
Genome-Wide Transcriptome Analysis of a Virulent sRNA, Trans217, in Xanthomonas oryzae pv. oryzae (Xoo), the Causative Agent of Rice Bacterial Blight

Abstract
:1. Introduction
2. Materials and Methods
2.1. Xoo RNA Sequencing
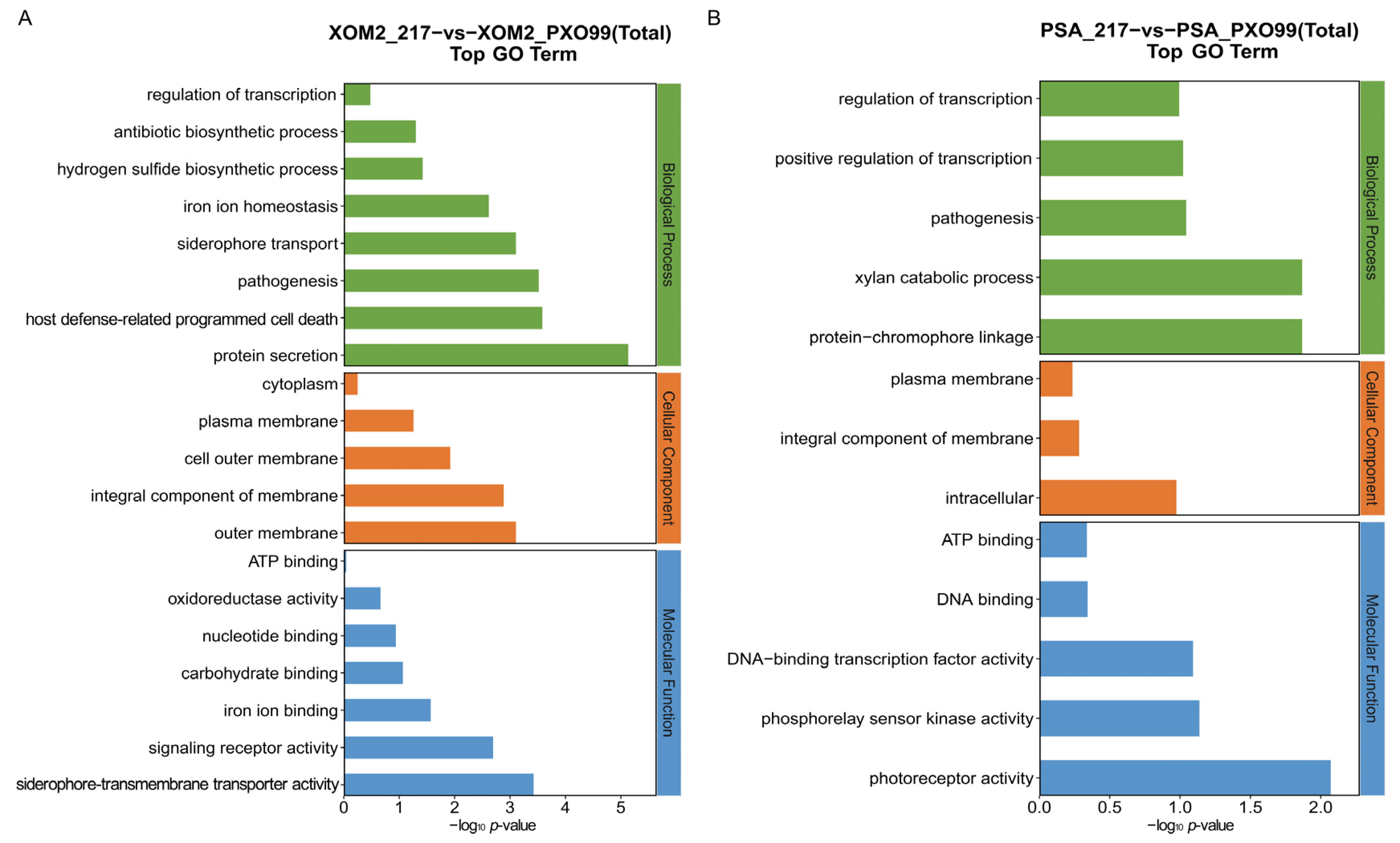
2.2. Enrichment Analysis of the DEGs
2.3. Verification of DEGs between the XOM2 and PSA Medium
2.4. Gene-Directed Mutagenesis and Complementation Assays
2.5. Virulence Assessment
2.6. Bacterial Growth Curve Determination
2.7. Oxidative Stress Assessment
3. Results
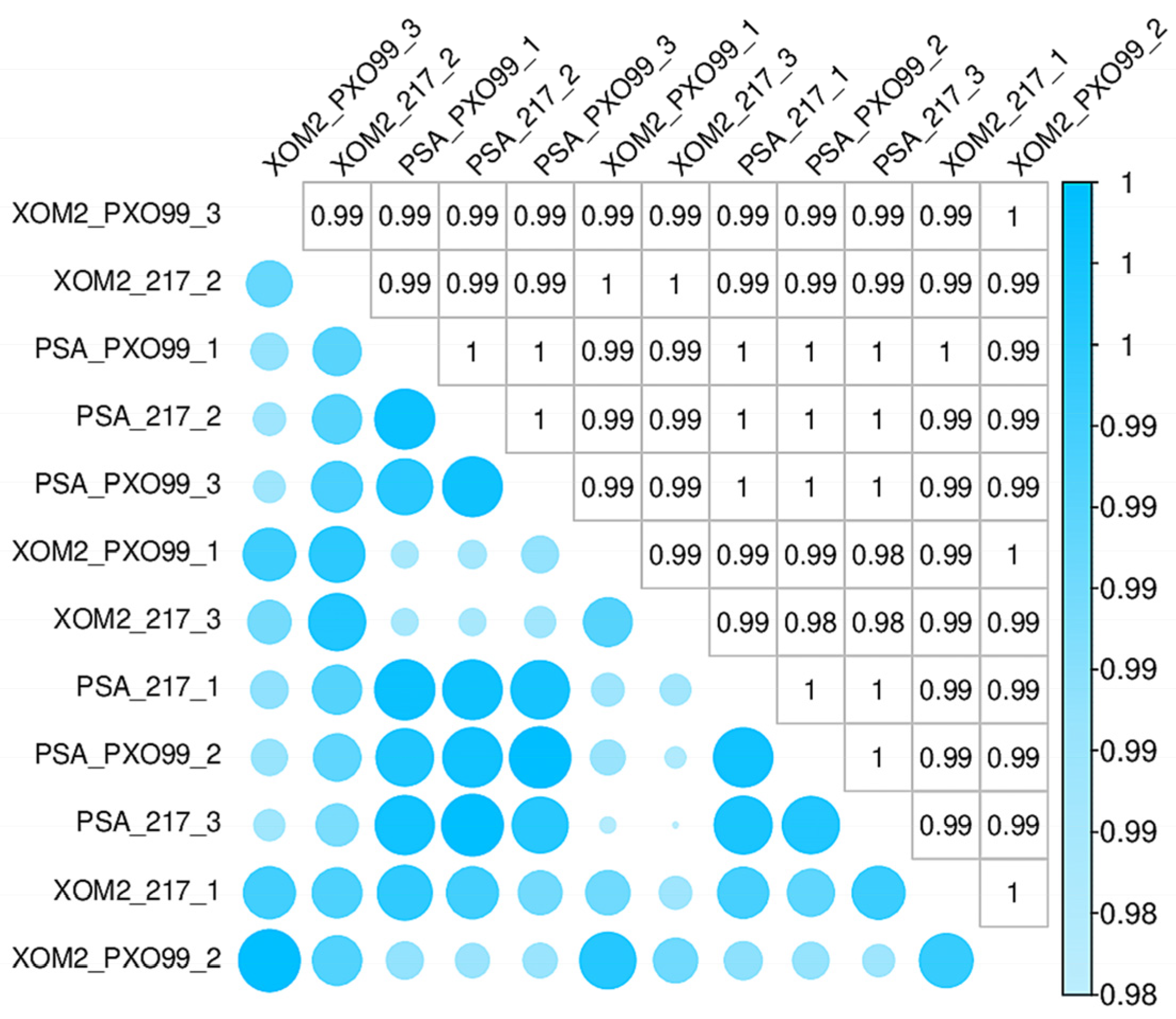
3.1. Overview of Comparative Transcriptomic Analyses
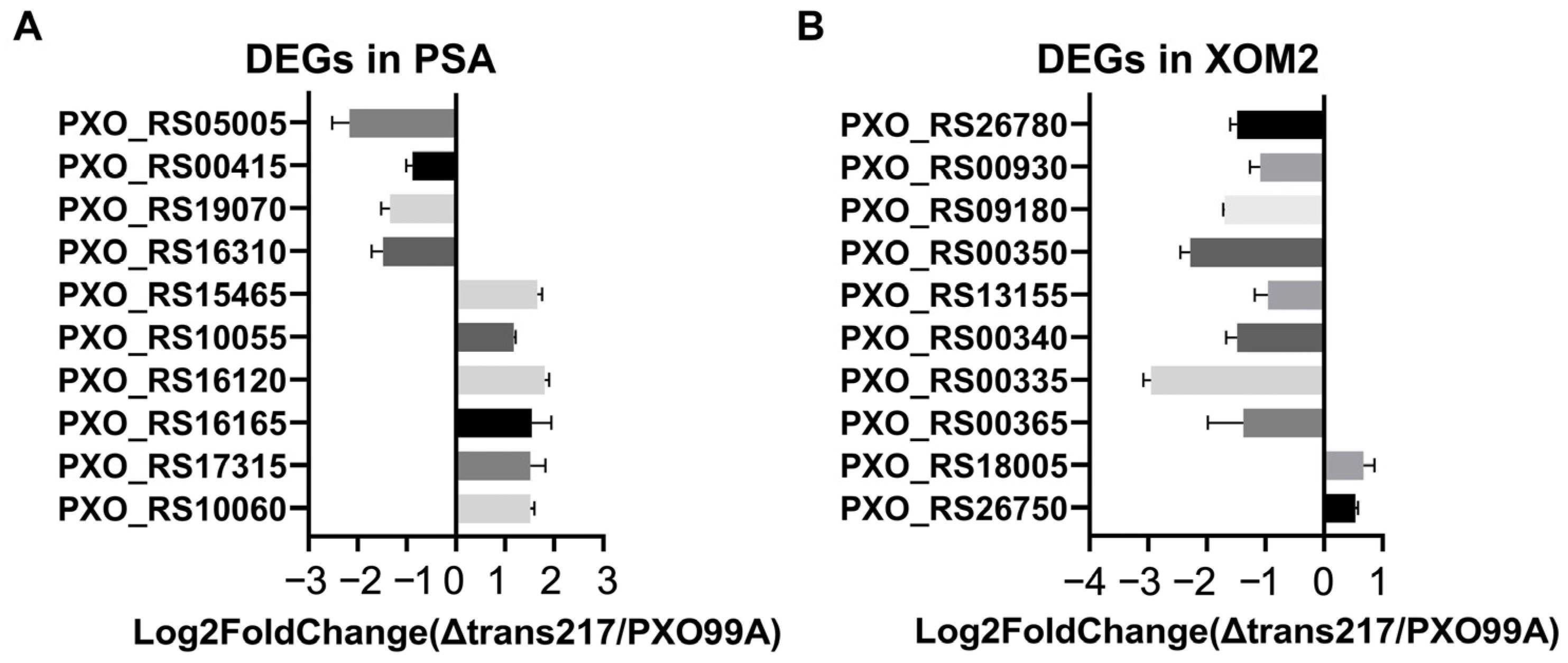
3.2. Validation of the Transcriptome Data with Quantitative Real-Time PCR
3.3. Quantitative and Categorical Analysis of the DEGs
3.4. Enrichment Analyses of DEGs
3.5. PXO_RS08490 Contributes to Bacterial Virulence
3.6. PXO_RS08490 Regulates Oxidative Stress
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Diallo, I.; Provost, P. RNA-sequencing analyses of small bacterial RNAs and their emergence as virulence factors in host-pathogen interactions. Int. J. Mol. Sci. 2020, 21, 1627. [Google Scholar] [CrossRef]
- Jogensen, M.G.; Pettersen, J.S.; Kallipolitis, B.H. sRNA-mediated control in bacteria: An increasing diversity of regulatory mechanisms. Biochim. Biophys. Acta Gene Regul. Mech. 2020, 1863, 194504. [Google Scholar]
- Felden, B.; Cattoir, V. Bacterial adaptation to antibiotics through regulatory RNAs. Antimicrob. Agents Chemother. 2018, 62, 2503–2517. [Google Scholar] [CrossRef]
- Dutcher, H.A.; Raghavan, R. Origin, evolution, and loss of bacterial small RNAs. Microbiol. Spectr. 2018, 6, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Massé, E.; Majdalani, N.; Gottesman, S. Regulatory roles for small RNAs in bacteria. Curr. Opin. Microbiol. 2003, 6, 120–124. [Google Scholar] [CrossRef]
- Bronesky, D.; Wu, Z.; Marzi, S.; Walter, P.; Geissmann, T.; Moreau, K.; Vandenesch, F.; Caldelari, I.; Romby, P. Staphylococcus aureus RNAIII and its regulon link quorum sensing, stress responses, metabolic adaptation, and regulation of virulence gene expression. Annu. Rev. Microbiol. 2016, 70, 299–316. [Google Scholar] [CrossRef] [PubMed]
- Urban, J.H.; Vogel, J. Two seemingly homologous noncoding RNAs act hierarchically to activate glmS mRNA translation. PLoS Biol. 2008, 6, e64. [Google Scholar] [CrossRef]
- Pfeiffer, V.; Kai, P.; Lucchini, S.; Hinton, J.C.; Vogel, J. Coding sequence targeting by MicC RNA reveals bacterial mRNA silencing downstream of translational initiation. Nat. Struct. Mol. Biol. 2009, 16, 840–846. [Google Scholar] [CrossRef]
- Sittka, A.; Sharma, C.M.; Rolle, K.; Vogel, J. Deep sequencing of Salmonella RNA associated with heterologous Hfq proteins in vivo reveals small RNAs as a major target class and identifies RNA processing phenotypes. RNA Biol. 2009, 6, 266–275. [Google Scholar] [CrossRef]
- Sonnleitner, E.; Wulf, A.; Campagne, S.; Pei, X.Y.; Wolfinger, M.T.; Forlani, G.; Prindl, K.; Abdou, L.; Resch, A.; Allain, F.H.; et al. Interplay between the catabolite repression control protein Crc, Hfq and RNA in Hfq-dependent translational regulation in Pseudomonas aeruginosa. Nucleic Acids Res. 2018, 46, 1470–1485. [Google Scholar] [CrossRef]
- Urbanowski, M.L.; Stauffer, L.T.; Stauffer, G.V. The gcvB gene encodes a small untranslated RNA involved in expression of the dipeptide and oligopeptide transport systems in Escherichia coli. Mol. Microbiol. 2010, 37, 856–868. [Google Scholar] [CrossRef]
- Lyu, Y.; Wu, J.; Shi, Y. Metabolic and physiological perturbations of Escherichia coli W3100 by bacterial small RNA RyhB. Biochimie 2019, 162, 144–155. [Google Scholar] [CrossRef]
- Calderón, P.F.; Morales, E.H.; Acuña, L.G.; Fuentes, D.N.; Gil, F.; Porwollik, S.; McClelland, M.; Saavedra, C.P.; Calderón, I.L. The small RNA RyhB homologs from Salmonella typhimurium participate in the response to S-nitrosoglutathione-induced stress. Biochem. Biophys. Res. Commun. 2014, 450, 641–645. [Google Scholar] [CrossRef] [PubMed]
- Porcheron, G.; Dozois, C.M. Interplay between iron homeostasis and virulence: Fur and RyhB as major regulators of bacterial pathogenicity. Vet. Microbiol. 2015, 179, 2–14. [Google Scholar] [CrossRef] [PubMed]
- Tronnet, S.; Garcie, C.; Brachmann, A.O.; Piel, J.; Oswald, E.; Martin, P. High iron supply inhibits the synthesis of the genotoxin colibactin by pathogenic Escherichia coli through a non-canonical Fur/RyhB-mediated pathway. Pathog. Dis. 2017, 75, ftx066. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Liu, S.; Wu, N.; Yuan, Y.; Zhang, W.; Zhang, Y. Small Non-coding RNA RyhB Mediates Persistence to Multiple Antibiotics and Stresses in Uropathogenic Escherichia coli by Reducing Cellular Metabolism. Front. Microbiol. 2018, 9, 136. [Google Scholar] [CrossRef]
- Wang, L.; Cai, X.; Wu, S.; Bomjan, R.; Nakayasu, E.S.; Händler, K.; Hinton, J.C.D.; Zhou, D.G. InvS coordinates expression of PrgH and FimZ, and is required for invasion of epithelial Cells by Salmonella enterica serovar Typhimurium. J. Bacteriol. 2017, 199, 10–1128. [Google Scholar] [CrossRef]
- Maki, K.; Morita, T.; Otaka, H.; Aiba, H. A minimal base-pairing region of a bacterial small RNA SgrS required for translational repression of ptsG mRNA. Mol. Microbiol. 2010, 76, 782–792. [Google Scholar] [CrossRef]
- Richards, G.R.; Patel, M.V.; Lloyd, C.R.; Vanderpool, C.K. Depletion of glycolytic intermediates plays a key role in glucose-phosphate stress in Escherichia coli. J. Bacteriol. 2013, 195, 4816–4825. [Google Scholar] [CrossRef]
- Ross, J.A.; Thorsing, M.; Lillebæk, E.M.S.; Teixeira, D.; Santos, P.; Kallipolitis, B.H. The LhrC sRNAs control expression of T cell-stimulating antigen TcsA in Listeria monocytogenes by decreasing tcsA mRNA stability. RNA Biol. 2019, 16, 270–281. [Google Scholar] [CrossRef]
- Dos Santos, P.T.; Menendez-Gil, P.; Sabharwal, D.; Christensen, J.H.; Brunhede, M.Z.; Lillebæk, E.M.S.; Kallipolitis, B.H. The small regulatory RNAs LhrC1–5 contribute to the response of Listeria monocytogenes to heme toxicity. Front. Microbiol. 2018, 9, 599. [Google Scholar] [CrossRef]
- Kathirvel, M.; Buchad, H.; Nair, M. Enhancement of the pathogenicity of Staphylococcus aureus strain Newman by a small noncoding RNA SprX1. Med. Microbiol. Immunol. 2016, 205, 563–574. [Google Scholar] [CrossRef]
- Schmidtke, C.; Findeiss, S.; Sharma, C.M.; Kuhfuss, J.; Hoffmann, S.; Vogel, J.; Stadler, P.F.; Bonas, U. Genome-wide transcriptome analysis of the plant pathogen Xanthomonas identifies sRNAs with putative virulence functions. Nucleic Acids Res. 2012, 40, 2020–2031. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Wang, S.; Nie, W.; Wang, P.; Fu, L.; Ahmad, I.; Zhu, B.; Chen, G. A key antisense sRNA modulates the oxidative stress response and virulence in Xanthomonas oryzae pv. oryzicola. PLoS Pathog. 2021, 17, e1009762. [Google Scholar] [CrossRef]
- Tang, D.J.; Chen, X.L.; Jia, Y.; Liang, Y.W.; He, Y.P.; Lu, T.T.; Zhu, C.R.; Han, B.; An, S.Q.; Tang, J.L. Genome-wide screen and functional analysis in Xanthomonas reveal a large number of mRNA-derived sRNAs, including the novel RsmA-sequester RsmU. Mol. Plant Pathol. 2020, 21, 1573–1590. [Google Scholar] [CrossRef]
- Schmidtke, C.; Abendroth, U.; Brock, J.; Serrania, J.; Becker, A.; Bonas, U. Small RNA sX13: A multifaceted regulator of virulence in the plant pathogen Xanthomonas. PLoS Pathog. 2013, 9, e1003626. [Google Scholar] [CrossRef]
- Chen, X.L.; Tang, D.J.; Jiang, R.P.; He, Y.Q.; Jiang, B.L.; Lu, G.T.; Tang, J.L. sRNA-Xcc1, an integron-encoded transposon- and plasmid-transferred trans-acting sRNA, is under the positive control of the key virulence regulators HrpG and HrpX of Xanthomonas campestris pathovar campestris. RNA Biol. 2011, 8, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Nino-Liu, D.O.; Ronald, P.C.; Bogdanove, A.J. Xanthomonas oryzae pathovars: Model pathogens of a model crop. Mol. Plant Pathol. 2006, 7, 303–324. [Google Scholar] [CrossRef]
- Sere, Y.; Onasanya, A.; Verdier, V.; Akator, K.; Ouedraogo, L.S.; Segda, Z.; Mbare, M.M.; Sido, A.Y.; Basso, A. Rice bacterial leaf blight in West Africa: Preliminary studies on disease in farmers’ fields and screening released varieties for resistance to the bacteria. Asian J. Plant Sci. 2005, 4, 577–579. [Google Scholar] [CrossRef]
- Hu, Y.; Zhang, L.; Wang, X.; Sun, F.; Kong, X.; Dong, H.; Xu, H. Two virulent sRNAs identified by genomic sequencing target the type III secretion system in rice bacterial blight pathogen. BMC Plant Biol. 2018, 18, 237. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Lozano, M.; Marvig, R.L.; Molin, S.; Long, K.S. Genome-wide identification of novel small RNAs in Pseudomonas aeruginosa. Environ. Microbiol. 2012, 14, 2006–2016. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Büttner, D.; Bonas, U. Getting across-bacterial type III effector proteins on their way to the plant cell. EMBO J. 2002, 21, 5313–5322. [Google Scholar] [CrossRef] [PubMed]
- White, F.F.; Yang, B. Host and pathogen factors controlling the rice-Xanthomonas oryzae interaction. Plant Physiol. 2009, 150, 1677–1686. [Google Scholar] [CrossRef] [PubMed]
- Grant, J.J.; Loake, G.J. Role of reactive oxygen intermediates and cognate redox signaling in disease resistance. Plant Physiol. 2000, 124, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Tayi, L.; Maku, R.V.; Patel, H.K.; Sonti, R.V. Identification of pectin degrading enzymes secreted by Xanthomonas oryzae pv. oryzae and determination of their role in virulence on rice. PLoS ONE 2016, 11, e0166396. [Google Scholar]
- Joe, A.; Stewart, V.; Ronald, P.C. The HrpX protein activates synthesis of the RaxX Sulfopeptide, required for activation of XA21-mediated immunity to Xanthomonas oryzae pv. oryzae. Mol. Plant Microbe Interact. 2021, 34, 1307–1315. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Q30 (%) | GC Content (%) | Total_Reads | Mapped_Reads | Gene Numbers |
|---|---|---|---|---|---|
| PSA_217_1 | 94.55 | 61.10 | 8,206,307 | 7,298,583 (89%) | 4503 |
| PSA_217_2 | 94.71 | 61.36 | 8,290,772 | 7,588,419 (92%) | 4500 |
| PSA_217_3 | 93.84 | 61.27 | 8,051,830 | 7,224,667 (90%) | 4485 |
| PSA_PXO99_1 | 94.55 | 60.65 | 7,812,714 | 6,333,425 (81%) | 4485 |
| PSA_PXO99_2 | 94.69 | 60.70 | 8,192,639 | 6,621,761 (81%) | 4493 |
| PSA_PXO99_3 | 94.64 | 60.61 | 7,992,108 | 6,561,197 (82%) | 4487 |
| XOM2_217_1 | 93.83 | 60.87 | 8,059,737 | 7,251,912 (90%) | 4505 |
| XOM2_217_2 | 93.79 | 60.89 | 8,199,489 | 7,352,156 (90%) | 4504 |
| XOM2_217_3 | 93.68 | 60.72 | 7,770,330 | 6,905,891 (89%) | 4508 |
| XOM2_PXO99_1 | 94.57 | 60.33 | 7,923,055 | 6,573,845 (83%) | 4507 |
| XOM2_PXO99_2 | 94.59 | 60.51 | 8,532,656 | 7,177,531 (84%) | 4506 |
| XOM2_PXO99_3 | 94.47 | 60.28 | 8,325,762 | 6,789,990 (82%) | 4499 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, Y.; Zhang, J.; Zhang, A. Genome-Wide Transcriptome Analysis of a Virulent sRNA, Trans217, in Xanthomonas oryzae pv. oryzae (Xoo), the Causative Agent of Rice Bacterial Blight. Microorganisms 2024, 12, 1684. https://doi.org/10.3390/microorganisms12081684
Hu Y, Zhang J, Zhang A. Genome-Wide Transcriptome Analysis of a Virulent sRNA, Trans217, in Xanthomonas oryzae pv. oryzae (Xoo), the Causative Agent of Rice Bacterial Blight. Microorganisms. 2024; 12(8):1684. https://doi.org/10.3390/microorganisms12081684
Chicago/Turabian StyleHu, Yiqun, Jianjian Zhang, and Aifang Zhang. 2024. "Genome-Wide Transcriptome Analysis of a Virulent sRNA, Trans217, in Xanthomonas oryzae pv. oryzae (Xoo), the Causative Agent of Rice Bacterial Blight" Microorganisms 12, no. 8: 1684. https://doi.org/10.3390/microorganisms12081684