
Molecular Evolutionary Analyses of the Fusion Genes in Human Parainfluenza Virus Type 4
 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Strains in This Study
2.2. Time-Scaled Phylogenetic Analyses and Estimation of Evolutionary Rate
2.3. Bayesian Skyline Plot (BSP) Analyses
2.4. Phylogenetic Distance Analyses
2.5. Selective Pressure Analyses
2.6. Construction of the Three-Dimensional (3D) Structure of Fusion Proteins
2.7. Conformational B-Cell Epitope Prediction
3. Results
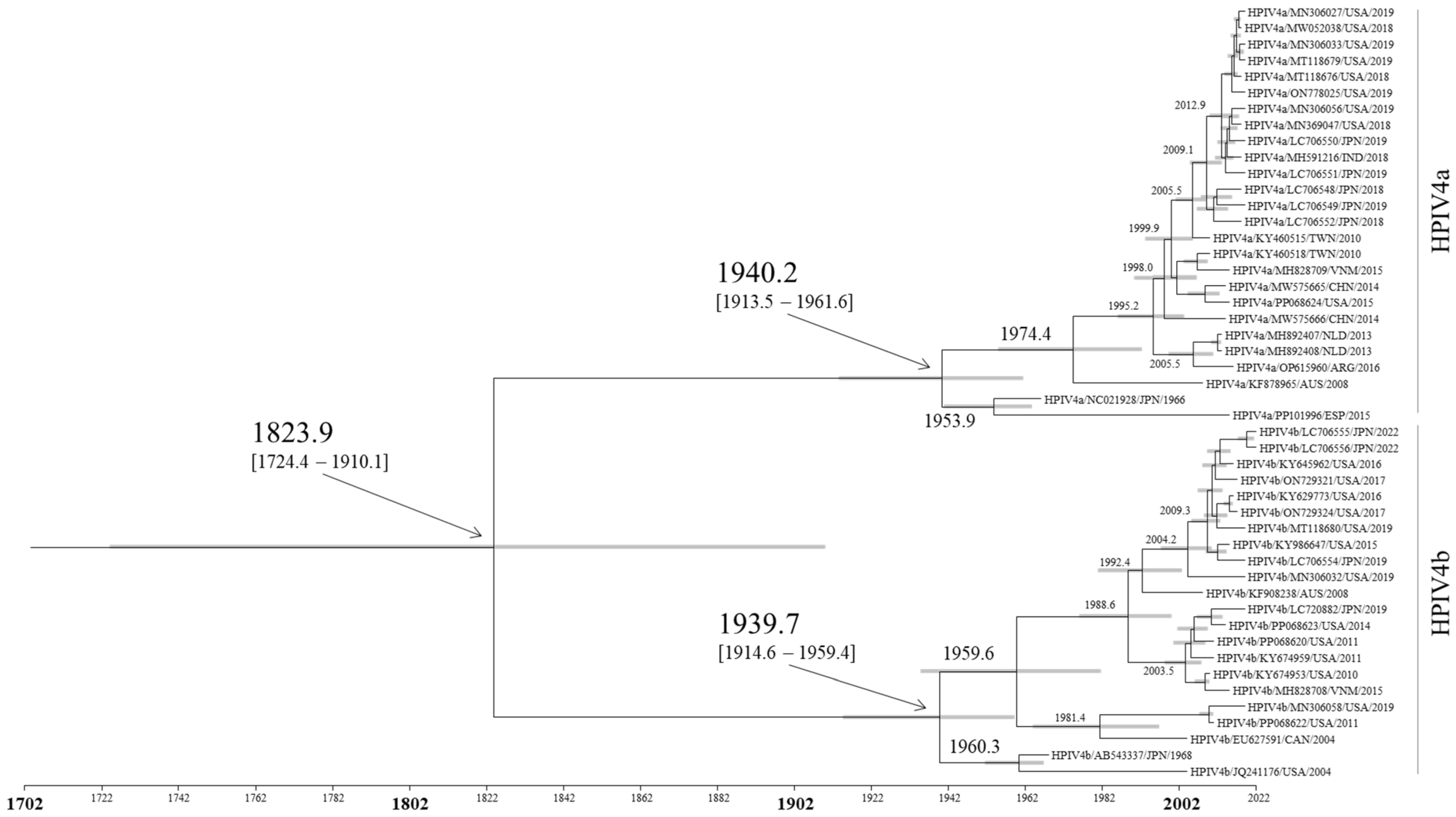
3.1. Time-Scaled Phylogeny of the F Gene in HPIV4
3.2. Evolutionary Rates of the F Gene in HPIV4
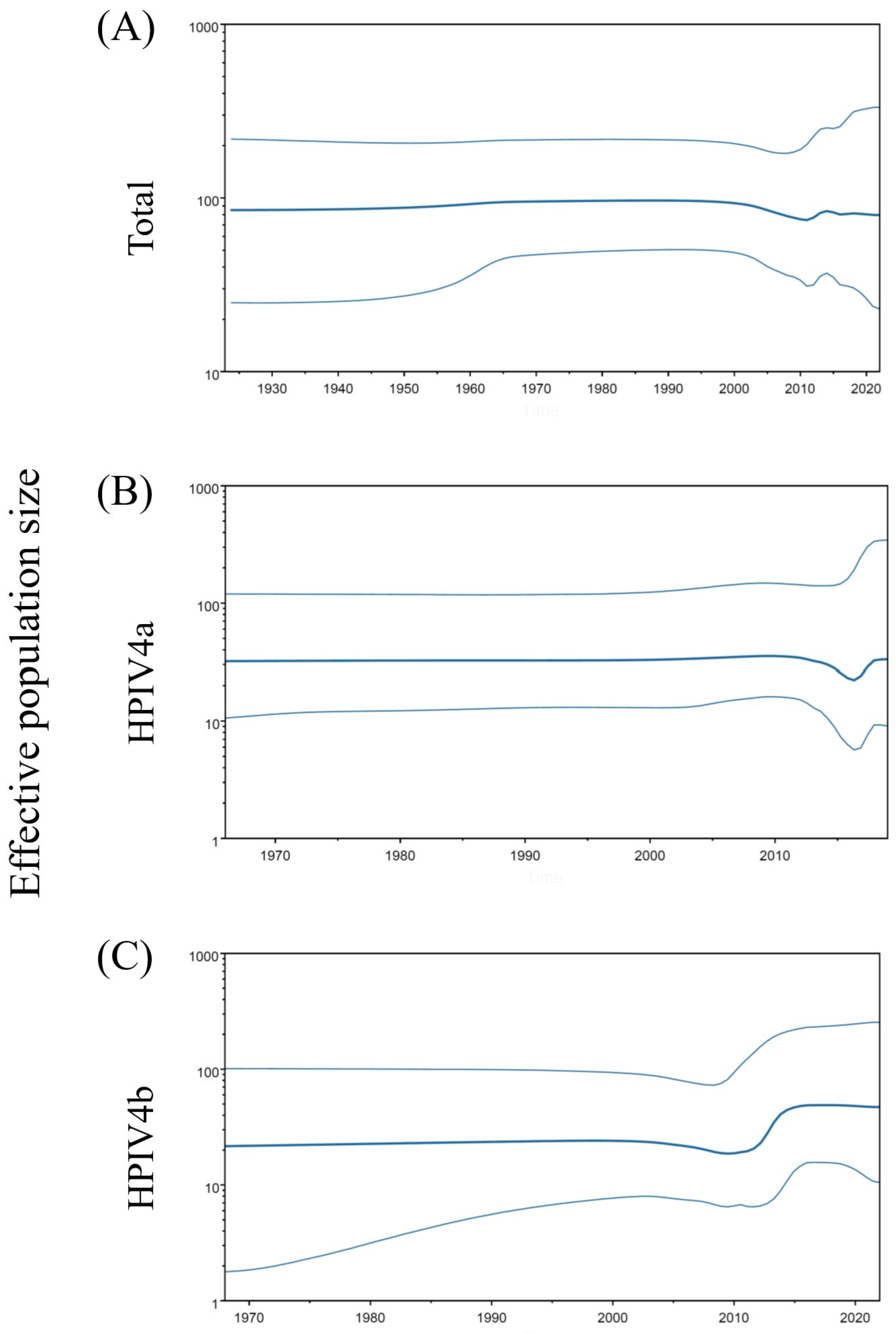
3.3. Phylodynamics of the F Gene in HPIV4
3.4. Phylogenetic Distances of the F Gene in HPIV4
3.5. Positive and Negative Selection Sites in the Fusion Protein
3.6. Three-Dimensional Mapping of Conformational B-Cell Epitopes in the Fusion Trimer Proteins of HPIV4
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Branche, A.R.; Falsey, A.R. Parainfluenza Virus Infection. Semin. Respir. Crit. Care Med. 2016, 37, 538–554. [Google Scholar] [CrossRef]
- Henrickson, K.J. Parainfluenza viruses. Clin. Microbiol. Rev. 2003, 16, 242–264. [Google Scholar] [CrossRef]
- Kimura, H.; Hayashi, Y.; Kitagawa, M.; Yoshizaki, M.; Saito, K.; Harada, K.; Okayama, K.; Miura, Y.; Kimura, R.; Shirai, T.; et al. Pathogen Profiles in Outpatients with Non-COVID-19 during the 7th Prevalent Period of COVID-19 in Gunma, Japan. Microorganisms 2023, 11, 2142. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.J.; Zhang, H.Y.; Ren, L.L.; Lu, Q.B.; Ren, X.; Zhang, C.H.; Wang, Y.F.; Lin, S.H.; Zhang, X.A.; Li, J.; et al. Etiological and epidemiological features of acute respiratory infections in China. Nat. Commun. 2021, 12, 5026. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, Y.; Deloria-Knoll, M.; Madhi, S.A.; Cohen, C.; Arguelles, V.L.; Basnet, S.; Bassat, Q.; Brooks, W.A.; Echavarria, M.; et al. Global burden of acute lower respiratory infection associated with human parainfluenza virus in children younger than 5 years for 2018: A systematic review and meta-analysis. Lancet Glob. Health 2021, 9, e1077–e1087. [Google Scholar] [CrossRef] [PubMed]
- Russell, E.; Ison, M.G. Parainfluenza Virus in the Hospitalized Adult. Clin. Infect. Dis. 2017, 65, 1570–1576. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.K.; Liu, Q.; Chen, D.H.; Liang, H.X.; Chen, X.K.; Huang, W.B.; Qin, S.; Yang, Z.F.; Zhou, R. Epidemiology and clinical presentation of the four human parainfluenza virus types. BMC Infect. Dis. 2013, 13, 28. [Google Scholar] [CrossRef] [PubMed]
- Vachon, M.L.; Dionne, N.; Leblanc, E.; Moisan, D.; Bergeron, M.G.; Boivin, G. Human parainfluenza type 4 infections, Canada. Emerg. Infect. Dis. 2006, 12, 1755–1758. [Google Scholar] [CrossRef] [PubMed]
- Slavin, K.A.; Passaro, D.J.; Hacker, J.K.; Hendry, R.M.; Kohl, S. Parainfluenza virus type 4: Case report and review of the literature. Pediatr. Infect. Dis. J. 2000, 19, 893–896. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, J.C.; Pérez-Breña, M.P.; García, M.L.; Cruz, N.; Erdman, D.D.; Echevarría, J.E. Detection and identification of human parainfluenza viruses 1, 2, 3, and 4 in clinical samples of pediatric patients by multiplex reverse transcription-PCR. J. Clin. Microbiol. 2000, 38, 1191–1195. [Google Scholar] [CrossRef] [PubMed]
- Lindquist, S.W.; Darnule, A.; Istas, A.; Demmler, G.J. Parainfluenza virus type 4 infections in pediatric patients. Pediatr. Infect. Dis. J. 1997, 16, 34–38. [Google Scholar] [CrossRef] [PubMed]
- Stewart-Jones, G.B.E.; Chuang, G.Y.; Xu, K.; Zhou, T.; Acharya, P.; Tsybovsky, Y.; Ou, L.; Zhang, B.; Fernandez-Rodriguez, B.; Gilardi, V.; et al. Structure-based design of a quadrivalent fusion glycoprotein vaccine for human parainfluenza virus types 1–4. Proc. Natl. Acad. Sci. USA 2018, 115, 12265–12270. [Google Scholar] [CrossRef] [PubMed]
- Mazur, N.I.; Terstappen, J.; Baral, R.; Bardají, A.; Beutels, P.; Buchholz, U.J.; Cohen, C.; Crowe, J.E., Jr.; Cutland, C.L.; Eckert, L.; et al. Respiratory syncytial virus prevention within reach: The vaccine and monoclonal antibody landscape. Lancet Infect. Dis. 2023, 23, e2–e21. [Google Scholar] [CrossRef] [PubMed]
- Battles, M.B.; McLellan, J.S. Respiratory syncytial virus entry and how to block it. Nat. Rev. Microbiol. 2019, 17, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Contreras, E.M.; Monreal, I.A.; Ruvalcaba, M.; Ortega, V.; Aguilar, H.C. Antivirals targeting paramyxovirus membrane fusion. Curr. Opin. Virol. 2021, 51, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Sarvmeili, J.; Baghban Kohnehrouz, B.; Gholizadeh, A.; Shanehbandi, D.; Ofoghi, H. Immunoinformatics design of a structural proteins driven multi-epitope candidate vaccine against different SARS-CoV-2 variants based on fynomer. Sci. Rep. 2024, 14, 10297. [Google Scholar] [CrossRef] [PubMed]
- Soria-Guerra, R.E.; Nieto-Gomez, R.; Govea-Alonso, D.O.; Rosales-Mendoza, S. An overview of bioinformatics tools for epitope prediction: Implications on vaccine development. J. Biomed. Inform. 2015, 53, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Bouckaert, R.; Vaughan, T.G.; Barido-Sottani, J.; Duchêne, S.; Fourment, M.; Gavryushkina, A.; Heled, J.; Jones, G.; Kühnert, D.; De Maio, N.; et al. BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2019, 15, e1006650. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed]
- Russel, P.M.; Brewer, B.J.; Klaere, S.; Bouckaert, R.R. Model Selection and Parameter Inference in Phylogenetics Using Nested Sampling. Syst. Biol. 2019, 68, 219–233. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Fourment, M.; Gibbs, M.J. PATRISTIC: A program for calculating patristic distances and graphically comparing the components of genetic change. BMC Evol. Biol. 2006, 6, 1. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Demšar, J.; Curk, T.; Erjavec, A.; Gorup, C.; Hočevar, T.; Milutinovič, M.; Možina, M.; Polajnar, M.; Toplak, M.; Starič, A.; et al. Orange: Data Mining Toolbox in Python. J. Mach. Learn. Technol. 2013, 14, 2349–2353. [Google Scholar]
- Weaver, S.; Shank, S.D.; Spielman, S.J.; Li, M.; Muse, S.V.; Kosakovsky Pond, S.L. Datamonkey 2.0: A Modern Web Application for Characterizing Selective and Other Evolutionary Processes. Mol. Biol. Evol. 2018, 35, 773–777. [Google Scholar] [CrossRef] [PubMed]
- Kosakovsky Pond, S.L.; Frost, S.D. Not so different after all: A comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef] [PubMed]
- Murrell, B.; Moola, S.; Mabona, A.; Weighill, T.; Sheward, D.; Kosakovsky Pond, S.L.; Scheffler, K. FUBAR: A fast, unconstrained bayesian approximation for inferring selection. Mol. Biol. Evol. 2013, 30, 1196–1205. [Google Scholar] [CrossRef] [PubMed]
- Murrell, B.; Wertheim, J.O.; Moola, S.; Weighill, T.; Scheffler, K.; Kosakovsky Pond, S.L. Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 2012, 8, e1002764. [Google Scholar] [CrossRef] [PubMed]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef] [PubMed]
- Meng, E.C.; Goddard, T.D.; Pettersen, E.F.; Couch, G.S.; Pearson, Z.J.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Tools for structure building and analysis. Protein Sci. 2023, 32, e4792. [Google Scholar] [CrossRef] [PubMed]
- Høie, M.H.; Gade, F.S.; Johansen, J.M.; Würtzen, C.; Winther, O.; Nielsen, M.; Marcatili, P. DiscoTope-3.0: Improved B-cell epitope prediction using inverse folding latent representations. Front. Immunol. 2024, 15, 1322712. [Google Scholar] [CrossRef] [PubMed]
- Ponomarenko, J.; Bui, H.H.; Li, W.; Fusseder, N.; Bourne, P.E.; Sette, A.; Peters, B. ElliPro: A new structure-based tool for the prediction of antibody epitopes. BMC Bioinform. 2008, 9, 514. [Google Scholar] [CrossRef] [PubMed]
- da Silva, B.M.; Myung, Y.; Ascher, D.B.; Pires, D.E.V. epitope3D: A machine learning method for conformational B-cell epitope prediction. Brief. Bioinform. 2022, 23, bbab423. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Chen, Z.; Zhang, L.; Yan, D.; Mao, T.; Tang, K.; Qiu, T.; Cao, Z. SEPPA 3.0-enhanced spatial epitope prediction enabling glycoprotein antigens. Nucleic Acids Res. 2019, 47, W388–W394. [Google Scholar] [CrossRef] [PubMed]
- Shashkova, T.I.; Umerenkov, D.; Salnikov, M.; Strashnov, P.V.; Konstantinova, A.V.; Lebed, I.; Shcherbinin, D.N.; Asatryan, M.N.; Kardymon, O.L.; Ivanisenko, N.V. SEMA: Antigen B-cell conformational epitope prediction using deep transfer learning. Front. Immunol. 2022, 13, 960985. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Akagawa, M.; Kimura, R.; Sada, M.; Shirai, T.; Okayama, K.; Hayashi, Y.; Kondo, M.; Takeda, M.; Ryo, A.; et al. Molecular evolutionary analyses of the fusion protein gene in human respirovirus 1. Virus Res. 2023, 333, 199142. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Zhu, Z.; Xu, J.; Sun, L.; Zhang, H.; Xu, H.; Zhang, F.; Wang, W.; Han, G.; Jiang, J.; et al. Molecular Evolution of Human Parainfluenza Virus Type 2 Based on Hemagglutinin-Neuraminidase Gene. Microbiol. Spectr. 2023, 11, e0453722. [Google Scholar] [CrossRef] [PubMed]
- Shao, N.; Liu, B.; Xiao, Y.; Wang, X.; Ren, L.; Dong, J.; Sun, L.; Zhu, Y.; Zhang, T.; Yang, F. Genetic Characteristics of Human Parainfluenza Virus Types 1-4 From Patients with Clinical Respiratory Tract Infection in China. Front. Microbiol. 2021, 12, 679246. [Google Scholar] [CrossRef] [PubMed]
- Aso, J.; Kimura, H.; Ishii, H.; Saraya, T.; Kurai, D.; Matsushima, Y.; Nagasawa, K.; Ryo, A.; Takizawa, H. Molecular Evolution of the Fusion Protein (F) Gene in Human Respirovirus 3. Front. Microbiol. 2020, 10, 3054. [Google Scholar] [CrossRef] [PubMed]
- Bose, M.E.; Shrivastava, S.; He, J.; Nelson, M.I.; Bera, J.; Fedorova, N.; Halpin, R.; Town, C.D.; Lorenzi, H.A.; Amedeo, P.; et al. Sequencing and analysis of globally obtained human parainfluenza viruses 1 and 3 genomes. PLoS ONE 2019, 14, e0220057. [Google Scholar] [CrossRef] [PubMed]
- Smielewska, A.; Emmott, E.; Ranellou, K.; Popay, A.; Goodfellow, I.; Jalal, H. UK circulating strains of human parainfluenza 3: An amplicon based next generation sequencing method and phylogenetic analysis. Wellcome Open Res. 2018, 3, 118. [Google Scholar] [CrossRef]
- Mao, N.; Ji, Y.; Xie, Z.; Wang, H.; Wang, H.; An, J.; Zhang, X.; Zhang, Y.; Zhu, Z.; Cui, A.; et al. Human parainfluenza virus-associated respiratory tract infection among children and genetic analysis of HPIV-3 strains in Beijing, China. PLoS ONE 2012, 7, e43893. [Google Scholar] [CrossRef] [PubMed]
- Boonyaratanakornkit, J.; Singh, S.; Weidle, C.; Rodarte, J.; Bakthavatsalam, R.; Perkins, J.; Stewart-Jones, G.B.E.; Kwong, P.D.; McGuire, A.T.; Pancera, M.; et al. Protective antibodies against human parainfluenza virus type 3 infection. mAbs 2021, 13, 1912884. [Google Scholar] [CrossRef] [PubMed]
- Suryadevara, N.; Otrelo-Cardoso, A.R.; Kose, N.; Hu, Y.X.; Binshtein, E.; Wolters, R.M.; Greninger, A.L.; Handal, L.S.; Carnahan, R.H.; Moscona, A.; et al. Functional and Structural Basis of Human Parainfluenza Virus Type 3 Neutralization with Human Monoclonal Antibodies. Nat. Microbiol. 2024, 9, 2128–2143. [Google Scholar] [CrossRef] [PubMed]
- Coelingh, K.V.; Winter, C.C. Naturally occurring human parainfluenza type 3 viruses exhibit divergence in amino acid sequence of their fusion protein neutralization epitopes and cleavage sites. J. Virol. 1990, 64, 1329–1334. [Google Scholar] [CrossRef] [PubMed]
- Sharon, J.; Rynkiewicz, M.J.; Lu, Z.; Yang, C.Y. Discovery of protective B-cell epitopes for development of antimicrobial vaccines and antibody therapeutics. Immunology 2014, 142, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Tsukagoshi, H.; Sada, M.; Sunagawa, S.; Shirai, T.; Okayama, K.; Sugai, T.; Tsugawa, T.; Hayashi, Y.; Ryo, A.; et al. Detailed Evolutionary Analyses of the F Gene in the Respiratory Syncytial Virus Subgroup A. Viruses 2021, 13, 2525. [Google Scholar] [CrossRef] [PubMed]
- Wolinsky, J.S.; Waxham, M.N.; Server, A.C. Protective effects of glycoprotein-specific monoclonal antibodies on the course of experimental mumps virus meningoencephalitis. J. Virol. 1985, 53, 727–734. [Google Scholar] [CrossRef] [PubMed]
- Taleb, S.A.; Al Thani, A.A.; Al Ansari, K.; Yassine, H.M. Human respiratory syncytial virus: Pathogenesis, immune responses, and current vaccine approaches. Eur. J. Clin. Microbiol. Infect. Dis. 2018, 37, 1817–1827. [Google Scholar] [CrossRef]
- Zhao, M.; Zheng, Z.Z.; Chen, M.; Modjarrad, K.; Zhang, W.; Zhan, L.T.; Cao, J.L.; Sun, Y.P.; McLellan, J.S.; Graham, B.S.; et al. Discovery of a Prefusion Respiratory Syncytial Virus F-Specific Monoclonal Antibody That Provides Greater In Vivo Protection than the Murine Precursor of Palivizumab. J. Virol. 2017, 91, e00176-17. [Google Scholar] [CrossRef]
- McLellan, J.S.; Ray, W.C.; Peeples, M.E. Structure and function of respiratory syncytial virus surface glycoproteins. Curr. Top. Microbiol. Immunol. 2013, 372, 83–104. [Google Scholar] [PubMed]
- Kwakkenbos, M.J.; Diehl, S.A.; Yasuda, E.; Bakker, A.Q.; van Geelen, C.M.; Lukens, M.V.; van Bleek, G.M.; Widjojoatmodjo, M.N.; Bogers, W.M.; Mei, H.; et al. Generation of stable monoclonal antibody-producing B cell receptor-positive human memory B cells by genetic programming. Nat. Med. 2010, 16, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Griffin, M.P.; Yuan, Y.; Takas, T.; Domachowske, J.B.; Madhi, S.A.; Manzoni, P.; Simões, E.A.F.; Esser, M.T.; Khan, A.A.; Dubovsky, F.; et al. Single-Dose Nirsevimab for Prevention of RSV in Preterm Infants. N. Engl. J. Med. 2020, 383, 415–425. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mizukoshi, F.; Kimura, H.; Sugimoto, S.; Kimura, R.; Nagasawa, N.; Hayashi, Y.; Hashimoto, K.; Hosoya, M.; Shirato, K.; Ryo, A. Molecular Evolutionary Analyses of the Fusion Genes in Human Parainfluenza Virus Type 4. Microorganisms 2024, 12, 1633. https://doi.org/10.3390/microorganisms12081633
Mizukoshi F, Kimura H, Sugimoto S, Kimura R, Nagasawa N, Hayashi Y, Hashimoto K, Hosoya M, Shirato K, Ryo A. Molecular Evolutionary Analyses of the Fusion Genes in Human Parainfluenza Virus Type 4. Microorganisms. 2024; 12(8):1633. https://doi.org/10.3390/microorganisms12081633
Chicago/Turabian StyleMizukoshi, Fuminori, Hirokazu Kimura, Satoko Sugimoto, Ryusuke Kimura, Norika Nagasawa, Yuriko Hayashi, Koichi Hashimoto, Mitsuaki Hosoya, Kazuya Shirato, and Akihide Ryo. 2024. "Molecular Evolutionary Analyses of the Fusion Genes in Human Parainfluenza Virus Type 4" Microorganisms 12, no. 8: 1633. https://doi.org/10.3390/microorganisms12081633