
Cholesterol Efflux Decreases TLR4-Target Gene Expression in Cultured Macrophages Exposed to T. brucei Ghosts
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Maintenance of Cells
2.2. Preparation and Characterization of BF-427 Parasite Ghosts (PGs)
2.3. Macrophage Exposure to PG
2.4. Isolation of Macrophage Lipid Rafts and Immunoblotting
2.5. Macrophage Total RNA Extraction and RT-qPCR
2.6. Statistical Analysis
3. Results
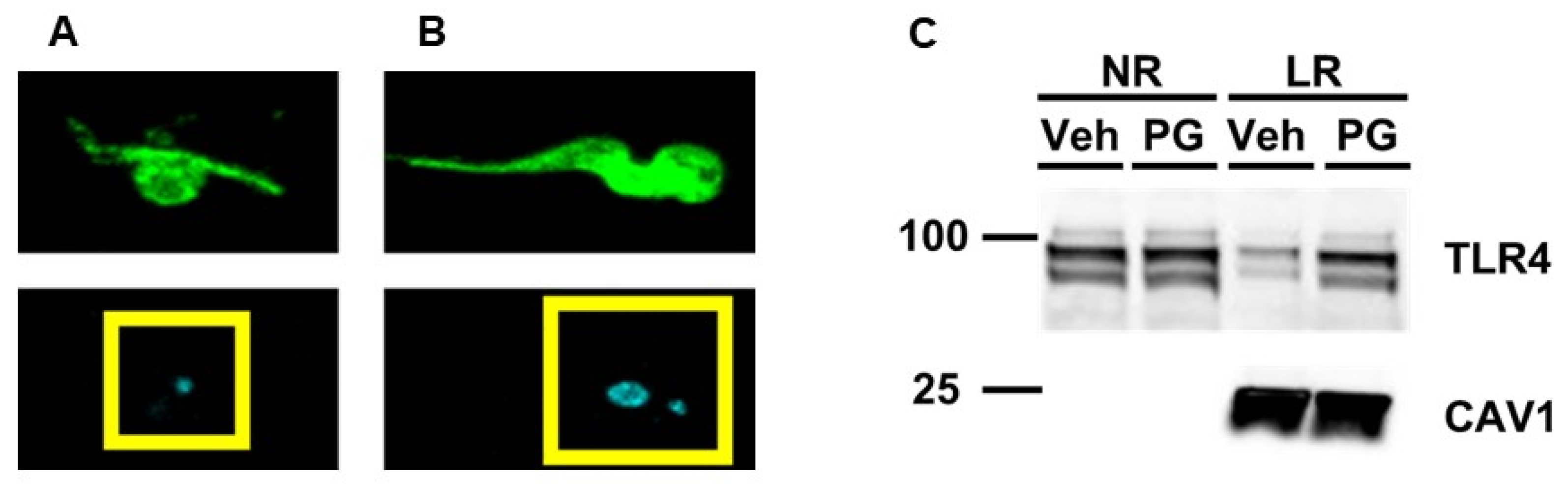
3.1. Exposing Cultured Macrophages to T. brucei “Ghosts” Triggers TLR4 Translocation to Lipid Rafts
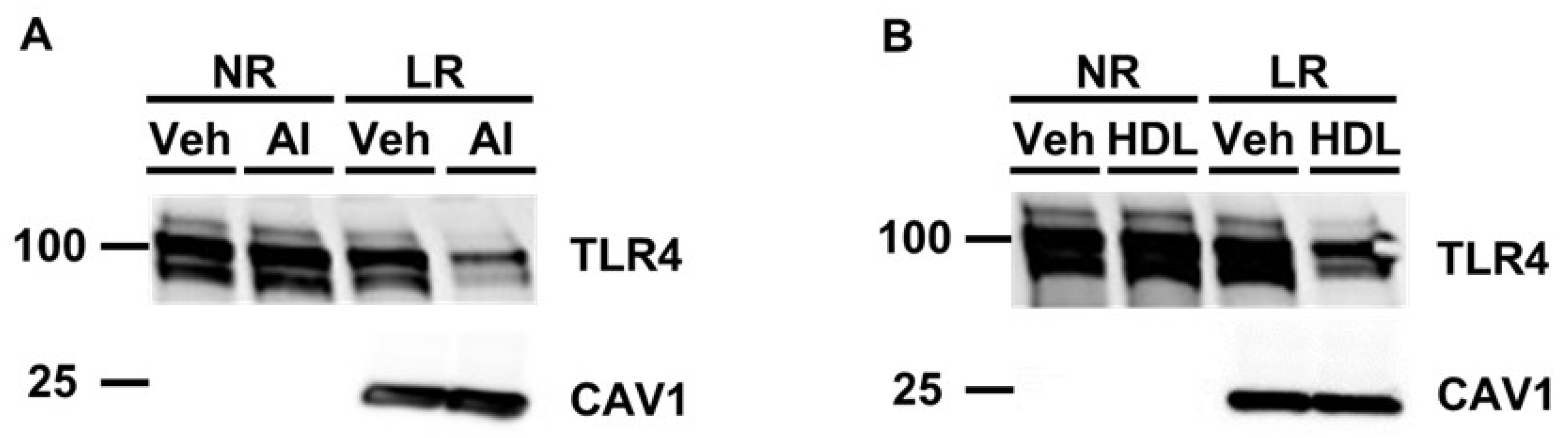
3.2. Pretreating Cultured Macrophages with Cholesterol Acceptors Reduces PG-Induced TLR4 Translocation to Lipid Rafts
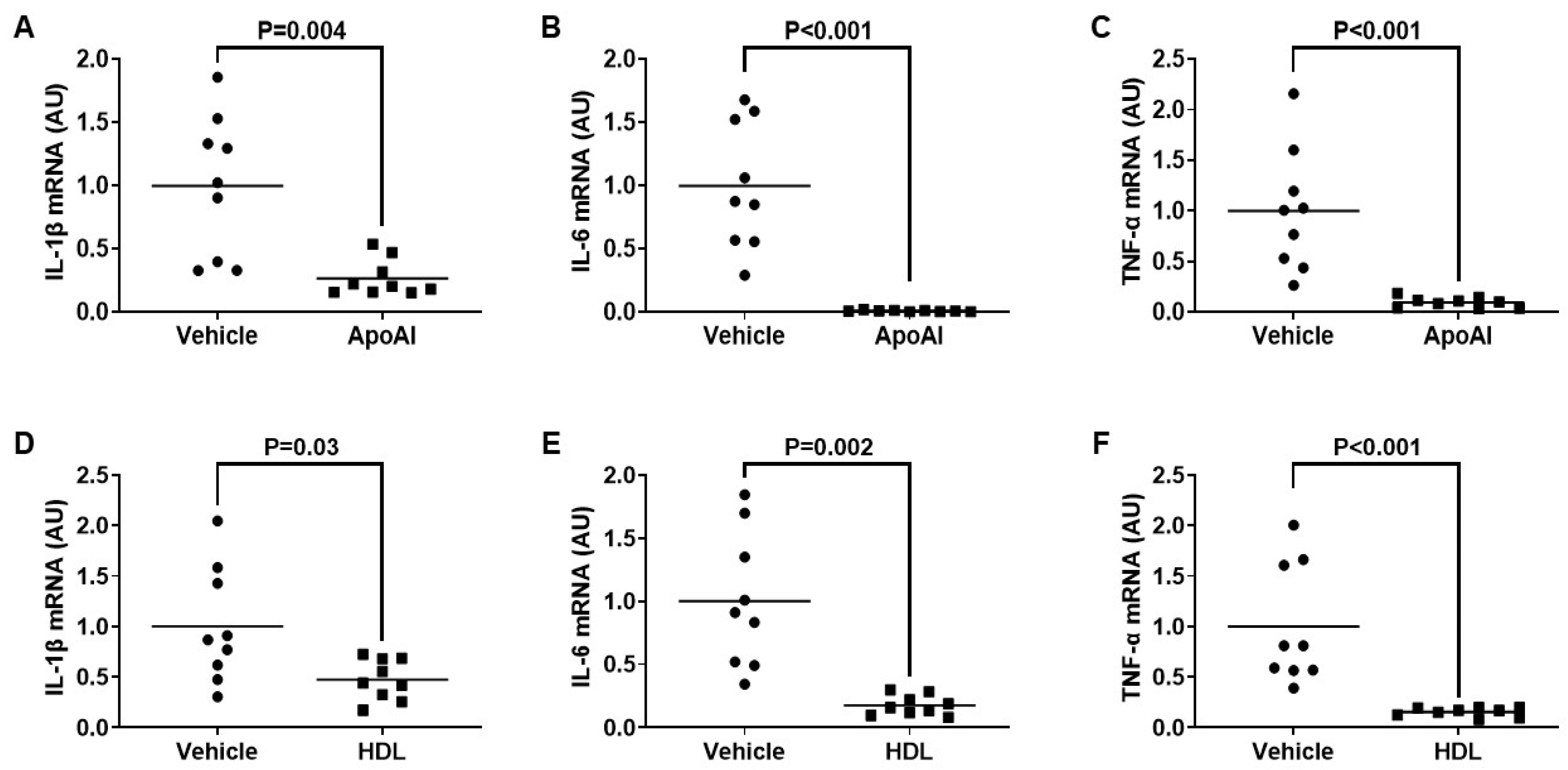
3.3. ApoAI- and HDL-Mediated Cholesterol Efflux Decreases TLR4-Target Gene Expression in Cultured Macrophages Exposed To PG
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Dean, S. Basic Biology of Trypanosoma brucei with Reference to the Development of Chemotherapies. Curr. Pharm. Des. 2021, 27, 1650–1670. [Google Scholar] [CrossRef] [PubMed]
- Malvy, D.; Chappuis, F. Sleeping sickness. Clin. Microbiol. Infect. 2011, 17, 986–995. [Google Scholar] [CrossRef]
- Hollingshead, C.M.; Bermudez, R. Human African Trypanosomiasis (Sleeping Sickness). In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Jacobs, R.T.; Nare, B.; Phillips, M.A. State of the art in African trypanosome drug discovery. Curr. Top. Med. Chem. 2011, 11, 1255–1274. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, J.; Ortiz, J.F.; Fabara, S.P.; Eissa-Garces, A.; Reddy, D.; Collins, K.D.; Tirupathi, R. Efficacy and Toxicity of Fexinidazole and Nifurtimox Plus Eflornithine in the Treatment of African Trypanosomiasis: A Systematic Review. Cureus 2021, 13, e16881. [Google Scholar] [CrossRef]
- Brun, R.; Schumacher, R.; Schmid, C.; Kunz, C.; Burri, C. The phenomenon of treatment failures in Human African Trypanosomiasis. Trop. Med. Int. Health 2001, 6, 906–914. [Google Scholar] [CrossRef]
- Bouteille, B.; Oukem, O.; Bisser, S.; Dumas, M. Treatment perspectives for human African trypanosomiasis. Fundam. Clin. Pharmacol. 2003, 17, 171–181. [Google Scholar] [CrossRef]
- Mulenga, G.M.; Henning, L.; Chilongo, K.; Mubamba, C.; Namangala, B.; Gummow, B. Insights into the Control and Management of Human and Bovine African Trypanosomiasis in Zambia between 2009 and 2019—A Review. Trop. Med. Infect. Dis. 2020, 5, 115. [Google Scholar] [CrossRef] [PubMed]
- Kargbo, A.; Jawo, E.; Amoutchi, A.I.; Koua, H.; Kuye, R.; Dabre, Z.; Bojang, A.; Vieira, R.F.C. Knowledge, Attitude, and Practice of Livestock Owners and Livestock Assistants towards African Trypanosomiasis Control in The Gambia. J. Parasitol. Res. 2022, 2022, 3379804. [Google Scholar] [CrossRef]
- Giordani, F.; Morrison, L.J.; Rowan, T.G.; HP, D.E.K.; Barrett, M.P. The animal trypanosomiases and their chemotherapy: A review. Parasitology 2016, 143, 1862–1889. [Google Scholar] [CrossRef]
- Desquesnes, M.; Gonzatti, M.; Sazmand, A.; Thevenon, S.; Bossard, G.; Boulange, A.; Gimonneau, G.; Truc, P.; Herder, S.; Ravel, S.; et al. A review on the diagnosis of animal trypanosomoses. Parasit. Vectors 2022, 15, 64. [Google Scholar] [CrossRef]
- Ralston, K.S.; Kabututu, Z.P.; Melehani, J.H.; Oberholzer, M.; Hill, K.L. The Trypanosoma brucei flagellum: Moving parasites in new directions. Annu. Rev. Microbiol. 2009, 63, 335–362. [Google Scholar] [CrossRef] [PubMed]
- Galvao-Castro, B.; Hochmann, A.; Lambert, P.H. The role of the host immune response in the development of tissue lesions associated with African trypanosomiasis in mice. Clin. Exp. Immunol. 1978, 33, 12–24. [Google Scholar] [PubMed]
- Seyfang, A.; Mecke, D.; Duszenko, M. Degradation, recycling, and shedding of Trypanosoma brucei variant surface glycoprotein. J. Protozool. 1990, 37, 546–552. [Google Scholar] [CrossRef]
- Cardoso de Almeida, M.L.; Turner, M.J. The membrane form of variant surface glycoproteins of Trypanosoma brucei. Nature 1983, 302, 349–352. [Google Scholar] [CrossRef]
- Sheader, K.; Vaughan, S.; Minchin, J.; Hughes, K.; Gull, K.; Rudenko, G. Variant surface glycoprotein RNA interference triggers a precytokinesis cell cycle arrest in African trypanosomes. Proc. Natl. Acad. Sci. USA 2005, 102, 8716–8721. [Google Scholar] [CrossRef] [PubMed]
- Baral, T.N. Immunobiology of African trypanosomes: Need of alternative interventions. J. Biomed. Biotechnol. 2010, 2010, 389153. [Google Scholar] [CrossRef]
- Onyilagha, C.; Uzonna, J.E. Host Immune Responses and Immune Evasion Strategies in African Trypanosomiasis. Front. Immunol. 2019, 10, 2738. [Google Scholar] [CrossRef]
- Dos-Santos, A.L.; Carvalho-Kelly, L.F.; Dick, C.F.; Meyer-Fernandes, J.R. Innate immunomodulation to trypanosomatid parasite infections. Exp. Parasitol. 2016, 167, 67–75. [Google Scholar] [CrossRef]
- Donelson, J.E.; Hill, K.L.; El-Sayed, N.M. Multiple mechanisms of immune evasion by African trypanosomes. Mol. Biochem. Parasitol. 1998, 91, 51–66. [Google Scholar] [CrossRef]
- Wang, Y.N.; Wang, M.; Field, M.C. Trypanosoma brucei: Trypanosome-specific endoplasmic reticulum proteins involved in variant surface glycoprotein expression. Exp. Parasitol. 2010, 125, 208–221. [Google Scholar] [CrossRef]
- Hertz, C.J.; Filutowicz, H.; Mansfield, J.M. Resistance to the African trypanosomes is IFN-gamma dependent. J. Immunol. 1998, 161, 6775–6783. [Google Scholar] [CrossRef] [PubMed]
- Drennan, M.B.; Stijlemans, B.; Van den Abbeele, J.; Quesniaux, V.J.; Barkhuizen, M.; Brombacher, F.; De Baetselier, P.; Ryffel, B.; Magez, S. The induction of a type 1 immune response following a Trypanosoma brucei infection is MyD88 dependent. J. Immunol. 2005, 175, 2501–2509. [Google Scholar] [CrossRef]
- Debierre-Grockiego, F. Glycolipids are potential targets for protozoan parasite diseases. Trends Parasitol. 2010, 26, 404–411. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Jiang, N.; Sang, X.; Feng, Y.; Chen, R.; Chen, Q. Trypanosoma brucei Lipophosphoglycan Induces the Formation of Neutrophil Extracellular Traps and Reactive Oxygen Species Burst via Toll-Like Receptor 2, Toll-Like Receptor 4, and c-Jun N-Terminal Kinase Activation. Front. Microbiol. 2021, 12, 713531. [Google Scholar] [CrossRef]
- Zhang, K.; Jiang, N.; Zhang, N.; Yu, L.; Sang, X.; Feng, Y.; Chen, R.; Chen, Q. Trypanosoma brucei Lipophosphoglycan Activates Host Immune Responses via the TLR-mediated p38 MAP Kinase and NF-κB Pathways. Zoonoses 2023, 3, 991. [Google Scholar] [CrossRef]
- Macaskill, J.A.; Holmes, P.H.; Whitelaw, D.D.; McConnell, I.; Jennings, F.W.; Urquhart, G.M. Immunological clearance of 75Se-labelled Trypanosoma brucei in mice. II. Mechanisms in immune animals. Immunology 1980, 40, 629–635. [Google Scholar]
- Magez, S.; Pinto Torres, J.E.; Obishakin, E.; Radwanska, M. Infections With Extracellular Trypanosomes Require Control by Efficient Innate Immune Mechanisms and Can Result in the Destruction of the Mammalian Humoral Immune System. Front. Immunol. 2020, 11, 382. [Google Scholar] [CrossRef]
- Fang, H.; Pengal, R.A.; Cao, X.; Ganesan, L.P.; Wewers, M.D.; Marsh, C.B.; Tridandapani, S. Lipopolysaccharide-induced macrophage inflammatory response is regulated by SHIP. J. Immunol. 2004, 173, 360–366. [Google Scholar] [CrossRef]
- Yvan-Charvet, L.; Wang, N.; Tall, A.R. Role of HDL, ABCA1, and ABCG1 transporters in cholesterol efflux and immune responses. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 139–143. [Google Scholar] [CrossRef]
- Zhu, X.; Owen, J.S.; Wilson, M.D.; Li, H.; Griffiths, G.L.; Thomas, M.J.; Hiltbold, E.M.; Fessler, M.B.; Parks, J.S. Macrophage ABCA1 reduces MyD88-dependent Toll-like receptor trafficking to lipid rafts by reduction of lipid raft cholesterol. J. Lipid Res. 2010, 51, 3196–3206. [Google Scholar] [CrossRef]
- Zhu, X.; Lee, J.Y.; Timmins, J.M.; Brown, J.M.; Boudyguina, E.; Mulya, A.; Gebre, A.K.; Willingham, M.C.; Hiltbold, E.M.; Mishra, N.; et al. Increased cellular free cholesterol in macrophage-specific Abca1 knock-out mice enhances pro-inflammatory response of macrophages. J. Biol. Chem. 2008, 283, 22930–22941. [Google Scholar] [CrossRef] [PubMed]
- Stamatikos, A.; Dronadula, N.; Ng, P.; Palmer, D.; Knight, E.; Wacker, B.K.; Tang, C.; Kim, F.; Dichek, D.A. ABCA1 Overexpression in Endothelial Cells In Vitro Enhances ApoAI-Mediated Cholesterol Efflux and Decreases Inflammation. Hum. Gene Ther. 2019, 30, 236–248. [Google Scholar] [CrossRef] [PubMed]
- Westerterp, M.; Tsuchiya, K.; Tattersall, I.W.; Fotakis, P.; Bochem, A.E.; Molusky, M.M.; Ntonga, V.; Abramowicz, S.; Parks, J.S.; Welch, C.L.; et al. Deficiency of ATP-Binding Cassette Transporters A1 and G1 in Endothelial Cells Accelerates Atherosclerosis in Mice. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1328–1337. [Google Scholar] [CrossRef]
- Cheng, A.M.; Handa, P.; Tateya, S.; Schwartz, J.; Tang, C.; Mitra, P.; Oram, J.F.; Chait, A.; Kim, F. Apolipoprotein A-I attenuates palmitate-mediated NF-kappaB activation by reducing Toll-like receptor-4 recruitment into lipid rafts. PLoS ONE 2012, 7, e33917. [Google Scholar] [CrossRef]
- Han, C.Y.; Tang, C.; Guevara, M.E.; Wei, H.; Wietecha, T.; Shao, B.; Subramanian, S.; Omer, M.; Wang, S.; O’Brien, K.D.; et al. Serum amyloid A impairs the antiinflammatory properties of HDL. J. Clin. Investig. 2016, 126, 266–281. [Google Scholar] [CrossRef] [PubMed]
- Umemoto, T.; Han, C.Y.; Mitra, P.; Averill, M.M.; Tang, C.; Goodspeed, L.; Omer, M.; Subramanian, S.; Wang, S.; Den Hartigh, L.J.; et al. Apolipoprotein AI and high-density lipoprotein have anti-inflammatory effects on adipocytes via cholesterol transporters: ATP-binding cassette A-1, ATP-binding cassette G-1, and scavenger receptor B-1. Circ. Res. 2013, 112, 1345–1354. [Google Scholar] [CrossRef]
- Vaure, C.; Liu, Y. A comparative review of toll-like receptor 4 expression and functionality in different animal species. Front. Immunol. 2014, 5, 316. [Google Scholar] [CrossRef]
- Kim, H.J.; Kim, H.; Lee, J.H.; Hwangbo, C. Toll-like receptor 4 (TLR4): New insight immune and aging. Immun. Ageing 2023, 20, 67. [Google Scholar] [CrossRef]
- Plociennikowska, A.; Hromada-Judycka, A.; Borzecka, K.; Kwiatkowska, K. Co-operation of TLR4 and raft proteins in LPS-induced pro-inflammatory signaling. Cell Mol. Life Sci. 2015, 72, 557–581. [Google Scholar] [CrossRef]
- Bi, X.; Vitali, C.; Cuchel, M. ABCA1 and Inflammation: From Animal Models to Humans. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1551–1553. [Google Scholar] [CrossRef]
- Paulnock, D.M.; Coller, S.P. Analysis of macrophage activation in African trypanosomiasis. J. Leukoc. Biol. 2001, 69, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Kuriakose, S.M.; Singh, R.; Uzonna, J.E. Host Intracellular Signaling Events and Pro-inflammatory Cytokine Production in African Trypanosomiasis. Front. Immunol. 2016, 7, 181. [Google Scholar] [CrossRef] [PubMed]
- Echesabal-Chen, J.; Huang, K.; Vojtech, L.; Oladosu, O.; Esobi, I.; Sachdeva, R.; Vyavahare, N.; Jo, H.; Stamatikos, A. Constructing Lipoparticles Capable of Endothelial Cell-Derived Exosome-Mediated Delivery of Anti-miR-33a-5p to Cultured Macrophages. Curr. Issues Mol. Biol. 2023, 45, 5631–5644. [Google Scholar] [CrossRef] [PubMed]
- Hirumi, H.; Hirumi, K. Continuous cultivation of Trypanosoma brucei blood stream forms in a medium containing a low concentration of serum protein without feeder cell layers. J. Parasitol. 1989, 75, 985–989. [Google Scholar] [CrossRef] [PubMed]
- Ezeh, I.O.; Ugwu, E.N.; Enemuo, O.V.; Obi, C.F.; Iheagwam, C.N.; Ezeokonkwo, R.C.; Onah, D.N. Efficacy of repeated doses of diminazene aceturate (Dinazene((R))) in the treatment of experimental Trypanosoma brucei infection of Albino rats. Iran. J. Vet. Res. 2016, 17, 124–129. [Google Scholar]
- Esobi, I.C.; Barksdale, C.; Heard-Tate, C.; Reigers Powell, R.; Bruce, T.F.; Stamatikos, A. MOVAS Cells: A Versatile Cell Line for Studying Vascular Smooth Muscle Cell Cholesterol Metabolism. Lipids 2021, 56, 413–422. [Google Scholar] [CrossRef]
- Esobi, I.C.; Oladosu, O.; Echesabal-Chen, J.; Powell, R.R.; Bruce, T.; Stamatikos, A. miR-33a Expression Attenuates ABCA1-Dependent Cholesterol Efflux and Promotes Macrophage-Like Cell Transdifferentiation in Cultured Vascular Smooth Muscle Cells. J. Lipids 2023, 2023, 8241899. [Google Scholar] [CrossRef]
- Oladosu, O.; Esobi, I.C.; Powell, R.R.; Bruce, T.; Stamatikos, A. Dissecting the Impact of Vascular Smooth Muscle Cell ABCA1 versus ABCG1 Expression on Cholesterol Efflux and Macrophage-like Cell Transdifferentiation: The Role of SR-BI. J. Cardiovasc. Dev. Dis. 2023, 10, 416. [Google Scholar] [CrossRef]
- Esobi, I.; Olanrewaju, O.; Echesabal-Chen, J.; Stamatikos, A. Utilizing the LoxP-Stop-LoxP System to Control Transgenic ABC-Transporter Expression In Vitro. Biomolecules 2022, 12, 679. [Google Scholar] [CrossRef]
- Oladosu, O.; Chin, E.; Barksdale, C.; Powell, R.R.; Bruce, T.; Stamatikos, A. Inhibition of miR-33a-5p in Macrophage-like Cells In Vitro Promotes apoAI-Mediated Cholesterol Efflux. Pathophysiology 2024, 31, 117–126. [Google Scholar] [CrossRef]
- Huang, K.; Garimella, S.; Clay-Gilmour, A.; Vojtech, L.; Armstrong, B.; Bessonny, M.; Stamatikos, A. Comparison of Human Urinary Exosomes Isolated via Ultracentrifugation Alone versus Ultracentrifugation Followed by SEC Column-Purification. J. Pers. Med. 2022, 12, 340. [Google Scholar] [CrossRef] [PubMed]
- Stamatikos, A.; Knight, E.; Vojtech, L.; Bi, L.; Wacker, B.K.; Tang, C.; Dichek, D.A. Exosome-Mediated Transfer of Anti-miR-33a-5p from Transduced Endothelial Cells Enhances Macrophage and Vascular Smooth Muscle Cell Cholesterol Efflux. Hum. Gene Ther. 2020, 31, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Jo, H.; Echesabal-Chen, J.; Stamatikos, A. Combined LXR and RXR Agonist Therapy Increases ABCA1 Protein Expression and Enhances ApoAI-Mediated Cholesterol Efflux in Cultured Endothelial Cells. Metabolites 2021, 11, 640. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Pitman, M.; Oladosu, O.; Echesabal-Chen, J.; Vojtech, L.; Esobi, I.; Larsen, J.; Jo, H.; Stamatikos, A. The Impact of MiR-33a-5p Inhibition in Pro-Inflammatory Endothelial Cells. Diseases 2023, 11, 88. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Pays, E.; Nolan, D.P. Genetic and immunological basis of human African trypanosomiasis. Curr. Opin. Immunol. 2021, 72, 13–20. [Google Scholar] [CrossRef]
- Stijlemans, B.; Caljon, G.; Van Den Abbeele, J.; Van Ginderachter, J.A.; Magez, S.; De Trez, C. Immune Evasion Strategies of Trypanosoma brucei within the Mammalian Host: Progression to Pathogenicity. Front. Immunol. 2016, 7, 233. [Google Scholar] [CrossRef]
- Yu, L.; Li, Q.; Jiang, N.; Fan, R.; Zhang, N.; Zhang, Y.; Sun, W.; Chen, R.; Feng, Y.; Sang, X.; et al. Toll-like receptor 9 signaling is associated with immune responses to Trypanosoma brucei infection. Int. Immunopharmacol. 2024, 134, 112250. [Google Scholar] [CrossRef]
- Latz, E.; Visintin, A.; Espevik, T.; Golenbock, D.T. Mechanisms of TLR9 activation. J. Endotoxin Res. 2004, 10, 406–412. [Google Scholar] [CrossRef]
- Lee, B.L.; Barton, G.M. Trafficking of endosomal Toll-like receptors. Trends Cell Biol. 2014, 24, 360–369. [Google Scholar] [CrossRef]
- Degirmenci, I.; Ozbayer, C.; Kebapci, M.N.; Kurt, H.; Colak, E.; Gunes, H.V. Common variants of genes encoding TLR4 and TLR4 pathway members TIRAP and IRAK1 are effective on MCP1, IL6, IL1beta, and TNFalpha levels in type 2 diabetes and insulin resistance. Inflamm. Res. 2019, 68, 801–814. [Google Scholar] [CrossRef] [PubMed]
- Varshney, P.; Yadav, V.; Saini, N. Lipid rafts in immune signalling: Current progress and future perspective. Immunology 2016, 149, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Sviridov, D.; Mukhamedova, N.; Miller, Y.I. Lipid rafts as a therapeutic target. J. Lipid Res. 2020, 61, 687–695. [Google Scholar] [CrossRef]
- Ponte-Sucre, A. An Overview of Trypanosoma brucei Infections: An Intense Host-Parasite Interaction. Front. Microbiol. 2016, 7, 2126. [Google Scholar] [CrossRef]
- Machado, H.; Bizarra-Rebelo, T.; Costa-Sequeira, M.; Trindade, S.; Carvalho, T.; Rijo-Ferreira, F.; Rentroia-Pacheco, B.; Serre, K.; Figueiredo, L.M. Trypanosoma brucei triggers a broad immune response in the adipose tissue. PLoS Pathog. 2021, 17, e1009933. [Google Scholar] [CrossRef]
- An, S.M.; Cho, S.H.; Yoon, J.C. Adipose Tissue and Metabolic Health. Diabetes Metab. J. 2023, 47, 595–611. [Google Scholar] [CrossRef]
- Coelho, M.; Oliveira, T.; Fernandes, R. Biochemistry of adipose tissue: An endocrine organ. Arch. Med. Sci. 2013, 9, 191–200. [Google Scholar] [CrossRef]
- Galic, S.; Oakhill, J.S.; Steinberg, G.R. Adipose tissue as an endocrine organ. Mol. Cell Endocrinol. 2010, 316, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.K. Adipocytes. Curr. Biol. 2014, 24, R988–R993. [Google Scholar] [CrossRef]
- Harvey, I.; Boudreau, A.; Stephens, J.M. Adipose tissue in health and disease. Open Biol. 2020, 10, 200291. [Google Scholar] [CrossRef]
- Nakao, K. Adiposcience and adipotoxicity. Nat. Clin. Pract. Endocrinol. Metab. 2009, 5, 63. [Google Scholar] [CrossRef]
- Richard, A.J.; White, U.; Elks, C.M.; Stephens, J.M. Adipose Tissue: Physiology to Metabolic Dysfunction. In Endotext; Feingold, K.R., Anawalt, B., Blackman, M.R., Boyce, A., Chrousos, G., Corpas, E., de Herder, W.W., Dhatariya, K., Dungan, K., Hofland, J., et al., Eds.; MDText.com, Inc.: Dartmouth, MA, USA, 2000; Available online: https://www.ncbi.nlm.nih.gov/books/NBK555602/ (accessed on 16 August 2024).
- Mabille, D.; Dirkx, L.; Thys, S.; Vermeersch, M.; Montenye, D.; Govaerts, M.; Hendrickx, S.; Takac, P.; Van Weyenbergh, J.; Pintelon, I.; et al. Impact of pulmonary African trypanosomes on the immunology and function of the lung. Nat. Commun. 2022, 13, 7083. [Google Scholar] [CrossRef]
- Carvalho, T.; Trindade, S.; Pimenta, S.; Santos, A.B.; Rijo-Ferreira, F.; Figueiredo, L.M. Trypanosoma brucei triggers a marked immune response in male reproductive organs. PLoS Negl. Trop. Dis. 2018, 12, e0006690. [Google Scholar] [CrossRef] [PubMed]
- Capewell, P.; Cren-Travaille, C.; Marchesi, F.; Johnston, P.; Clucas, C.; Benson, R.A.; Gorman, T.A.; Calvo-Alvarez, E.; Crouzols, A.; Jouvion, G.; et al. The skin is a significant but overlooked anatomical reservoir for vector-borne African trypanosomes. Elife 2016, 5, e17716. [Google Scholar] [CrossRef]
- Quintana, J.F.; Sinton, M.C.; Chandrasegaran, P.; Lestari, A.N.; Heslop, R.; Cheaib, B.; Ogunsola, J.; Ngoyi, D.M.; Kuispond Swar, N.R.; Cooper, A.; et al. gammadelta T cells control murine skin inflammation and subcutaneous adipose wasting during chronic Trypanosoma brucei infection. Nat. Commun. 2023, 14, 5279. [Google Scholar] [CrossRef] [PubMed]
- Casas-Sanchez, A.; Acosta-Serrano, A. Skin deep. Elife 2016, 5, e21506. [Google Scholar] [CrossRef]
- Reuter, C.; Hauf, L.; Imdahl, F.; Sen, R.; Vafadarnejad, E.; Fey, P.; Finger, T.; Jones, N.G.; Walles, H.; Barquist, L.; et al. Vector-borne Trypanosoma brucei parasites develop in artificial human skin and persist as skin tissue forms. Nat. Commun. 2023, 14, 7660. [Google Scholar] [CrossRef]
- Quintana, J.F.; Chandrasegaran, P.; Sinton, M.C.; Briggs, E.M.; Otto, T.D.; Heslop, R.; Bentley-Abbot, C.; Loney, C.; de Lecea, L.; Mabbott, N.A.; et al. Single cell and spatial transcriptomic analyses reveal microglia-plasma cell crosstalk in the brain during Trypanosoma brucei infection. Nat. Commun. 2022, 13, 5752. [Google Scholar] [CrossRef] [PubMed]
- Tesoriero, C.; Xu, Y.Z.; Mumba Ngoyi, D.; Bentivoglio, M. Neural Damage in Experimental Trypanosoma brucei gambiense Infection: The Suprachiasmatic Nucleus. Front. Neuroanat. 2018, 12, 6. [Google Scholar] [CrossRef]
- Laperchia, C.; Palomba, M.; Seke Etet, P.F.; Rodgers, J.; Bradley, B.; Montague, P.; Grassi-Zucconi, G.; Kennedy, P.G.; Bentivoglio, M. Trypanosoma brucei Invasion and T-Cell Infiltration of the Brain Parenchyma in Experimental Sleeping Sickness: Timing and Correlation with Functional Changes. PLoS Negl. Trop. Dis. 2016, 10, e0005242. [Google Scholar] [CrossRef]
- Mogk, S.; Meiwes, A.; Bosselmann, C.M.; Wolburg, H.; Duszenko, M. The lane to the brain: How African trypanosomes invade the CNS. Trends Parasitol. 2014, 30, 470–477. [Google Scholar] [CrossRef]
- Frevert, U.; Movila, A.; Nikolskaia, O.V.; Raper, J.; Mackey, Z.B.; Abdulla, M.; McKerrow, J.; Grab, D.J. Early invasion of brain parenchyma by African trypanosomes. PLoS ONE 2012, 7, e43913. [Google Scholar] [CrossRef]
- Alfituri, O.A.; Quintana, J.F.; MacLeod, A.; Garside, P.; Benson, R.A.; Brewer, J.M.; Mabbott, N.A.; Morrison, L.J.; Capewell, P. To the Skin and Beyond: The Immune Response to African Trypanosomes as They Enter and Exit the Vertebrate Host. Front. Immunol. 2020, 11, 1250. [Google Scholar] [CrossRef]
- Mabille, D.; Caljon, G. Inflammation following trypanosome infection and persistence in the skin. Curr. Opin. Immunol. 2020, 66, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Amin, D.N.; Vodnala, S.K.; Masocha, W.; Sun, B.; Kristensson, K.; Rottenberg, M.E. Distinct Toll-like receptor signals regulate cerebral parasite load and interferon alpha/beta and tumor necrosis factor alpha-dependent T-cell infiltration in the brains of Trypanosoma brucei-infected mice. J. Infect. Dis. 2012, 205, 320–332. [Google Scholar] [CrossRef] [PubMed]
- Venturelli, A.; Tagliazucchi, L.; Lima, C.; Venuti, F.; Malpezzi, G.; Magoulas, G.E.; Santarem, N.; Calogeropoulou, T.; Cordeiro-da-Silva, A.; Costi, M.P. Current Treatments to Control African Trypanosomiasis and One Health Perspective. Microorganisms 2022, 10, 1298. [Google Scholar] [CrossRef] [PubMed]
- Priotto, G.; Pinoges, L.; Fursa, I.B.; Burke, B.; Nicolay, N.; Grillet, G.; Hewison, C.; Balasegaram, M. Safety and effectiveness of first line eflornithine for Trypanosoma brucei gambiense sleeping sickness in Sudan: Cohort study. BMJ 2008, 336, 705–708. [Google Scholar] [CrossRef]
- Yun, O.; Priotto, G.; Tong, J.; Flevaud, L.; Chappuis, F. NECT is next: Implementing the new drug combination therapy for Trypanosoma brucei gambiense sleeping sickness. PLoS Negl. Trop. Dis. 2010, 4, e720. [Google Scholar] [CrossRef]
- Jamabo, M.; Mahlalela, M.; Edkins, A.L.; Boshoff, A. Tackling Sleeping Sickness: Current and Promising Therapeutics and Treatment Strategies. Int. J. Mol. Sci. 2023, 24, 12529. [Google Scholar] [CrossRef] [PubMed]
- Papagni, R.; Novara, R.; Minardi, M.L.; Frallonardo, L.; Panico, G.G.; Pallara, E.; Cotugno, S.; Ascoli Bartoli, T.; Guido, G.; De Vita, E.; et al. Human African Trypanosomiasis (sleeping sickness): Current knowledge and future challenges. Front. Trop. Dis. 2023, 4, 1087003. [Google Scholar] [CrossRef]
- Snijders, R.; Fukinsia, A.; Claeys, Y.; Mpanya, A.; Hasker, E.; Meheus, F.; Miaka, E.; Boelaert, M. Cost of a new method of active screening for human African trypanosomiasis in the Democratic Republic of the Congo. PLoS Negl. Trop. Dis. 2020, 14, e0008832. [Google Scholar] [CrossRef] [PubMed]
- Antillon, M.; Huang, C.I.; Crump, R.E.; Brown, P.E.; Snijders, R.; Miaka, E.M.; Keeling, M.J.; Rock, K.S.; Tediosi, F. Cost-effectiveness of sleeping sickness elimination campaigns in five settings of the Democratic Republic of Congo. Nat. Commun. 2022, 13, 1051. [Google Scholar] [CrossRef]
- De Koning, H.P. The Drugs of Sleeping Sickness: Their Mechanisms of Action and Resistance, and a Brief History. Trop. Med. Infect. Dis. 2020, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- Hasker, E.; Hope, A.; Bottieau, E. Gambiense human African trypanosomiasis: The bumpy road to elimination. Curr. Opin. Infect. Dis. 2022, 35, 384–389. [Google Scholar] [CrossRef] [PubMed]
- Dickie, E.A.; Giordani, F.; Gould, M.K.; Maser, P.; Burri, C.; Mottram, J.C.; Rao, S.P.S.; Barrett, M.P. New Drugs for Human African Trypanosomiasis: A Twenty First Century Success Story. Trop. Med. Infect. Dis. 2020, 5, 29. [Google Scholar] [CrossRef]
- Lindner, A.K.; Lejon, V.; Chappuis, F.; Seixas, J.; Kazumba, L.; Barrett, M.P.; Mwamba, E.; Erphas, O.; Akl, E.A.; Villanueva, G.; et al. New WHO guidelines for treatment of gambiense human African trypanosomiasis including fexinidazole: Substantial changes for clinical practice. Lancet Infect. Dis. 2020, 20, e38–e46. [Google Scholar] [CrossRef]
- Pfarr, K.M.; Krome, A.K.; Al-Obaidi, I.; Batchelor, H.; Vaillant, M.; Hoerauf, A.; Opoku, N.O.; Kuesel, A.C. The pipeline for drugs for control and elimination of neglected tropical diseases: 1. Anti-infective drugs for regulatory registration. Parasit. Vectors 2023, 16, 82. [Google Scholar] [CrossRef]
- Muraca, G.; Berti, I.R.; Sbaraglini, M.L.; Favaro, W.J.; Duran, N.; Castro, G.R.; Talevi, A. Trypanosomatid-Caused Conditions: State of the Art of Therapeutics and Potential Applications of Lipid-Based Nanocarriers. Front. Chem. 2020, 8, 601151. [Google Scholar] [CrossRef]
- De Rycker, M.; Wyllie, S.; Horn, D.; Read, K.D.; Gilbert, I.H. Anti-trypanosomatid drug discovery: Progress and challenges. Nat. Rev. Microbiol. 2023, 21, 35–50. [Google Scholar] [CrossRef]
- Steketee, P.C.; Giordani, F.; Vincent, I.M.; Crouch, K.; Achcar, F.; Dickens, N.J.; Morrison, L.J.; MacLeod, A.; Barrett, M.P. Transcriptional differentiation of Trypanosoma brucei during in vitro acquisition of resistance to acoziborole. PLoS Negl. Trop. Dis. 2021, 15, e0009939. [Google Scholar] [CrossRef]
- Bouazizi-Ben Messaoud, H.; Guichard, M.; Lawton, P.; Delton, I.; Azzouz-Maache, S. Changes in Lipid and Fatty Acid Composition During Intramacrophagic Transformation of Leishmania donovani Complex Promastigotes into Amastigotes. Lipids 2017, 52, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, D.; Banerjee, S.; Sen, A.; Banerjee, K.K.; Das, P.; Roy, S. Leishmania donovani affects antigen presentation of macrophage by disrupting lipid rafts. J. Immunol. 2005, 175, 3214–3224. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, J.; Bose, M.; Roy, S.; Bhattacharyya, S.N. Leishmania donovani targets Dicer1 to downregulate miR-122, lower serum cholesterol, and facilitate murine liver infection. Cell Host Microbe 2013, 13, 277–288. [Google Scholar] [CrossRef]
- Lal, C.S.; Kumar, A.; Kumar, S.; Pandey, K.; Kumar, N.; Bimal, S.; Sinha, P.K.; Das, P. Hypocholesterolemia and increased triglyceride in pediatric visceral leishmaniasis. Clin. Chim. Acta 2007, 382, 151–153. [Google Scholar] [CrossRef]
- Majumder, S.; Dey, R.; Bhattacharjee, S.; Rub, A.; Gupta, G.; Bhattacharyya Majumdar, S.; Saha, B.; Majumdar, S. Leishmania-induced biphasic ceramide generation in macrophages is crucial for uptake and survival of the parasite. J. Infect. Dis. 2012, 205, 1607–1616. [Google Scholar] [CrossRef]
- Pucadyil, T.J.; Chattopadhyay, A. Cholesterol: A potential therapeutic target in Leishmania infection? Trends Parasitol. 2007, 23, 49–53. [Google Scholar] [CrossRef]
- Roy, K.; Mandloi, S.; Chakrabarti, S.; Roy, S. Cholesterol Corrects Altered Conformation of MHC-II Protein in Leishmania donovani Infected Macrophages: Implication in Therapy. PLoS Negl. Trop. Dis. 2016, 10, e0004710. [Google Scholar] [CrossRef] [PubMed]
- Semini, G.; Paape, D.; Paterou, A.; Schroeder, J.; Barrios-Llerena, M.; Aebischer, T. Changes to cholesterol trafficking in macrophages by Leishmania parasites infection. Microbiologyopen 2017, 6, e00469. [Google Scholar] [CrossRef]
- Winberg, M.E.; Holm, A.; Sarndahl, E.; Vinet, A.F.; Descoteaux, A.; Magnusson, K.E.; Rasmusson, B.; Lerm, M. Leishmania donovani lipophosphoglycan inhibits phagosomal maturation via action on membrane rafts. Microbes Infect. 2009, 11, 215–222. [Google Scholar] [CrossRef]
- Garzon, E.; Holzmuller, P.; Bras-Goncalves, R.; Vincendeau, P.; Cuny, G.; Lemesre, J.L.; Geiger, A. The Trypanosoma brucei gambiense secretome impairs lipopolysaccharide-induced maturation, cytokine production, and allostimulatory capacity of dendritic cells. Infect. Immun. 2013, 81, 3300–3308. [Google Scholar] [CrossRef]
- Stijlemans, B.; Leng, L.; Brys, L.; Sparkes, A.; Vansintjan, L.; Caljon, G.; Raes, G.; Van Den Abbeele, J.; Van Ginderachter, J.A.; Beschin, A.; et al. MIF contributes to Trypanosoma brucei associated immunopathogenicity development. PLoS Pathog. 2014, 10, e1004414. [Google Scholar] [CrossRef] [PubMed]
- Olsson, S.; Sundler, R. The role of lipid rafts in LPS-induced signaling in a macrophage cell line. Mol. Immunol. 2006, 43, 607–612. [Google Scholar] [CrossRef]
- Park, Y.; Pham, T.X.; Lee, J. Lipopolysaccharide represses the expression of ATP-binding cassette transporter G1 and scavenger receptor class B, type I in murine macrophages. Inflamm. Res. 2012, 61, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Facchin, B.M.; Dos Reis, G.O.; Vieira, G.N.; Mohr, E.T.B.; da Rosa, J.S.; Kretzer, I.F.; Demarchi, I.G.; Dalmarco, E.M. Inflammatory biomarkers on an LPS-induced RAW 264.7 cell model: A systematic review and meta-analysis. Inflamm. Res. 2022, 71, 741–758. [Google Scholar] [CrossRef] [PubMed]
- Funk, J.L.; Feingold, K.R.; Moser, A.H.; Grunfeld, C. Lipopolysaccharide stimulation of RAW 264.7 macrophages induces lipid accumulation and foam cell formation. Atherosclerosis 1993, 98, 67–82. [Google Scholar] [CrossRef] [PubMed]
- Kaur, G.; Dufour, J.M. Cell lines: Valuable tools or useless artifacts. Spermatogenesis 2012, 2, 1–5. [Google Scholar] [CrossRef]
- Zhang, X.; Goncalves, R.; Mosser, D.M. The isolation and characterization of murine macrophages. Curr. Protoc. Immunol. 2008, 14, 14.1.1–14.1.14. [Google Scholar] [CrossRef]
- Hoshino, K.; Takeuchi, O.; Kawai, T.; Sanjo, H.; Ogawa, T.; Takeda, Y.; Takeda, K.; Akira, S. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: Evidence for TLR4 as the Lps gene product. J. Immunol. 1999, 162, 3749–3752. [Google Scholar] [CrossRef]
- Takeuchi, O.; Hoshino, K.; Kawai, T.; Sanjo, H.; Takada, H.; Ogawa, T.; Takeda, K.; Akira, S. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity 1999, 11, 443–451. [Google Scholar] [CrossRef]
- Wang, X.; Collins, H.L.; Ranalletta, M.; Fuki, I.V.; Billheimer, J.T.; Rothblat, G.H.; Tall, A.R.; Rader, D.J. Macrophage ABCA1 and ABCG1, but not SR-BI, promote macrophage reverse cholesterol transport in vivo. J. Clin. Investig. 2007, 117, 2216–2224. [Google Scholar] [CrossRef]
- Marquart, T.J.; Allen, R.M.; Ory, D.S.; Baldan, A. miR-33 links SREBP-2 induction to repression of sterol transporters. Proc. Natl. Acad. Sci. USA 2010, 107, 12228–12232. [Google Scholar] [CrossRef] [PubMed]
- Wacker, B.K.; Dronadula, N.; Bi, L.; Stamatikos, A.; Dichek, D.A. Apo A-I (Apolipoprotein A-I) Vascular Gene Therapy Provides Durable Protection Against Atherosclerosis in Hyperlipidemic Rabbits. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 206–217. [Google Scholar] [CrossRef] [PubMed]
- Brewer, H.B., Jr.; Schaefer, E.J.; Foldyna, B.; Ghoshhajra, B.B. High-density lipoprotein infusion therapy: A review. J. Clin. Lipidol. 2024, 18, e374–e383. [Google Scholar] [CrossRef]
- Gibson, C.M.; Duffy, D.; Korjian, S.; Bahit, M.C.; Chi, G.; Alexander, J.H.; Lincoff, A.M.; Heise, M.; Tricoci, P.; Deckelbaum, L.I.; et al. Apolipoprotein A1 Infusions and Cardiovascular Outcomes after Acute Myocardial Infarction. N. Engl. J. Med. 2024, 390, 1560–1571. [Google Scholar] [CrossRef] [PubMed]
- Rader, D.J. Apolipoprotein A-I Infusion Therapies for Coronary Disease: Two Outs in the Ninth Inning and Swinging for the Fences. JAMA Cardiol. 2018, 3, 799–801. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernando, L.; Echesabal-Chen, J.; Miller, M.; Powell, R.R.; Bruce, T.; Paul, A.; Poudyal, N.; Saliutama, J.; Parman, K.; Paul, K.S.; et al. Cholesterol Efflux Decreases TLR4-Target Gene Expression in Cultured Macrophages Exposed to T. brucei Ghosts. Microorganisms 2024, 12, 1730. https://doi.org/10.3390/microorganisms12081730
Fernando L, Echesabal-Chen J, Miller M, Powell RR, Bruce T, Paul A, Poudyal N, Saliutama J, Parman K, Paul KS, et al. Cholesterol Efflux Decreases TLR4-Target Gene Expression in Cultured Macrophages Exposed to T. brucei Ghosts. Microorganisms. 2024; 12(8):1730. https://doi.org/10.3390/microorganisms12081730
Chicago/Turabian StyleFernando, Lawrence, Jing Echesabal-Chen, Murphy Miller, Rhonda Reigers Powell, Terri Bruce, Apurba Paul, Nava Poudyal, Joshua Saliutama, Kristina Parman, Kimberly S. Paul, and et al. 2024. "Cholesterol Efflux Decreases TLR4-Target Gene Expression in Cultured Macrophages Exposed to T. brucei Ghosts" Microorganisms 12, no. 8: 1730. https://doi.org/10.3390/microorganisms12081730