine Culture Collection of China (MCCC), respectively. These type strains were used for comparison of phenotypic, physiological, and chemotaxonomic characteristics with the isolated strain LZ166T in this study.
Additionally, strain LZ166T has been deposited in multiple culture collections, including the MCCC (accession number = MCCC M28807T), the KACC (accession number = KACC 23148T), and the Korean Culture Type Collection (KCTC, accession number = KCTC 82889T).
2.2. Morphology and Physiology
The cell size, morphology, and flagella pattern during the mid-exponential growth phase were examined using a transmission electron microscope (model HT7800, Hitachi, Tokyo, Japan) after cells being negatively stained with uranyl acetate. The Gram-staining experiment was carried out using a commercial Gram-staining kit (Qingdao Hope Bio-Technology Co., Ltd., Qingdao, China) following the manufacturer’s protocol. The motility of strain LZ166T was assessed both microscopically and by the semi-solid agar puncture method in MA medium containing 0.5% (w/v) agar. The growth of strain LZ166T was examined in Marine Broth 2216 (MB, BD Difco) medium under a range of temperatures (4, 10, 15, 25, 28, 30, 34, 37, 41, 45, 55, and 60 °C), NaCl concentrations (0, 0.5, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 12, 15, 18, and 20%, w/v), and pH conditions (3.0, 4.0, 5.0, 6.0, 7.0, 8.0, 9.0, 10.0, 11.0, and 12.0), respectively. For NaCl concentrations, 0–2% and 3–20% mediums were prepared based on 2216E and MB medium, respectively. The pH of MB medium was adjusted using various buffers to cover the desired pH range: 10 mM acetate/acetic acid buffer for pH 3.0–5.5, MES buffer for pH 5.0–6.5, PIPES buffer for pH 6.0–7.0, HEPES buffer for pH 7.0–8.0, and Tris and CAPSO buffer for pH values greater than 8.0 [12]. The anaerobic growth was determined in an anaerobic chamber with pure N2 using MB medium. Growth on nutrient agar (NA), R2A agar (BD Difco), and LB agar plates was also tested at 28 °C.
Catalase activity was assessed by the formation of oxygen bubbles when the cells were exposed to a 3% (w/v) H2O2 solution [1]. Oxidase activity was determined by the oxidation of 1% (w/v) N,N,N,N-tetramethyl-1,4-phenylenediamine (BioMérieux, Lyon, France). The hydrolysis of skimmed milk, starch, cellulose, Tween 20, 40, 60, and 80 was examined on MA plates supplemented with the corresponding substrates. Additional biochemical properties were identified using API ZYM [13] and 20NE strips (BioMérieux, Lyon, France) following the manufacturer’s protocol. Utilization of various carbon sources by strain LZ166T was assessed with the Biolog GEN III Microplates (Biolog, Hayward, CA, USA) following the manufacturer’s instructions.
2.3. Chemotaxonomy
To analyze the cellular fatty acid and polar lipids, strain LZ166T and closely related species were cultivated in MA medium at 28 °C for 72 h. Fatty acids were extracted, saponified, methylated, and then determined using gas chromatography (Agilent Technologies 6850, Santa Clara, CA, USA) as described by Li et al. [12]. The composition of isoprenoid quinones from freeze-dried strains was extracted using chloroform/methanol (2:1, v:v), separated by thin-layer chromatography (TLC) on silica gel GF254 plates (10 × 20 cm, Qingdao Haiyang Chemical Co., Ltd., Qingdao, China), and redissolved in methanol. Redissolved isoprenoid quinones were then analyzed using high-performance liquid chromatography (HPLC, Waters 2695, Milford, MA, USA) [14]. Polar lipids were extracted and separated using two-dimensional TLC on silica gel 60 F254 plates (10 × 10 cm, Merck) with a first phase of chloroform/methanol/water (65:25:4, v:v:v) and a second phase of chloroform/acetic acid/methanol/water (80:15:12:5, v:v:v:v). The polar lipids were visualized using molybdatophosphoric acid, ninhydrin, molybdenum blue reagent, and 1-naphthol-sulphuric acid and identified based on their relative positions under different chromogenic agents [14].
2.4. 16S rRNA Gene Phylogeny
The genomic DNA of strain LZ166T was extracted using a bacterial genomic DNA extraction kit (SBS Genetech Co., Ltd., Shanghai, China) according to the manufacturer’s instructions. The purified genomic DNA was quantified by NanoDrop 2000 spectrophotometer (Thermo Scientific, Waltham, MA, USA). The complete 16S rRNA gene was amplified using universal primers 27F (5′-AGAGTTTGATCMTGGCTCAG-3′) and 1492R (5′-GGTTACCTTGTTACGACTT-3′). Further, 16S rRNA genes were sequenced via the Sanger method (Applied BiosystemsTM 3730XL, Waltham, MA, USA) by Sangon Biotech (Shanghai) Co., Ltd., Shanghai, China. The 16S rRNA gene sequence of LZ166T was subjected to BLAST analysis on the EzBioCloud platform (https://www.ezbiocloud.net/identify accessed on 20 June 2024) [15]. Fifty 16S rRNA gene sequences of reference species were downloaded from the NCBI database. All 16S rRNA gene sequences were trimmed, aligned, and then used to construct phylogenetic trees using MEGA software (v 7.0.21) [16]. The trees were generated with 1000 bootstrap iterations and clustering using maximum likelihood (ML) [17], neighbor joining (NJ) [18], and minimum evolution (ME) [19] methods. Evolutionary distances were calculated using Kimura 2-parameter model [20]. The phylogenetic trees were visualized and decorated using ChiPlot (https://www.chiplot.online/ accessed on 1 July 2024) [21]. The complete 16S rRNA gene sequence was deposited in the GenBank database under accession number PP812687.
2.5. Genome Sequencing, Phylogenomic Analysis, and Comparative Analysis
The genome DNA of strain LZ166T was sequenced using the pair-end method on the Illumina HiSeq X Ten sequencing platform by Shanghai Majorbio Bio-Pharm Technology Co., Ltd. (Shanghai, China). The high-quality reads (Q30) were assembled using SPAdes software (v 3.13.0) [22]. Gene prediction was performed using Prokka (v 1.13) [23]. In addition, rRNA and tRNA genes were identified using RNAmmer (v 1.2) [24] and ARAGORN (v 1.2.41) [25]. Genome sequences of reference species were obtained from NCBI. The G+C content of the chromosomal DNA was determined directly from the genome sequence. A phylogenomic tree based on 13 genome sequences was constructed using PhyloPhlAn 3.0 (v 3.0.58) [26]. Average nucleotide identity (ANI) and average amino identity (AAI) among strain LZ166T and reference strains were calculated using PYANI (v 0.2.9) [27] and CompareM (v 0.0.32, https://github.com/donovan-h-parks/CompareM accessed on 20 June 2024), respectively. Digital DNA–DNA hybridization (dDDH) estimated values were calculated using the GGDC 3.0 on TYGS (https://ggdc.dsmz.de/ggdc.php accessed on 20 June 2024) [28]. The genomic overview was presented based on rapid annotation using the RAST server (https://rast.nmpdr.org/rast.cgi accessed on 20 June 2024) [29]. The genome of strain LZ166T was annotated against clusters of orthologous groups (COG) and carbohydrate-active enzyme (CAZy) databases using Eggnog-mapper (v 2.0) [30]. The carbon, nitrogen, and sulfur metabolic pathways were annotated using METABOLIC (v 4.0) [31] and KofamKOALA [32] against the KEGG database. The draft genome sequence of strain LZ166T was deposited in GenBank under accession number JBDPGJ000000000.
3. Results and Discussion
3.1. Morphology and Physiology
Strain LZ166T is Gram-stain-negative, facultatively anaerobic, and catalase- and oxidase-positive. The cells are short rod-shaped (1.3–1.7 μm long; 1.0–1.1 μm wide), non-spore-forming, and motile by a lateral flagella (Figure S1). After incubation for 3 days on an MA plate at 28 °C, circular (1–2 mm in diameter), convex, smooth, creamy-white, and non-transparent colonies were observed. Growth was observed at temperatures ranging from 10 to 45 °C (optimum 34–37 °C), in NaCl concentrations of 0–5% (w/v) (optimum 1–3%), and at a pH range of 5–10 (optimum pH 6–8). The physiological and biochemical differences among strain LZ166T, A. microcysteis NIBR3T, and A. oceanicum B7T are detailed in Table 1. The strain was positive for skimmed milk hydrolysis but negative for cellulose, starch, Tween 20, 40, 60, and 80 hydrolysis. Additionally, the R2A and LB plates supported the heterotrophic growth of LZ166T but the NA plate did not. Anaerobic growth was observed in the MB medium. While most of the API ZYM results were consistent between LZ166T and the reference species, differences were noted in the activity of alkaline phosphatase and the utilization of D-mannitol, potassium gluconate, and malic acid. Moreover, the Biolog test suggested that LZ166T was capable of utilizing a range of organic carbon sources, including dextrin, D-cellobiose, gentiobiose, D-turanose, α-D-glucose, and D-fucose, for heterotrophic growth (Table S1).

Table 1.
Comparison of the phenotypic characteristics of strain LZ166T with reference strains of the genus Aquibium.
3.2. Chemotaxonomy
The respiratory quinones of strain LZ166T were determined as Q-10 (100%). The predominant respiratory quinones are consistent with the ubiquinone systems characteristic of the genus Aquibium [1]. The polar lipids of LZ166T were composed of diphosphatidylglycerol (DPG), phosphatidylethanolamine (PE), glycolipid (GL), and phosphatidylglycerol (PG) (Figure S2). DPG, PG, and PE were also detected in A. microcysteis NIBR3T, A. carbonis B2.3T, and A. oceanicum B7T [1], which supported the affiliation of strain LZ166T with Aquibium. However, the presence of GL can be used to differentiate the novel strain from the reference species as it is unique to strain LZ166T. The major cellular fatty acids (>5%) of strain LZ166T are summed feature 8 (C18:1ω7c and/or C18:1ω6c, 39.3%), iso-C17:0 (13.3%), 11-methyl C18:1ω7c (12.0%), C19:0 cyclo ω8c (9.3%), and C16:0 (5.5%) (Table 2 and Table S2). Although the fatty acid profiles of strain LZ166T are similar to those of the reference strains in Aquibium, their proportions differ from each other [1].
Table 2.
Comparison of major fatty acids (>5%) between strain LZ166T and closely related Aquibium species.
3.3. 16S rRNA Gene Phylogeny
The 16S rRNA gene sequence analysis indicated that LZ166T exhibited the highest sequence similarity of 98.58% with A. oceanicum B7T, followed by A. carbonis B2.7T (97.94%) and A. mycrocysteis NIBR3T (96.71%), which were lower than the cut-off value of 98.65% [33]. A detailed ML tree (Figure S3) and a concise ML tree (Figure 1) based on the 16S rRNA gene sequences were constructed and both of them clearly revealed that strain LZ166T formed a separate lineage within the genus Aquibium. This topology was further confirmed by the ME (Figure S4) and NJ methods (Figure S5). The collective findings from the 16S rRNA gene phylogeny suggest that strain LZ166T could represent a potential novel species within Aquibium.
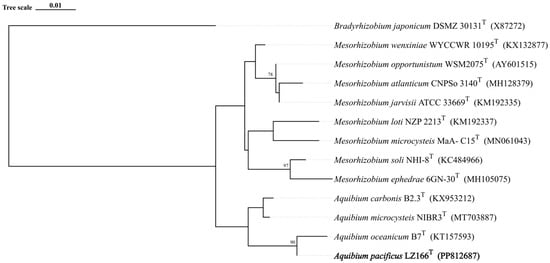
Figure 1.
Maximum likelihood phylogenetic tree based on 13 16S rRNA gene sequences showing the positions between strain LZ166T and other closely related phylogenetic neighbors. Bootstrap numbers (>70%) were shown with 1000 calculations. The bold font represents the novel species identified in this study. Bradyrhizobium japonicum DSMZ_30131T (X87272) was used as the out group. Bar, 0.01 substitutions per nucleotide position.
3.4. Genomic Features
A total of 10,595,752 raw reads were produced with 258X sequencing depth. The assembled genome size of strain LZ166T is 6,119,659 bp with a chromosomal DNA G+C content of 64.7 mol%, which is similar to the other species within the genus Aquibium, which ranges from 65.1–67.9 mol% [1]. Moreover, fifty tRNA genes for twenty amino acids as well as one gene each for 5S rRNA, 16S rRNA, and 23S rRNA were also identified in the genome of LZ166T. A total of 5513 protein-coding sequences (CDSs) were predicted, with the majority (5220/5513, 94.7%) assigned to a putative function based on the COG categories, while the rest were annotated as hypothetical proteins. Detailed information on the gene classification according to the RAST, COG, and CAZy databases is provided in Figure S6.
The ANI and AAI values between LZ166T and the other closely related species in the genus Aquibium are in the ranges of 76.79–90.73% and 79.03–88.50%, respectively (Table S3). These values are below the 95–96% cut-off thresholds previously proposed for species delineation [34,35,36]. The dDDH estimated values between LZ166T and the other three species, A. microcysteis NIBR3T, A. oceanicum B7T, and A. carbonis B2.3T, were 22.8%, 36.1%, and 22.2%, respectively (Table S3), all of which are below the dDDH standard cut-off value of 70% [36,37]. Furthermore, a whole-genome phylogenomic tree (Figure 2) was also constructed, which corroborated the phylogenetic relationships derived from the 16S rRNA genes’ phylogeny and genetic relatedness, as depicted in Figure 1. Altogether, these results suggest that strain LZ166T represents a novel species within the genus Aquibium.
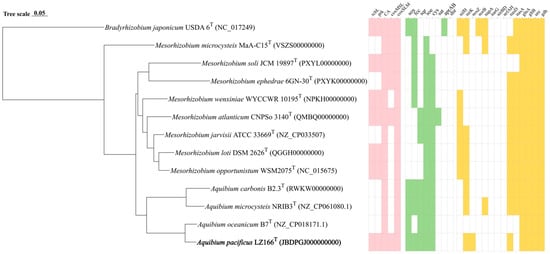
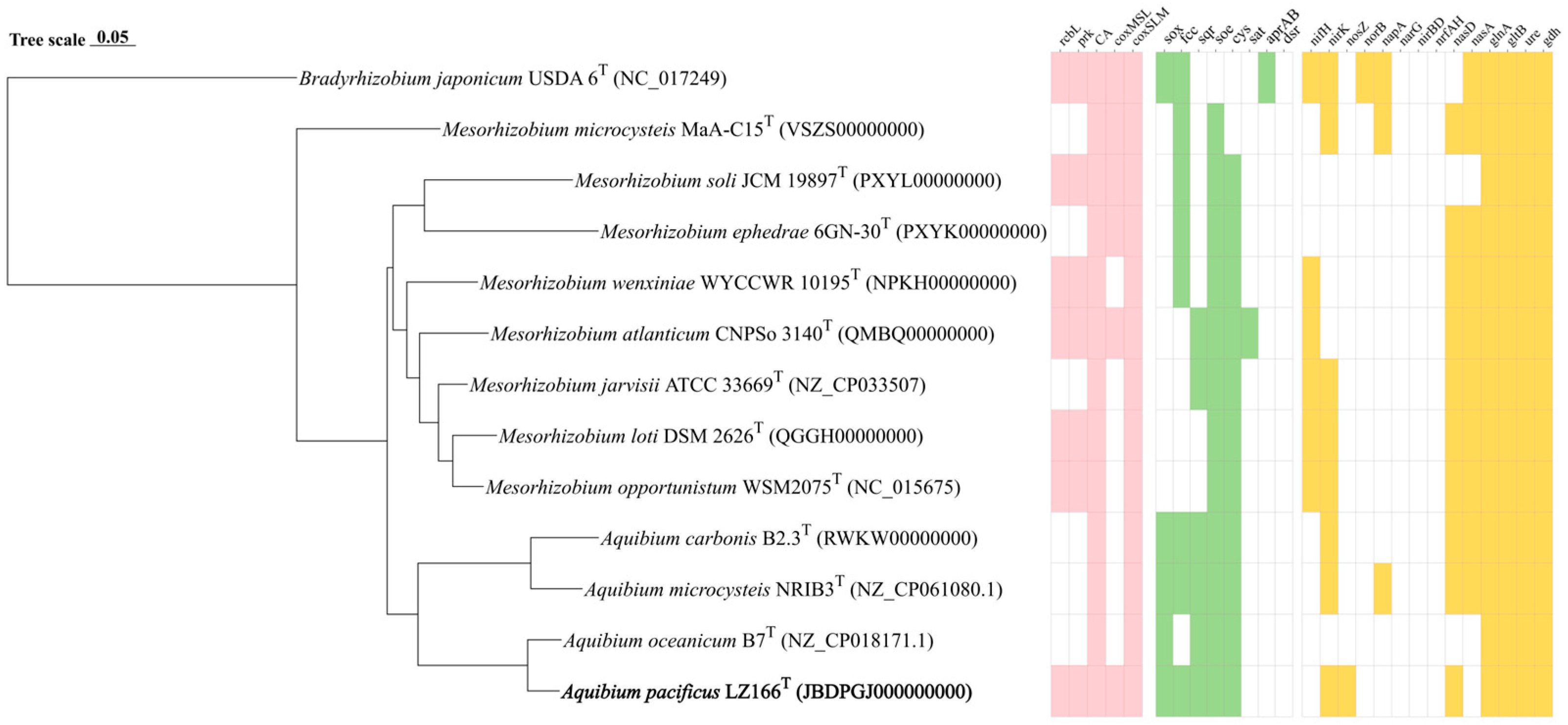
Figure 2.
Phylogenomic tree of LZ166T and its functional genes involved in carbon, sulfur, and nitrogen metabolism in comparison with closely related species. The bold font represents the novel species identified in this study. Bradyrhizobium japonicum USDA 6T (GFC_000284375.1) was used as the out group. Bar, 0.05 substitutions per nucleotide position. Pink, green, and yellow blocks, presence of corresponding carbon, sulfur, and nitrogen functional genes, respectively. White blocks, absence or partial presence of corresponding functional genes. rcbL, ribulose-1,5-bisphosphate carboxylase/oxygenase gene large chain. prk, phosphoribulokinase gene. CA, carbonic anhydrase gene. coxMSL, carbon monoxide dehydrogenase (form I) gene. coxSLM, carbon monoxide dehydrogenase (form II) gene. sox, thiosulfate oxidation genes cluster (soxABCDXYZ). fcc, flavocytochrome c-sulfide dehydrogenase gene. sqr, sulfide:quinone oxidoreductases gene. soe, sulfite dehydrogenase (quinone) gene. cys, assimilatory sulfate reduction genes cluster (cysNDCHJIK). sat, sulfate adenylyltransferase gene. aprAB, adenylylsulfate reductase gene. dsr, dissimilatory sulfite reductase gene. nifH, nitrogenase gene. nirK, copper-containing nitrite reductase (denitrification) gene. nosZ, nitrous oxide reductase gene. norB nitric oxide reductase gene. napA, periplasmic dissimilatory nitrate reductase gene. narG, membrane-bound dissimilatory nitrate reductase gene. nirBD, dissimilatory nitrite reductase (NADH) gene. nrfAH, dissimilatory nitrite reductase (cytochrome c-552). nasD, assimilatory nitrite reductase gene. nasA, assimilatory nitrate reductase. glnA, glutamine synthase gene. gltB, glutamate synthase gene. ure, urease gene. gdh, glutamate dehydrogenase gene.
According to the RAST annotation, 1398 genes were detected in the genome of strain LZ166T and could be assigned to 279 subsystems belonging to 25 categories. Among the 25 categories, amino acids and derivatives (241) was the most common, followed by carbohydrates (193), protein metabolism (172), and cofactors, vitamins, prosthetic groups, and pigments (107). Most of the genes in the amino acids and derivatives category are connected to branched-chain amino acids. Most of the genes in the carbohydrates category were relevant to the TCA cycle belonging to the central carbohydrate metabolism subcategory (Figure S6A). The COG analysis suggested that, except those with unknown functions (S, 22.45%), amino acid transport and metabolism (E, 12.53%) was the most abundant category, followed by carbohydrate transport and metabolism (G, 7.91%), transcription (K, 7.78%), energy production and conversion (C, 7.45%), and lipid transport and metabolism (I, 6.84%) (Figure S6B). Additionally, a total of 71 genes in the genome of strain LZ166T matched the CAZy families. Glycosyl transferases (GTs) was the largest CAZy family with thirty-eight genes, followed by glycoside hydrolases (GHs, twenty-nine genes), carbohydrate-binding modules (CBMs, six genes), carbohydrate esterases (CEs, three genes), and polysaccharide lyases (PLs, one gene). However, auxiliary activities (AAs) were not detected (Figure S6C).
3.5. Genomic Functional Analysis
3.5.1. Carbon Metabolism
The draft genome sequence of strain LZ166T was utilized to infer its metabolic profiles, which was subsequently compared with those of three other Aquibium species (Figure 2). LZ166T is capable of obtaining carbon from both inorganic and organic sources. A distinctive feature of the LZ166T genome is its potential for carbon dioxide fixation via the Calvin–Benson–Bassham (CBB) cycle. The genome contains a complete set of genes necessary for the CBB cycle, including those encoding key enzymes such as ribulose-1,5-bisphosphate carboxylase (Rubisco) and phosphoribulokinase (PRK) [38]. The Rubisco enzyme catalyzes the carboxylation of ribulose-1,5-bisphosphate, resulting in the formation of 3-phosphoglycerate [39]. To date, four forms of Rubisco have been identified, with form I being the most prevalent [40]. The genome of LZ166T encodes form I RubisCO (RcbL and RcbS), which shows the highest similarity (88.8% and 84.7%, respectively) to the enzymes from Mesorhizobium mediterraneum. Although the rcbL and rcbS genes were predicted in the genome of LZ166T, no complete carbon fixation pathways have been identified in the genomes of the other Aquibium species. This indicates that the rcbL, rcbS, and prk genes may serve as genetic markers to distinguish strain LZ166T from the other members of the genus. The presence of the CBB pathway hints that LZ166T may be capable of autotrophic metabolism. Additionally, the genome of strain LZ166T also encodes the genes involved in the Embden–Meyerhof pathway (EMP), hexose monophosphate pathway (HMP), Entner–Doudoroff pathway (ED), and tricarboxylic acid cycle (TCA), indicating a heterotrophic lifestyle that relies on organic matter for carbon and energy [41]. Overall, the presence of both carbon fixation and organic matter breakdown genes in the genome of LZ166T suggests a mixotrophic strategy, potentially allowing it to adapt to the dynamic and variable conditions of the marine environment [11].
The genome of strain LZ166T, in common with those of the other Aquibium species, includes all the key genes that encode the aerobic carbon monoxide dehydrogenase (CoxSLM). While the other Aquibium species only contain the form II cox gene, LZ166T distinctively harbors both the form I and form II cox genes (Figure 2 and Figure S7). Moreover, at least three copies of cox gene clusters could be found in the genome of LZ166T. This dual presence of cox genes indicates that LZ166T might have the capacity to oxidize carbon monoxide (CO). The co-existence of the CBB pathway for carbon fixation and the CO oxidation pathway in the genome of LZ166T suggests that this strain could utilize the energy derived from CO oxidation. This capability might, in turn, facilitate its autotrophic growth [42]. It seems not surprising as some CO oxidizers have been confirmed to fix CO2 through the CBB cycle by using the energy obtained from CO oxidation [42].
3.5.2. Sulfur Metabolism
Sulfur metabolism is crucial for bacteria, providing not only the essential sulfur element but also a source of energy. The genome of strain LZ166T encodes the complete set of genes for the SOX pathway (soxABCDXYZ), which is responsible for oxidizing thiosulfate to sulfate [43]. In addition to the SOX system, the LZ166T genome also includes genes encoding flavocytochrome c-sulfide dehydrogenase (fcc) and sulfide:quinone oxidoreductase (sqr) genes, indicating that this strain potentially possesses the capacity to oxidize sulfide to elemental sulfur [43]. The presence of the SOX system, fcc, and sqr in the other Aquibium species suggests that sulfur oxidation is probably a common feature of this genus. However, an exception is A. oceanicum B7T, which lacks the fcc gene despite its phylogenetic proximity to strain LZ166T. Furthermore, the Aquibium species, including LZ166T, encodes the sulfite dehydrogenases (soe) gene, which plays a role in sulfite oxidation [43,44]. Strain LZ166T and the other Aquibium species possessed a complete assimilatory sulfate reduction pathway (cysND, cysC, cysH, and cysJI), indicating that they can reduce sulfate into sulfide, even to L-cysteine (cysK) [45]. The presence of multiple sulfur oxidation pathways in the genome of LZ166T underscores its capacity to oxidize sulfide, sulfite, or thiosulfate, thereby conserving energy and potentially supporting chemoautotrophic growth.
3.5.3. Nitrogen Metabolism
The analysis of nitrogen metabolism using KEGG annotation revealed the absence of nitrogenase (nifH) genes in strain LZ166T as well as in the other Aquibium species, a distinguishing feature between the genera Aquibium and Mesorhizobium [1]. Strain LZ166T was found to harbor an incomplete denitrification pathway, characterized by the presence of key genes of nirK and nosZ, which encode nitrite reductase [46] and nitrous oxide reductase [47], respectively. This genetic profile implies the potential for LZ166T to utilize nitrite or nitrous oxide as electron acceptors. However, neither the nitric oxide reductase genes (norB) [48] nor the dissimilatory nitrate reductase genes (napA and narG) [46] and the nitrite reductase genes (nrfAH and nirBD) [49] could be identified in the genome. Additionally, the assimilatory nitrate reduction pathway was annotated in the genome of LZ166T, which contained the nasDE genes, responsible for the conversion of nitrite to ammonia [50]. However, the assimilatory nitrate reductase gene, nasA [51], is absent in the LZ166T genome. The genome of strain LZ166T encodes the glutamine synthase (glnA) and glutamate synthase (gltB) genes, enabling the assimilation of ammonium [51]. Furthermore, the presence of the urease (ure) and glutamate dehydrogenase (gdh) genes provided genomic evidence for strain LZ166T to transform nitrogen from organic to inorganic forms [46,51].
A comprehensive overview of the metabolic pathways involved in the carbon, nitrogen, and sulfur cycles for strain LZ166T (Figure 3) and the reference strains of the genus Aquibium is presented in Figure 2. Consequently, the genomic functional analysis underscores the metabolic versatility of strain LZ166T.
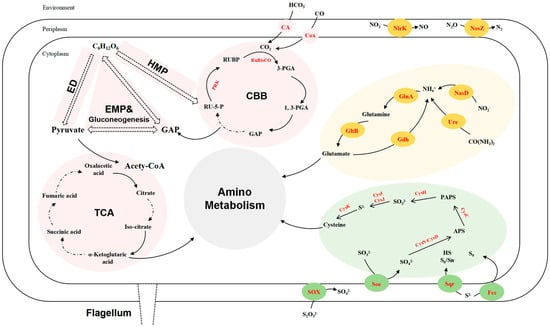
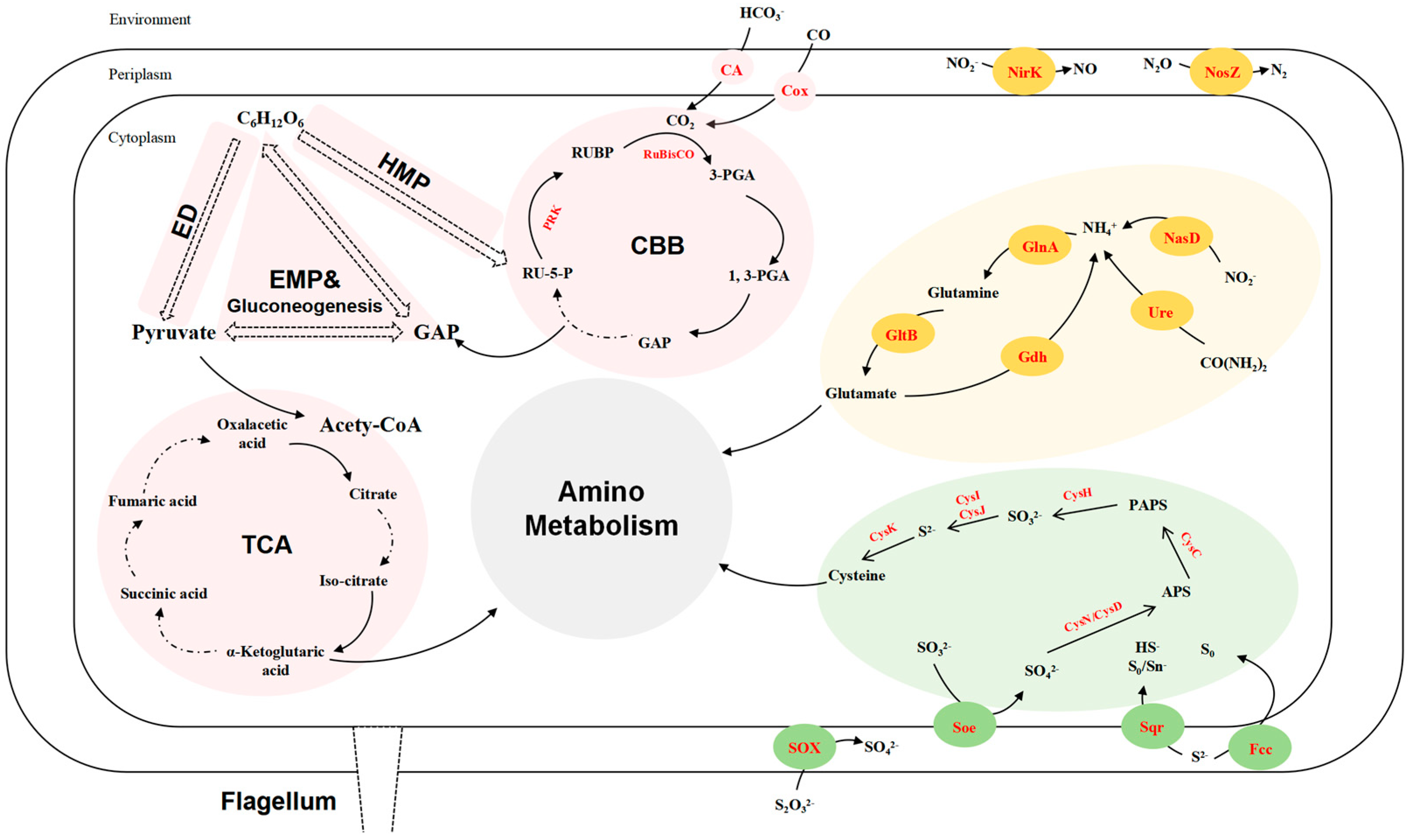
Figure 3.
Reconstructed carbon (pink), sulfur (green), and nitrogen (yellow) metabolism of strain LZ166T based on functional genes (corresponding enzymes are in red). ED, Entner–Doudoroff pathway. EMP, Embden–Meyerhof–Parnas pathway. HMP, hexose monophosphate pathway. CBB, Calvin–Benson–Bassham cycle. TCA, tricarboxylic acid cycle. GAP, glyceraldehyde 3–phosphate. RU–5–P, ribulose–5–phosphate. PRK, phosphoribulokinase. RUBP, ribulose–1,5–bisphosphate. RuBisCO, ribulose–1,5–bisphosphate carboxylase/oxygenase. 3–PGA, 3–phosphoglycerate. 1, 3–PGA, 1, 3–diphosphoglycerate. PAPS, phosphoadenosine phosphosulfate. APS, adenosine 5′–phosphosulfate. CA, carbonic anhydrase. Cox, carbon monoxide dehydrogenase. NirK, copper-containing nitrite reductase. NosZ, nitrous oxide reductase. NasD, assimilatory nitrite reductase. GlnA, glutamine synthase. GltB, glutamate synthase. Gdh, glutamate dehydrogenase. Ure, urease. CysK, cysteine synthase. CysI, sulfite reductase (NADPH) hemoprotein beta-component. CysJ, sulfite reductase (NADPH) flavoprotein alpha-component. CysH, PAPS reductase. CysC, adenylylsulfate kinase. CysN, sulfate adenylyltransferase subunit 1. CysD, sulfate adenylyltransferase subunit 2. SOX, thiosulfate oxidation enzymes. Soe sulfite dehydrogenase. Sqr, sulfide:quinone oxidoreductases. Fcc, flavocytochrome c-sulfide dehydrogenase.
3.6. Description of Aquibium pacificus sp. nov.
Aquibium pacifica (pa.ci’ fi.cus. L. gen. adj. Pacificus, pacific, pertaining to the Pacific Ocean).
The cells are short rod-shaped (1.3–1.7 μm long; 1.0–1.1 μm wide) with a lateral flagella, facultatively anaerobic, motile, and Gram-negative. The colonies are 1–2 mm in diameter, convex, smooth, creamy-white, and non-transparent after incubation on an MA plate for 3 days. The strains can grow in the R2A medium and LB medium but not in the NA medium. Growth can be observed at 10–45 °C (optimum 34–37 °C), 0–5% (w/v) NaCl (optimum 1–3%), and pH 5–10 (optimum pH 6–8). Catalase, oxidase, esterase (C4), esterase lipase (C8), leucine aminopeptidase, trypsin, and α-glucosidase are positive. Alkaline phosphatase, lipase (C14), valine aminopeptidase, cystine aminopeptidase, α-chymotryp, acid phosphatase, and naphtol-AS-B1-phosphoamidase are weakly positive. α-galactosidase, β-galactosidase, α-glucosidase, β-glucosidase, N-acetyl-β-glucosaminidase, α-mannosidase, and α-fucosidase are negative. The strain is able to hydrolyze or assimilate urea, D-glucose, L-arabinose, and D-mannitol and is weakly positive for aesculin hydrolysis. It is unable to reduce nitrate to nitrite, produce indole, ferment D-glucose, hydrolyze arginine or gelatin, or assimilate D-mannose, D-maltose, potassium gluconate, capric acid, adipic acid, malic acid, trisodium citrate, or phenylacetic acid. Skimmed milk utilization is observed. The major fatty acids (>5%) are summed feature 8 (C18:1ω7c and/or C18:1ω6c), iso-C17:0 and 11-methyl C18:1ω7c, C19:0 cyclo ω8c, and C16:0. DPG, PE, GL, and PG are the major polar lipids. The predominant respiratory quinone is Q-10.
The type strain designated as LZ166T (=MCCC M28807T = KACC 23148T = KCTC 82889T) was isolated from deep seawater in the western Pacific Ocean. The genome size is 6,154,543 bp with 64.7% G+C content. The GenBank accession numbers of the 16S rRNA gene sequence and genome sequence of strain LZ166T are PP812687 and JBDPGJ000000000, respectively.
4. Conclusions
The study presents the detailed characterization and taxonomic classification of Aquibium pacificus sp. nov., a newly identified mixotrophic bacterium isolated from the western Pacific Ocean’s bathypelagic seawater. The 16S rRNA gene sequence analysis and genomic features, including the ANI, AAI, and dDDH values, distinctly place LZ166T within the Aquibium genus, yet as a separate lineage. The physiological and biochemical characteristics of LZ166T were also found to be distinct from the other species within the genus, highlighting its novelty. It hints at a versatile metabolic profile, capable of both autotrophic and heterotrophic growth, with a genome that supports carbon fixation via the CBB cycle and inorganic sulfur oxidation. Notably, LZ166T lacks nitrogenase genes, indicating a divergence from the nitrogen-fixing capabilities observed in other closely related genera. Its metabolic versatility, as evidenced by the presence of genes for carbon fixation and organic matter breakdown, positions it as a mixotrophic organism. The findings of this study extend the knowledge of the Aquibium species and their ecological roles in the marine environment, emphasizing the adaptability of LZ166T to the dynamic deep-sea ecosystem.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/microorganisms12081584/s1. Figure S1: Transmission electron micrograph in exponential growth phase showing the cell morphology of strain LZ166T. Figure S2: Polar lipids of strain LZ166T following separation by two-dimensional TLC. Figure S3: Maximum likelihood phylogenetic tree based on 51 16S rRNA gene sequences showing the position between strain LZ166T and other closely related phylogenetic neighbors. Figure S4: Minimum evolution phylogenetic tree based on 51 16S rRNA gene sequences showing the position between strain LZ166T and other closely related phylogenetic neighbors. Figure S5: Neighbor joining phylogenetic tree based on 51 16S rRNA gene sequences showing the position between strain LZ166T and other closely related phylogenetic neighbors. Figure S6: Genes classification of strain LZ166T against the RAST, COG and CAZy databases. Figure S7: Phylogenetic analysis of form I and form II putative CoxL partial amino acid sequences based on alignments using ClustalW and analysis using MEGA7 with a neighbour joining algorithm. Table S1: The Biolog GNIII test of strain LZ166T. Table S2: Cellular fatty acid compositions of strain LZ166T and its reference strains. Table S3: The average nucleotide identity(ANI), average amino identity(AAI) and digital DNA-DNA hybridization (dDDH) value (%) between strain LZ166T and its close-related strains in Aquibium.
Author Contributions
F.J.: validation, formal analysis, investigation, data curation, writing—original draft preparation, and visualization. X.H.: validation and investigation. D.L.: formal analysis. X.Z.: investigation and data curation. J.H.: investigation and data curation. Q.L.: resources. J.W.: investigation. L.W.: conceptualization, resources, writing—review and editing, supervision, project administration, and funding acquisition. Z.S.: conceptualization, resources, writing—review and editing, supervision, project administration, and funding acquisition. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by National Natural Science Foundation of China (No. 42030412, Zongze Shao); the Natural Science Foundation of Fujian Province (No. 2023J011379, Liping Wang); the COMRA Program (No. DY135-B2-01, Zongze Shao); the fund of National Microbial Resource Center (No. NMRC-2024-9, Zongze Shao); and the Scientific Research Foundation of Third Institute of Oceanography, MNR (No. 2019021, Zongze Shao).
Data Availability Statement
The 16S rRNA gene sequence and genome sequence of strain LZ166T are available in GenBank under accession numbers PP812687 and JBDPGJ000000000, respectively.
Acknowledgments
We thank all participants of the Marine Culture Collection of China for kind help during experiment.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Kim, M.; Kim, W.; Park, W. Aquibium microcysteis gen. nov., sp. nov., isolated from a Microcystis aeruginosa culture and reclassification of Mesorhizobium carbonis as Aquibium carbonis comb. nov. and Mesorhizobium oceanicum as Aquibium oceanicum comb. nov. Int. J. Syst. Evol. Microbiol. 2022, 72, 005230. [Google Scholar] [CrossRef] [PubMed]
- Parte, A.C. LPSN—List of prokaryotic names with standing in nomenclature. Nucleic Acids Res. 2014, 42, D613–D616. [Google Scholar] [CrossRef] [PubMed]
- Oren, A.; Garrity, G.M. Notification that new names of prokaryotes, new combinations, and new taxonomic opinions have appeared in volume 72, part 1 of the IJSEM. Int. J. Syst. Evol. Microbiol. 2022, 72, 005292. [Google Scholar]
- Li, J.; Xin, W.; Xu, Z.; Xiang, F.; Zhang, J.; Xi, L.; Qu, J.; Liu, J. Mesorhizobium carbonis sp. nov., isolated from coal bed water. Antonie Leeuwenhoek 2019, 112, 1221–1229. [Google Scholar] [CrossRef] [PubMed]
- Fu, G.; Yu, X.; Zhang, C.; Zhao, Z.; Wu, D.; Su, Y.; Wang, R.; Han, S.; Wu, M.; Sun, C. Mesorhizobium oceanicum sp. nov., isolated from deep seawater. Int. J. Syst. Evol. Microbiol. 2017, 67, 2739–2745. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Kim, W.; Park, Y.; Jung, J.; Park, W. Lineage-specific evolution of Aquibium, a close relative of Mesorhizobium, during habitat adaptation. Appl. Environ. Microbiol. 2024, 90, e02091-23. [Google Scholar] [CrossRef] [PubMed]
- Eiler, A. Evidence for the ubiquity of mixotrophic bacteria in the upper ocean: Implications and consequences. Appl. Environ. Microbiol. 2006, 72, 7431–7437. [Google Scholar] [CrossRef]
- Muñoz-Marín, M.d.C. Mixotrophy in depth. Nat. Microbiol. 2022, 7, 1949–1950. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, Z.; Zeng, L.; Dong, C.; Shao, Z. The oxidation of hydrocarbons by diverse heterotrophic and mixotrophic bacteria that inhabit deep-sea hydrothermal ecosystems. ISME J. 2020, 14, 1994–2006. [Google Scholar] [CrossRef]
- Li, J.; Xiang, S.; Li, Y.; Cheng, R.; Lai, Q.; Wang, L.; Li, G.; Dong, C.; Shao, Z. Arcobacteraceae are ubiquitous mixotrophic bacteria playing important roles in carbon, nitrogen, and sulfur cycling in global oceans. Environ. Microbiol. 2024, 9, e00513-24. [Google Scholar] [CrossRef]
- Moran, M.A. The global ocean microbiome. Science 2015, 350, aac8455. [Google Scholar] [CrossRef]
- Li, Y.; Wang, S.; Ji, B.; Yuan, Q.; Wei, S.; Lai, Q.; Wu, K.; Jiang, L.; Shao, Z. Sulfurovum mangrovi sp. nov., an obligately chemolithoautotrophic, hydrogen-oxidizing bacterium isolated from coastal marine sediments. Int. J. Syst. Evol. Microbiol. 2023, 73, 006142. [Google Scholar] [CrossRef] [PubMed]
- Humble, M.W.; King, A.; Phillips, I. API ZYM: A simple rapid system for the detection of bacterial enzymes. J. Clin. Pathol. 1977, 30, 275–277. [Google Scholar] [CrossRef] [PubMed]
- Minnikin, D.E.; O’Donnell, A.G.; Goodfellow, M.; Alderson, G.; Athalye, M.; Schaal, A.; Parlett, J.H. An integrated procedure for the extraction of bacterial isoprenoid quinones and polar lipids. J. Microbiol. Methods 1984, 2, 233–241. [Google Scholar] [CrossRef]
- Yoon, S.; Ha, S.; Kwon, S.; Lim, J.; Kim, Y.; Seo, H.; Chun, J. Introducing EzBioCloud: A taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 2017, 67, 1613–1617. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J. Evolutionary trees from DNA sequences: A maximum likelihood approach. J. Mol. Evol. 1981, 17, 368–376. [Google Scholar] [CrossRef]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [PubMed]
- Rzhetsky, A.; Nei, M. A simple method for estimating and testing minimum-evolution trees. Mol. Biol. Evol. 1992, 9, 945–967. [Google Scholar]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef]
- Xie, J.; Chen, Y.; Cai, G.; Cai, R.; Hu, Z.; Wang, H. Tree Visualization By One Table (tvBOT): A web application for visualizing, modifying and annotating phylogenetic trees. Nucleic Acids Res. 2023, 51, W587–W592. [Google Scholar] [CrossRef] [PubMed]
- Prjibelski, A.; Antipov, D.; Meleshko, D.; Lapidus, A.; Korobeynikov, A. Using SPAdes de novo assembler. Curr. Protoc. Bioinform. 2020, 70, e102. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Lagesen, K.; Hallin, P.; Rødland, E.A.; Stærfeldt, H.-H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef] [PubMed]
- Laslett, D.; Canback, B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004, 32, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Asnicar, F.; Thomas, A.M.; Beghini, F.; Mengoni, C.; Manara, S.; Manghi, P.; Zhu, Q.; Bolzan, M.; Cumbo, F.; May, U.; et al. Precise phylogenetic analysis of microbial isolates and genomes from metagenomes using PhyloPhlAn 3.0. Nat. Commun. 2020, 11, 2500. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, L.; Glover, R.H.; Humphris, S.; Elphinstone, J.G.; Toth, I.K. Genomics and taxonomy in diagnostics for food security: Soft-rotting enterobacterial plant pathogens. Anal. Methods 2016, 8, 12–24. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Carbasse, J.S.; Peinado-Olarte, R.L.; Göker, M. TYGS and LPSN: A database tandem for fast and reliable genome-based classification and nomenclature of prokaryotes. Nucleic Acids Res. 2022, 50, D801–D807. [Google Scholar] [CrossRef] [PubMed]
- Brettin, T.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Olsen, G.J.; Olson, R.; Overbeek, R.; Parrello, B.; Pusch, G.D.; et al. RASTtk: A modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 2015, 5, 8365. [Google Scholar] [CrossRef]
- Cantalapiedra, C.P.; Hernández-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J.; Tamura, K. eggNOG-mapper v2: Functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol. Biol. Evol. 2021, 38, 5825–5829. [Google Scholar] [CrossRef]
- Zhou, Z.; Tran, P.Q.; Breister, A.M.; Liu, Y.; Kieft, K.; Cowley, E.S.; Karaoz, U.; Anantharaman, K. METABOLIC: High-throughput profiling of microbial genomes for functional traits, metabolism, biogeochemistry, and community-scale functional networks. Microbiome 2022, 10, 33. [Google Scholar] [CrossRef] [PubMed]
- Aramaki, T.; Blanc-Mathieu, R.; Endo, H.; Ohkubo, K.; Kanehisa, M.; Goto, S.; Ogata, H. KofamKOALA: KEGG ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics 2019, 36, 2251–2252. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Oh, H.; Park, S.; Chun, J. Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int. J. Syst. Evol. Microbiol. 2014, 64, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.; Rosselló-Móra, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 2009, 106, 19126–19131. [Google Scholar] [CrossRef] [PubMed]
- Konstantinidis, K.T.; Tiedje, J.M. Towards a genome-based taxonomy for prokaryotes. J. Bacteriol. 2005, 187, 6258–6264. [Google Scholar] [CrossRef] [PubMed]
- Thompson, C.C.; Chimetto, L.; Edwards, R.A.; Swings, J.; Stackebrandt, E.; Thompson, F. Microbial genomic taxonomy. BMC Genom. 2013, 14, 913. [Google Scholar] [CrossRef] [PubMed]
- Auch, A.F.; von Jan, M.; Klenk, H.-P.; Göker, M. Digital DNA-DNA hybridization for microbial species delineation by means of genome-to-genome sequence comparison. Stand. Genom. Sci. 2010, 2, 117–134. [Google Scholar] [CrossRef]
- Asplund-Samuelsson, J.; Hudson, E.P. Wide range of metabolic adaptations to the acquisition of the Calvin cycle revealed by comparison of microbial genomes. PLoS Comput. Biol. 2021, 17, e1008742. [Google Scholar] [CrossRef]
- Hügler, M.; Sievert, S.M. Beyond the Calvin cycle: Autotrophic carbon fixation in the ocean. Annu. Rev. Mar. Sci. 2011, 3, 261–289. [Google Scholar] [CrossRef]
- Tabita, F.R.; Hanson, T.E.; Li, H.; Satagopan, S.; Singh, J.; Chan, S. Function, structure, and evolution of the RubisCO-like proteins and their RubisCO homologs. Microbiol. Mol. Biol. Rev. 2007, 71, 576–599. [Google Scholar] [CrossRef]
- Patwardhan, S.; Phan, J.; Smedile, F.; Vetriani, C. The genome of Varunaivibrio sulfuroxidans strain TC8T, a metabolically versatile alphaproteobacterium from the Tor Caldara gas vents in the Tyrrhenian sea. Microorganisms 2023, 11, 1366. [Google Scholar] [CrossRef] [PubMed]
- King, G.M.; Weber, C.F. Distribution, diversity and ecology of aerobic CO-oxidizing bacteria. Nat. Rev. Microbiol. 2007, 5, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Liu, F.; Fang, W.; Yang, T.; Chen, G.-H.; He, Z.; Wang, S. Microbial sulfur metabolism and environmental implications. Sci. Total Environ. 2021, 778, 146085. [Google Scholar] [CrossRef]
- Boughanemi, S.; Infossi, P.; Giudici-Orticoni, M.-T.; Schoepp-Cothenet, B.; Guiral, M. Sulfite oxidation by the quinone-reducing molybdenum sulfite dehydrogenase SoeABC from the bacterium Aquifex aeolicus. BBA-Bioenerg. 2020, 1861, 148279. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Ou, P.; Zhao, B.; Zhang, W.; Yu, K.; Xie, K.; Zhuang, W.-Q. Assimilatory and dissimilatory sulfate reduction in the bacterial diversity of biofoulant from a full-scale biofilm-membrane bioreactor for textile wastewater treatment. Sci. Total Environ. 2021, 772, 145464. [Google Scholar] [CrossRef] [PubMed]
- Kuypers, M.M.M.; Marchant, H.K.; Kartal, B. The microbial nitrogen-cycling network. Nat. Rev. Microbiol. 2018, 16, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.D.; Han, H.; Yun, T.; Song, M.J.; Terada, A.; Laureni, M.; Yoon, S. Identification of nosZ-expressing microorganisms consuming trace N2O in microaerobic chemostat consortia dominated by an uncultured Burkholderiales. ISME J. 2022, 16, 2087–2098. [Google Scholar] [CrossRef] [PubMed]
- Braker, G.; Tiedje, J.M. Nitric oxide reductase (norB) genes from pure cultures and environmental samples. Appl. Environ. Microbiol. 2003, 69, 3476–3483. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; He, T.; Zhang, M.; Zheng, C.; Yang, L.; Yang, L. Review of the mechanisms involved in dissimilatory nitrate reduction to ammonium and the efficacies of these mechanisms in the environment. Environ. Pollut. 2024, 345. [Google Scholar] [CrossRef]
- Shi, W.; Lu, W.; Liu, Q.; Zhi, Y.; Zhou, P. The identification of the nitrate assimilation related genes in the novel Bacillus megaterium NCT-2 accounts for its ability to use nitrate as its only source of nitrogen. Funct. Integr. Genom. 2013, 14, 219–227. [Google Scholar] [CrossRef]
- Pajares, S.; Ramos, R. Processes and microorganisms involved in the marine nitrogen cycle: Knowledge and gaps. Front. Mar. Sci. 2019, 6, 739. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).