
Analysis of Midgut Bacterial Communities in Larvae and Adult Mosquitoes of Aedes aegypti Invaded by Three Different Microorganisms

 ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Preparation and Mosquito Rearing
2.2. Preparation of Microorganisms
2.3. PCR Amplification and Sequencing
2.4. 16S-rDNA Sequencing Data Analysis
3. Results
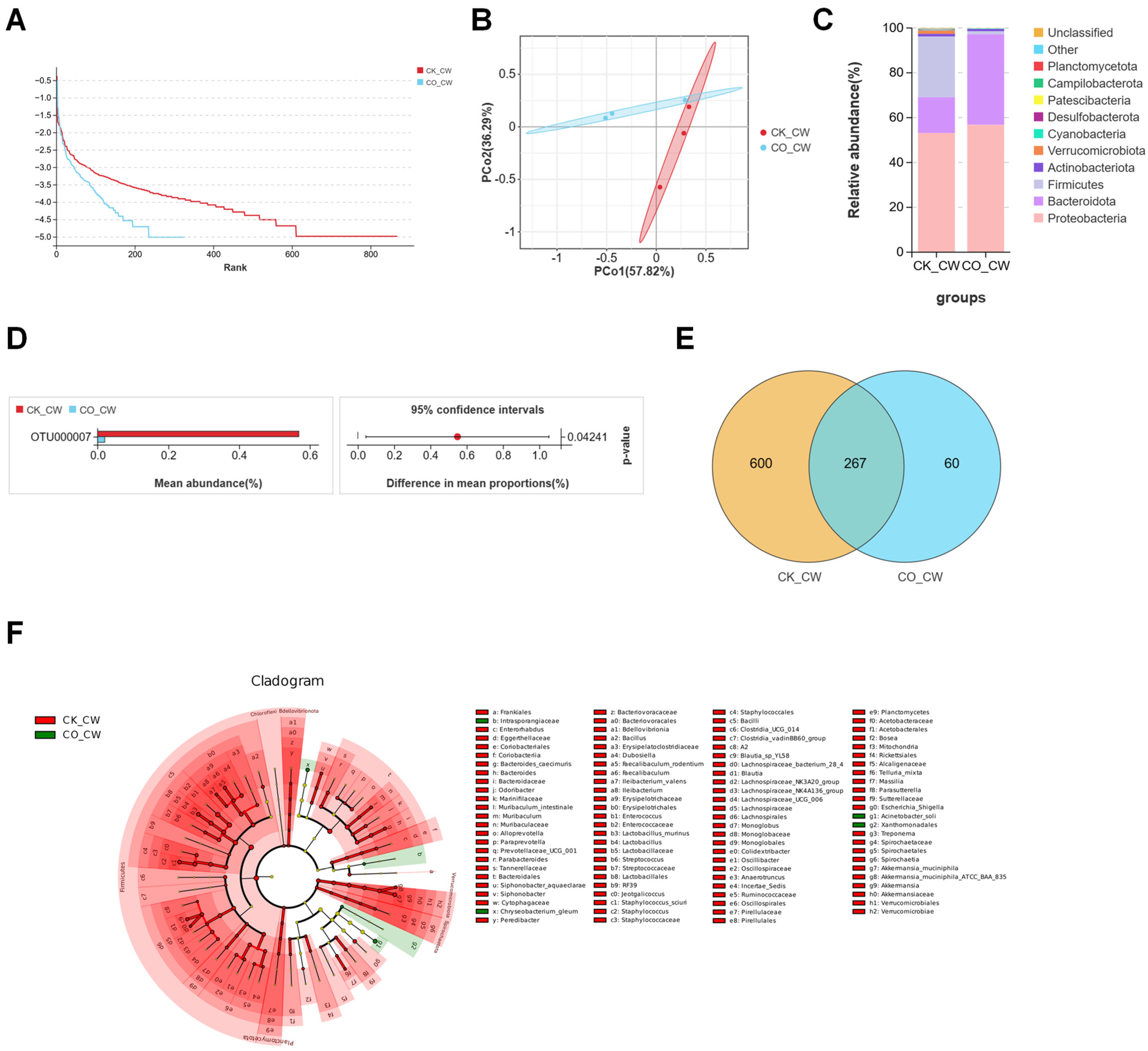
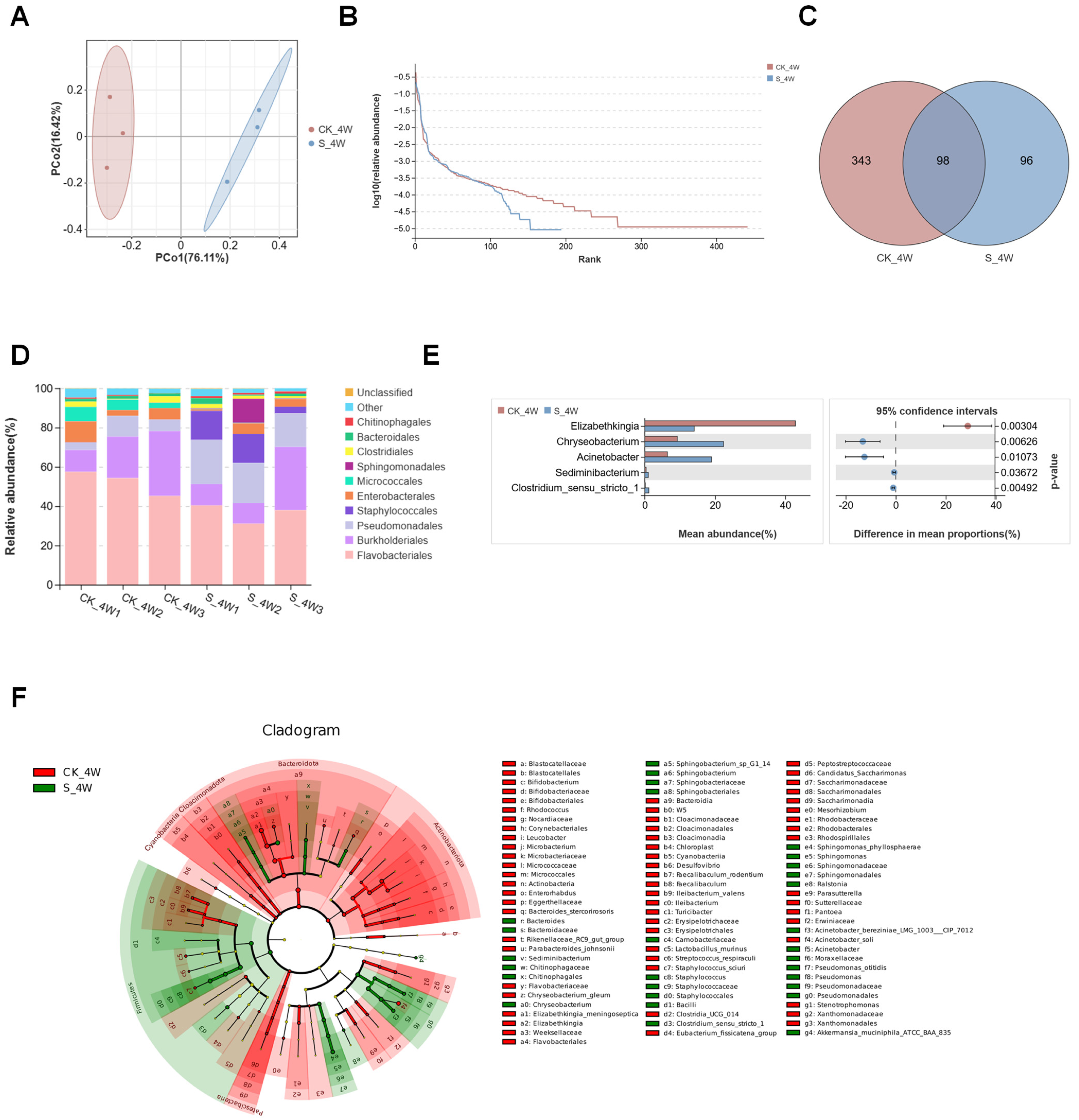
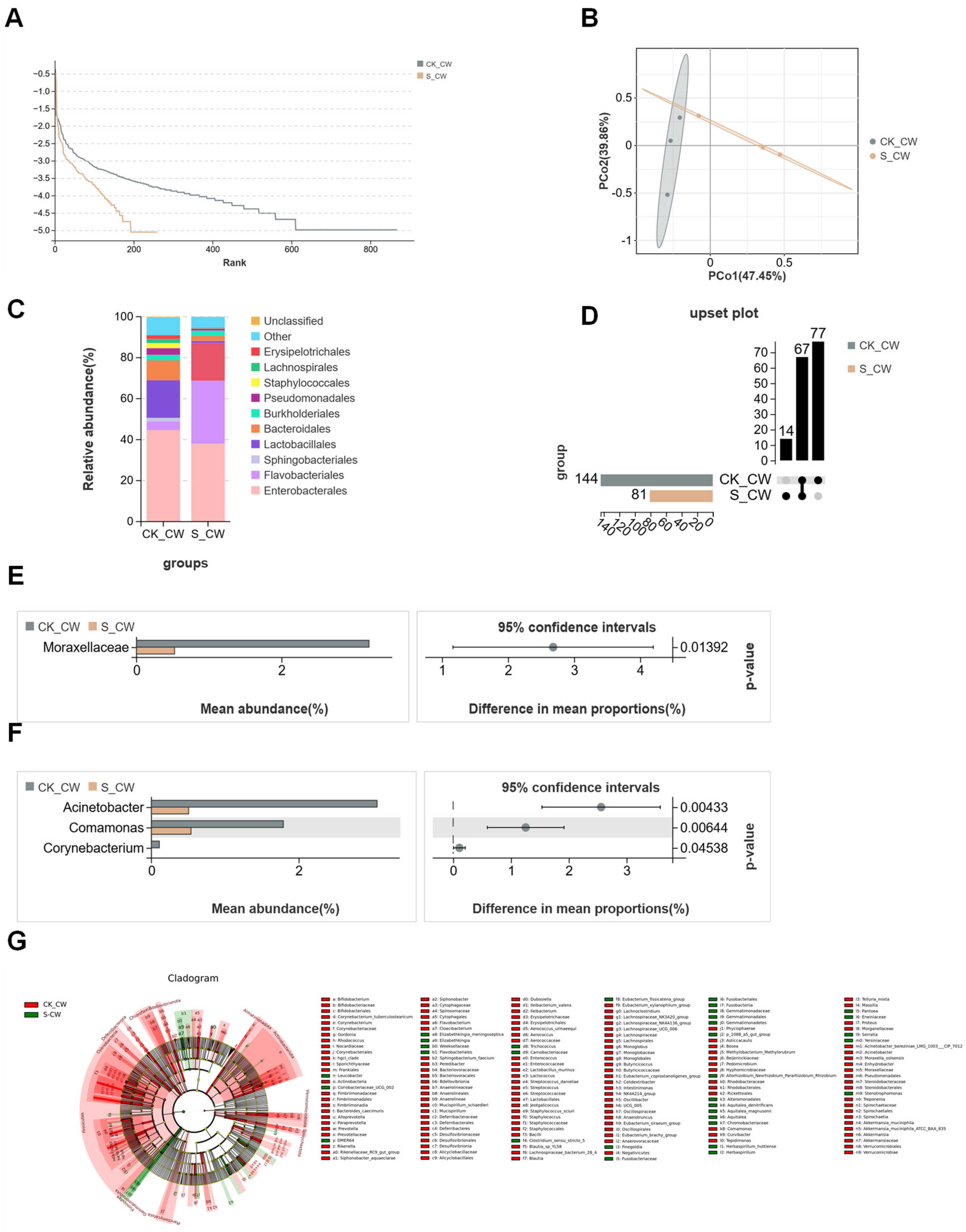
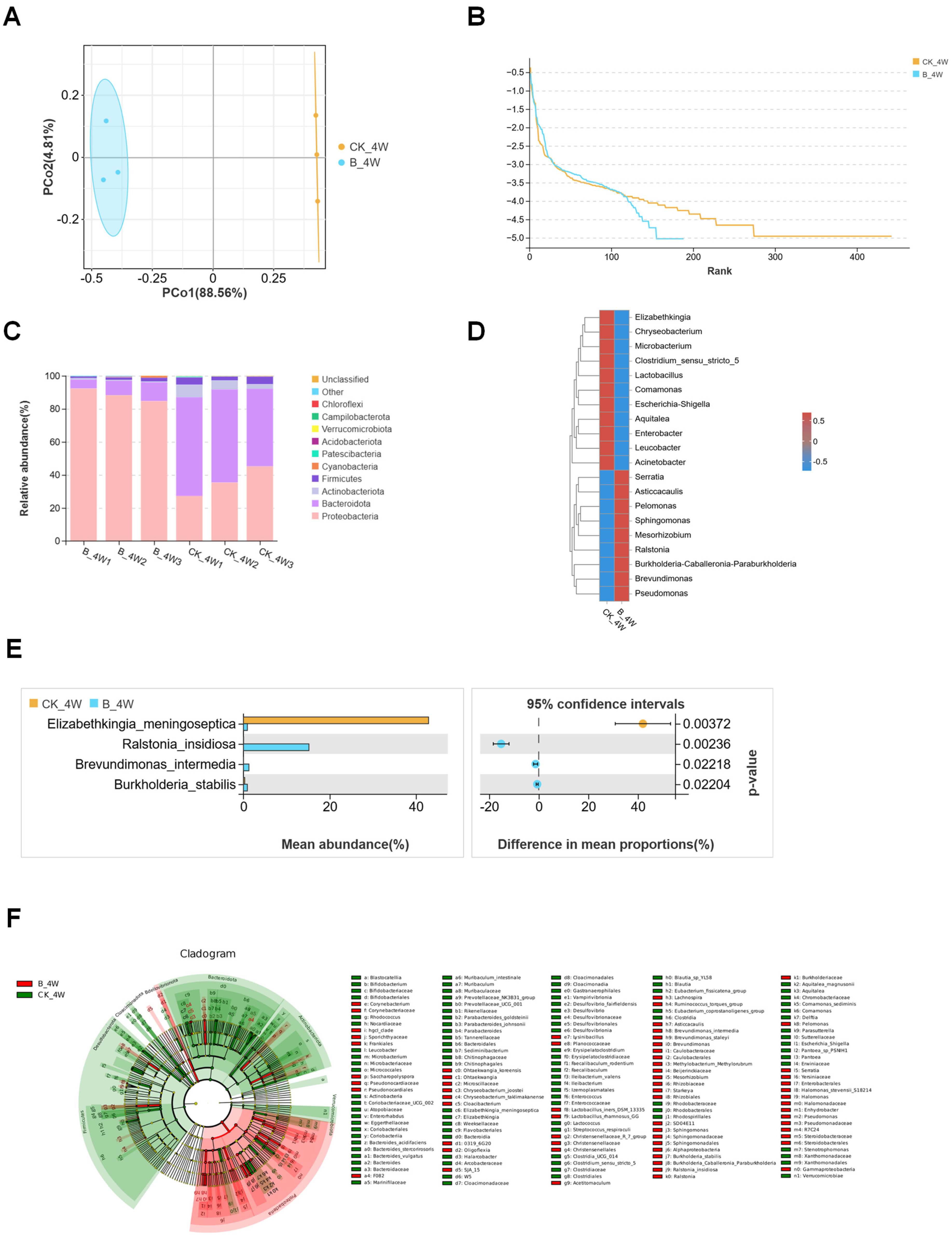
3.1. Structures of Midgut Bacterial Community in Larvae and Adults Colonized with E. coli
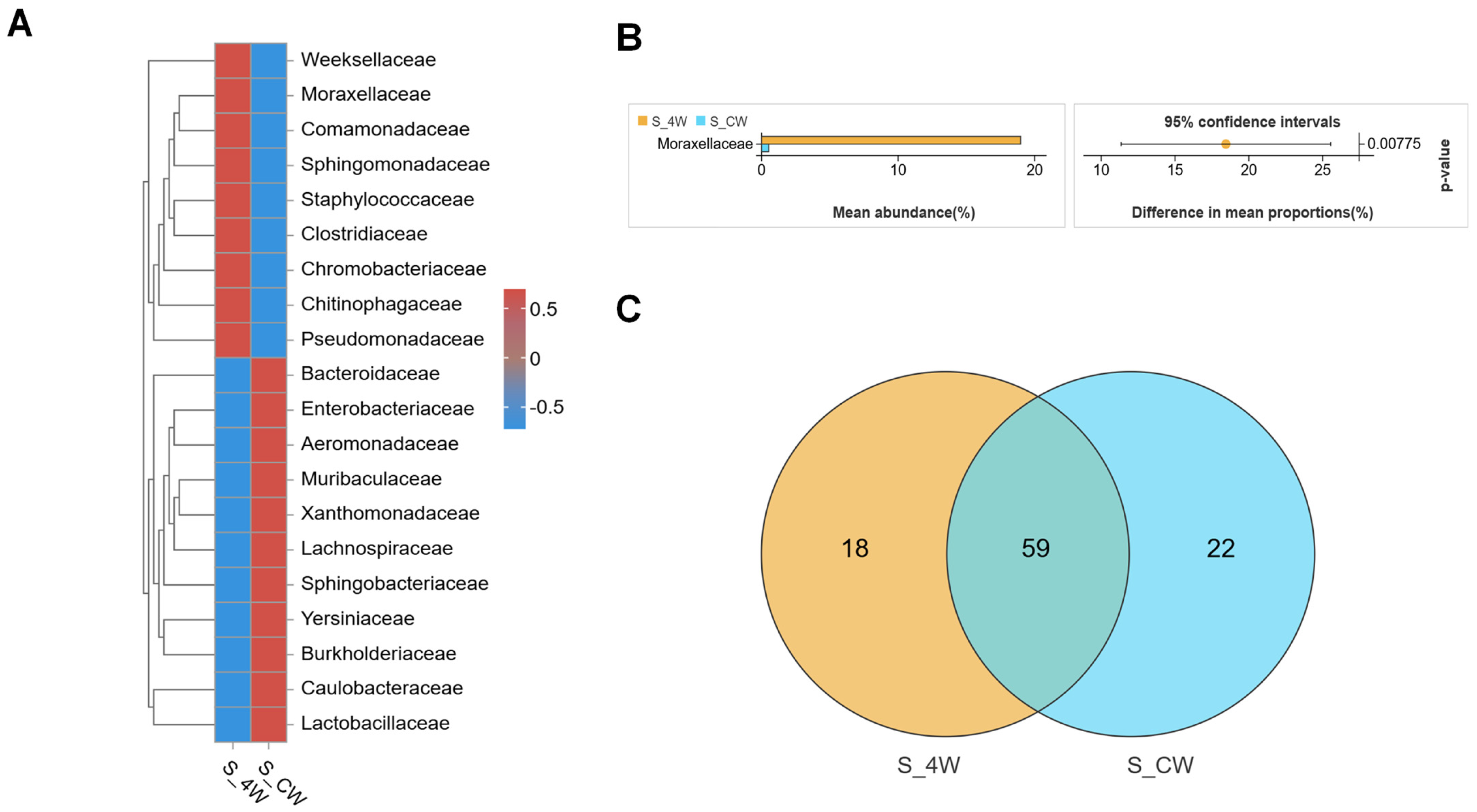
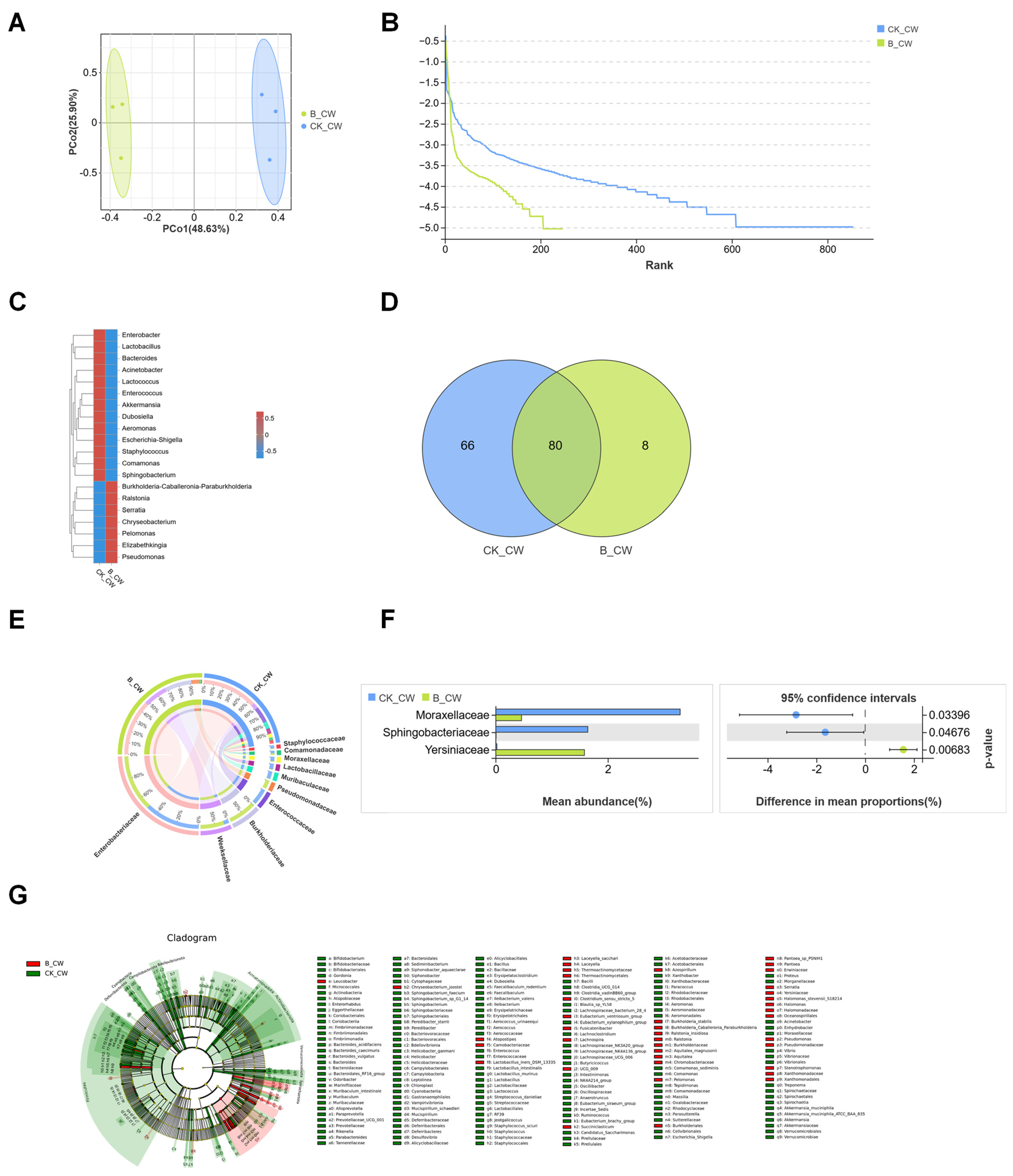
3.2. Structures of Midgut Bacterial Community in Larvae and Adults Colonized with S. aureus
3.3. Structures of Midgut Bacterial Community in Larvae and Adults Infected with B. bassiana
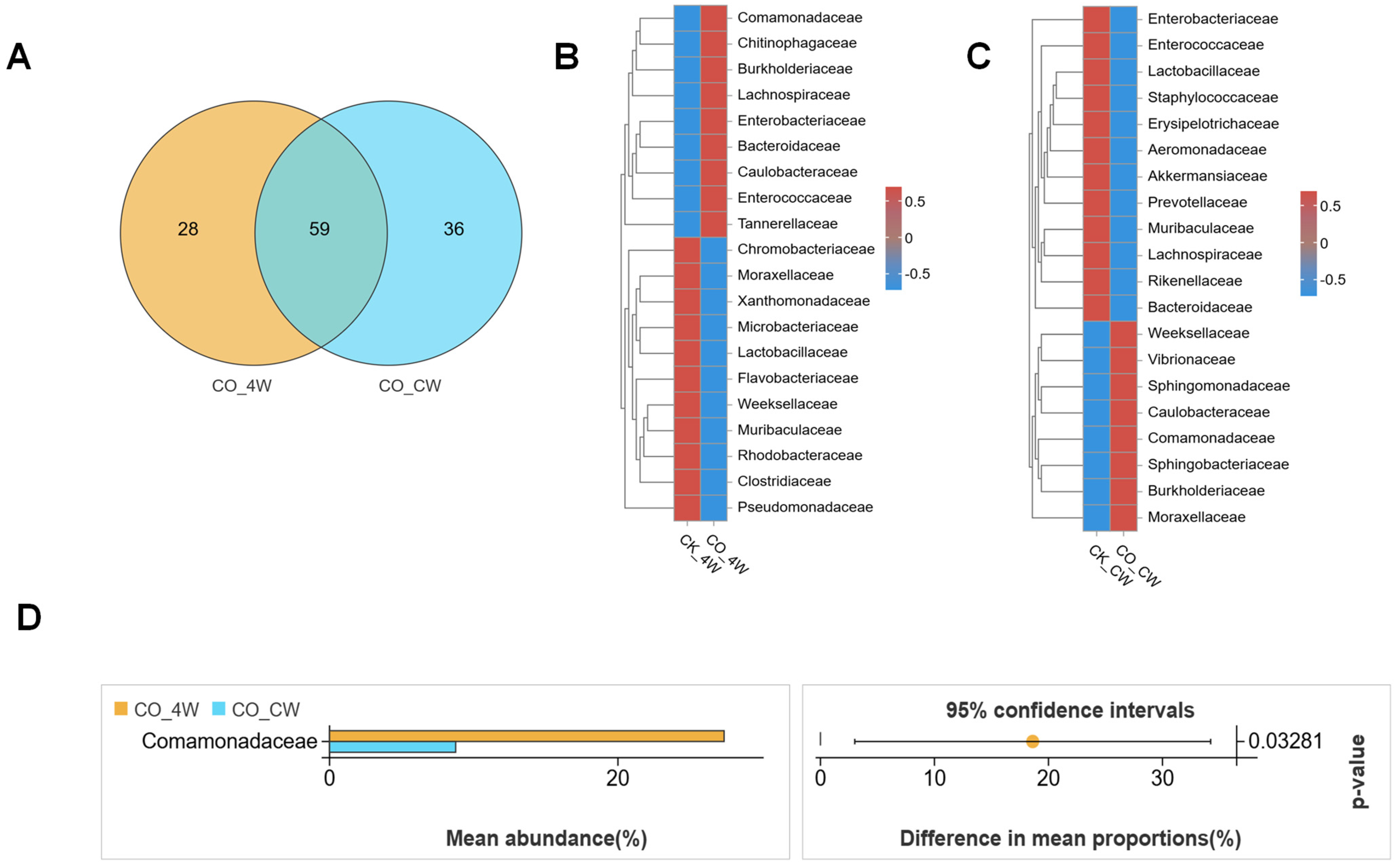
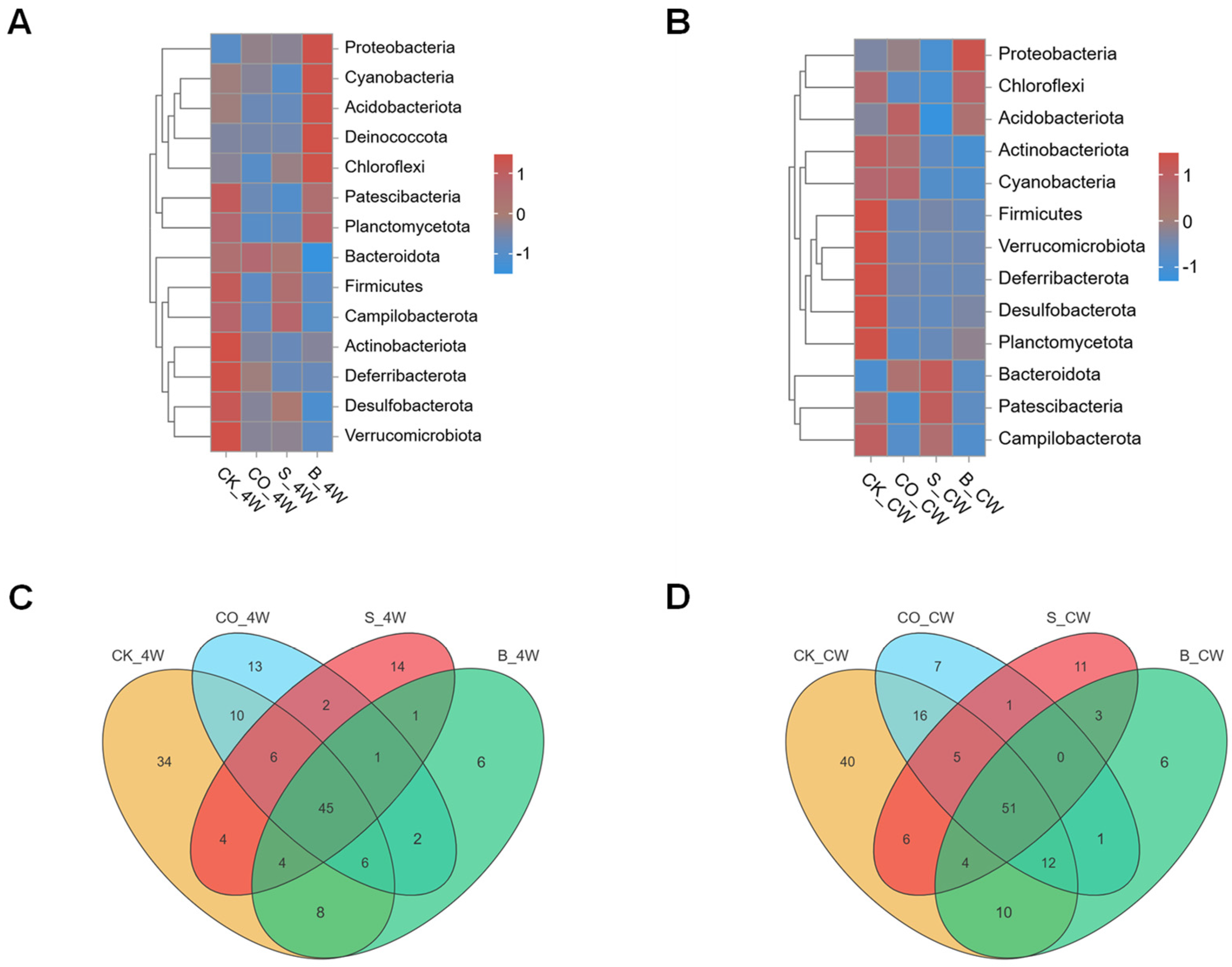
3.4. Comparative Analysis of the Composition of Fourth-Instar Larvae and Adult Mosquito of Midgut Microbiota in Ae. aegypti Mosquitoes
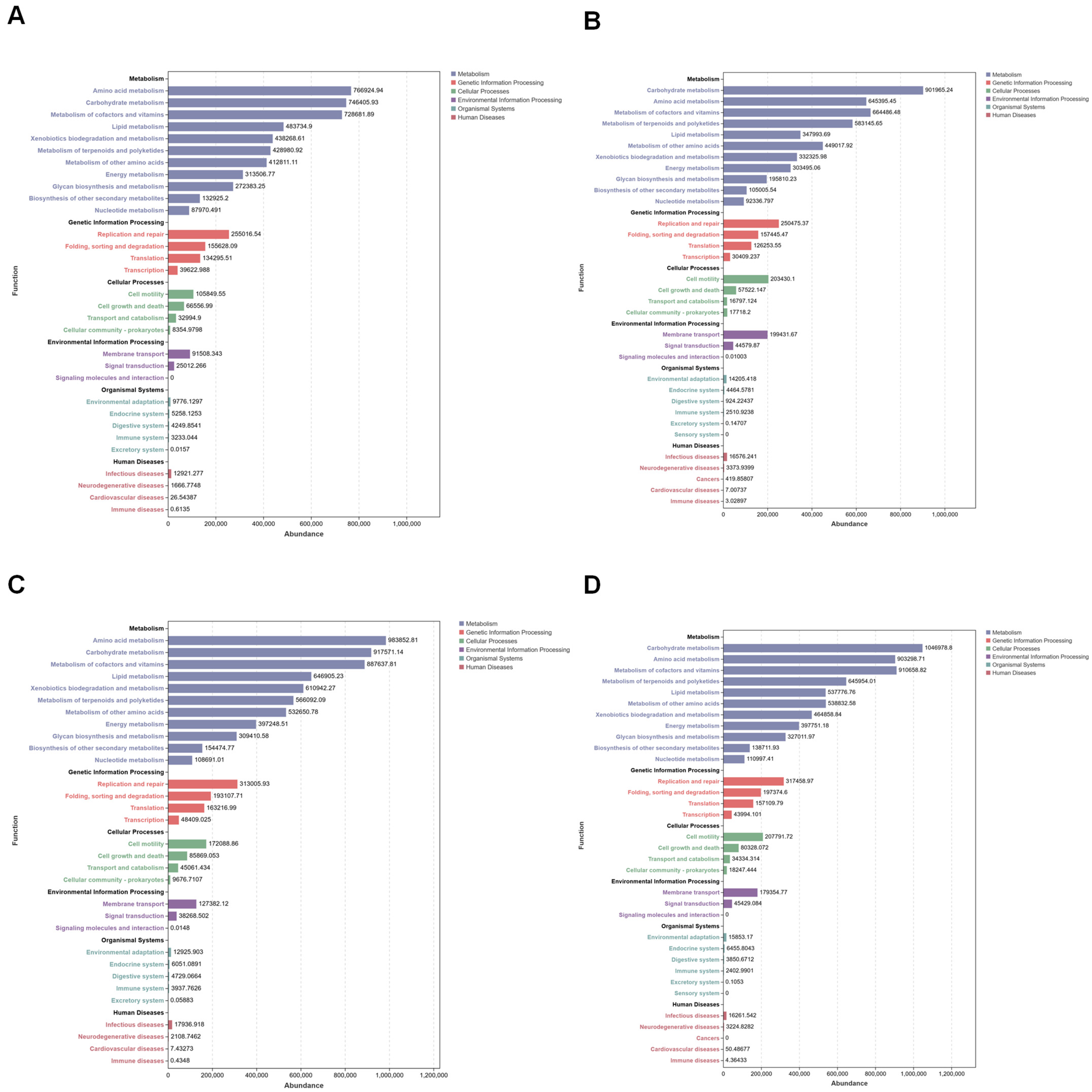
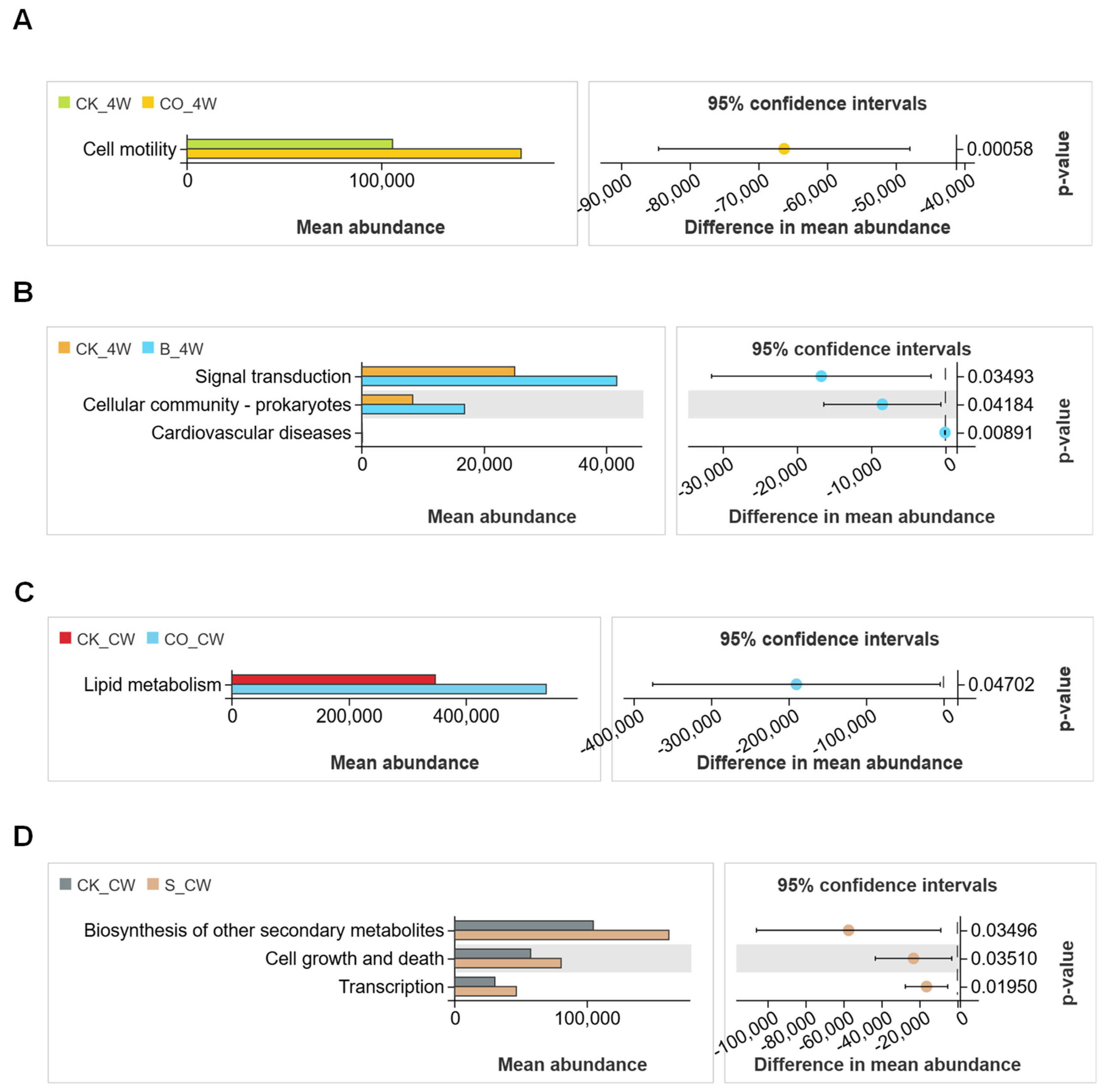
3.5. Functional Analysis of Mosquito Midgut Microbiota During Microorganism Colonization
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Coon, K.L.; Brown, M.R.; Strand, M.R. Gut bacteria differentially affect egg production in the anautogenous mosquito Aedes aegypti and facultatively autogenous mosquito Aedes atropalpus (Diptera: Culicidae). Parasite Vectors 2016, 9, 375. [Google Scholar] [CrossRef]
- Glaser, R.L.; Meola, M.A. The native Wolbachia endosymbionts of Drosophila melanogaster and Culex quinquefasciatus increase host resistance to West Nile virus infection. PLoS ONE 2010, 5, e11977. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Yu, X.; Cheng, G. Impact of the microbiome on mosquito-borne diseases. Protein Cell. 2023, 14, 743–761. [Google Scholar] [CrossRef] [PubMed]
- Boissière, A.; Tchioffo, M.T.; Bachar, D.; Abate, L.; Marie, A.; Nsango, S.E.; Shahbazkia, H.R.; Awono-Ambene, P.H.; Levashina, E.A.; Christen, R.; et al. Midgut microbiota of the malaria mosquito vector Anopheles gambiae and interactions with Plasmodium falciparum infection. PLoS Pathog. 2012, 8, e1002742. [Google Scholar] [CrossRef]
- Osei-Poku, J.; Mbogo, C.M.; Palmer, W.J.; Jiggins, F.M. Deep sequencing reveals extensive variation in the gut microbiota of wild mosquitoes from Kenya. Mol. Ecol. 2012, 21, 5138–5150. [Google Scholar] [CrossRef]
- Thongsripong, P.; Chandler, J.A.; Green, A.B.; Kittayapong, P.; Wilcox, B.A.; Kapan, D.D.; Bennett, S.N. Mosquito vector-associated microbiota: Metabarcoding bacteria and eukaryotic symbionts across habitat types in Thailand endemic for dengue and other arthropod-borne diseases. Ecol. Evol. 2017, 8, 1352–1368. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Sun, P.; Nie, K.; Zhu, Y.; Shi, M.; Xiao, C.; Liu, H.; Liu, Q.; Zhao, T.; Chen, X.; et al. A gut commensal bacterium promotes mosquito permissiveness to arboviruses. Cell Host Microbe 2019, 25, 101–112.e5. [Google Scholar] [CrossRef] [PubMed]
- Möhlmann, T.W.R.; Vogels, C.B.F.; Göertz, G.P.; Pijlman, G.P.; Ter Braak, C.J.F.; Te Beest, D.E.; Hendriks, M.; Nijhuis, E.H.; Warris, S.; Drolet, B.S.; et al. Impact of gut bacteria on the infection and transmission of pathogenic arboviruses by biting midges and mosquitoe. Microb. Ecol. 2020, 80, 703–717. [Google Scholar] [CrossRef] [PubMed]
- Frankel-Bricker, J. Shifts in the microbiota associated with male mosquitoes (Aedes aegypti) exposed to an obligate gut fungal symbiont (Zancudomyces culisetae). Sci. Rep. 2020, 10, 12886. [Google Scholar] [CrossRef] [PubMed]
- Tenaillon, O.; Skurnik, D.; Picard, B.; Denamur, E. The population genetics of commensal Escherichia coli. Nat. Rev. Microbiol. 2010, 8, 207–217. [Google Scholar] [CrossRef]
- Russo, T.A.; Johnson, J.R. Medical and economic impact of extraintestinal infections due to Escherichia coli: Focus on an increasingly important endemic problem. Microbes Infect. 2003, 5, 449–456. [Google Scholar] [CrossRef]
- Tchioffo, M.T.; Abate, L.; Boissière, A.; Nsango, S.E.; Gimonneau, G.; Berry, A.; Oswald, E.; Dubois, D.; Morlais, I. An epidemiologically successful Escherichia coli sequence type modulates Plasmodium falciparum infection in the mosquito midgut. Infect. Genet. Evol. 2016, 43, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Tchioffo, M.T.; Boissière, A.; Churcher, T.S.; Abate, L.; Gimonneau, G.; Nsango, S.E.; Awono-Ambéné, P.H.; Christen, R.; Berry, A.; Morlais, I. Modulation of malaria infection in Anopheles gambiae mosquitoes exposed to natural midgut bacteria. PLoS ONE 2013, 8, e81663. [Google Scholar] [CrossRef]
- Apte-Deshpande, A.; Paingankar, M.; Gokhale, M.D.; Deobagkar, D.N. Serratia odorifera a midgut inhabitant of Aedes aegypti mosquito enhances its susceptibility to Dengue-2 virus. PLoS ONE 2012, 7, e40401. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Manfredini, F.; Dimopoulos, G. Implication of the mosquito midgut microbiota in the defense against malaria parasites. PLoS Pathog. 2009, 5, e1000423. [Google Scholar] [CrossRef]
- Guégan, M.; Zouache, K.; Démichel, C.; Minard, G.; Tran Van, V.; Potier, P.; Mavingui, P.; Valiente Moro, C. The mosquito holobiont: Fresh insight into mosquito-microbiota interactions. Microbiome 2018, 6, 49. [Google Scholar] [CrossRef] [PubMed]
- Stathopoulos, S.; Neafsey, D.E.; Lawniczak, M.K.; Muskavitch, M.A.; Christophides, G.K. Genetic dissection of Anopheles gambiae gut epithelial responses to Serratia marcescens. PLoS Pathog. 2014, 10, e1003897. [Google Scholar] [CrossRef]
- Gabrieli, P.; Caccia, S.; Varotto-Boccazzi, I.; Arnoldi, I.; Barbieri, G.; Comandatore, F.; Epis, S. Mosquito Trilogy: Microbiota, Immunity and Pathogens, and Their Implications for the Control of Disease Transmission. Front. Microbiol. 2021, 12, 630438. [Google Scholar] [CrossRef] [PubMed]
- Knols, B.G.; Bukhari, T.; Farenhorst, M. Entomopathogenic fungi as the next-generation control agents against malaria mosquitoes. Future Microbiol. 2010, 5, 339–341. [Google Scholar] [CrossRef] [PubMed]
- Blanford, S.; Chan, B.H.; Jenkins, N.; Sim, D.; Turner, R.J.; Read, A.F.; Thomas, M.B. Fungal pathogen reduces potential for malaria transmission. Science 2005, 308, 1638–1641. [Google Scholar] [CrossRef] [PubMed]
- Wei, G.; Lai, Y.; Wang, G.; Chen, H.; Li, F.; Wang, S. Insect pathogenic fungus interacts with the gut microbiota to accelerate mosquito mortality. Proc. Natl. Acad. Sci. USA 2017, 114, 5994–5999. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, J.L.; Dunlap, C.A.; Muturi, E.J.; Barletta, A.B.F.; Rooney, A.P. Entomopathogenic fungal infection leads to temporospatial modulation of the mosquito immune system. PLoS Negl. Trop. Dis. 2018, 12, e0006433. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Wu, X.; Xu, L.; Guo, S.; Chen, G.; Zhang, X. Repressed Beauveria bassiana infections in Delia antiqua due to associated microbiota. Pest Manag. Sci. 2019, 75, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Xu, Z.; Li, L.; Zhang, Y.; Diao, J.; Cao, J.; Xu, L.; Ma, L. Gut bacterial microbiota of Lymantria dispar asiatica and its involvement in Beauveria bassiana infection. J. Invertebr. Pathol. 2023, 197, 107897. [Google Scholar] [CrossRef] [PubMed]
- Patterson, E.I.; Villinger, J.; Muthoni, J.N.; Dobel-Ober, L.; Hughes, G.L. Exploiting insect-specific viruses as a novel strategy to control vector-borne disease. Curr. Opin. Insect Sci. 2020, 39, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Tawidian, P.; Rhodes, V.L.; Michel, K. Mosquito-fungus interactions and antifungal immunity. Insect Biochem. Mol. Biol. 2019, 111, 103182. [Google Scholar] [CrossRef] [PubMed]
- Suo, P.; Wang, K.; Yu, H.; Fu, X.; An, L.; Bhowmick, B.; Zhang, J.; Han, Q. Seasonal Variation of Midgut Bacterial Diversity in Culexquinquefasciatus Populations in Haikou City, Hainan Province, China. Biology 2022, 11, 1166. [Google Scholar] [CrossRef] [PubMed]
- Souza, R.S.; Virginio, F.; Riback, T.I.S.; Suesdek, L.; Barufi, J.B.; Genta, F.A. Microorganism-based larval diets affect mosquito development, size and nutritional reserves in the yellow fever mosquito Aedes aegypti (Diptera: Culicidae). Front. Physiol. 2019, 10, 152. [Google Scholar] [CrossRef]
- Singer, E.; Bushnell, B.; Coleman-Derr, D.; Bowman, B.; Bowers, R.M.; Levy, A.; Gies, E.A.; Cheng, J.F.; Copeland, A.; Klenk, H.P.; et al. High-resolution phylogenetic microbial community profiling. ISME J. 2016, 10, 2020–2032. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Subramanian, S.; Faith, J.J.; Gevers, D.; Gordon, J.I.; Knight, R.; Mills, D.A.; Caporaso, J.G. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 2013, 10, 57–59. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed]
- Pruesse, E.; Quast, C.; Knittel, K.; Fuchs, B.M.; Ludwig, W.; Peplies, J.; Glöckner, F.O. SILVA: A comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007, 35, 7188–7196. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Gaby, J.C.; Rishishwar, L.; Valderrama-Aguirre, L.C.; Green, S.J.; Valderrama-Aguirre, A.; Jordan, I.K.; Kostka, J.E. Diazotroph community characterization via a high-throughput nifH amplicon sequencing and analysis pipeline. Appl. Environ. Microbiol. 2018, 84, e01512–e01517. [Google Scholar] [CrossRef] [PubMed]
- Antonelli, P.; Grizard, S.; Tran, F.H.; Lejon, D.; Bellemain, A.; Van Mavingui, P.; Roiz, D.; Simard, F.; Martin, E.; Abrouk, D.; et al. Bioaccumulation of polycyclic aromatic hydrocarbons and microbiota dynamics across developmental stages of the Asian tiger mosquito, Aedes albopictus exposed to urban pollutants. Ecotoxicol. Environ. Saf. 2024, 286, 117214. [Google Scholar] [CrossRef] [PubMed]
- Duguma, D.; Hall, M.W.; Smartt, C.T.; Neufeld, J.D. Temporal variations of microbiota associated with the immature stages of two florida Culex mosquito vectors. Microb. Ecol. 2017, 74, 979–989. [Google Scholar] [CrossRef]
- Trzebny, A.; Slodkowicz-Kowalska, A.; Björkroth, J.; Dabert, M. Microsporidian infection in mosquitoes (Culicidae) is associated with gut microbiome composition and predicted gut microbiome functional content. Microb. Ecol. 2023, 85, 247–263. [Google Scholar] [CrossRef] [PubMed]
- Arellano, A.A.; Coon, K.L. Bacterial communities in carnivorous pitcher plants colonize and persist in inquiline mosquitoes. Anim. Microbiome 2022, 4, 13. [Google Scholar] [CrossRef]
- Caragata, E.P.; Otero, L.M.; Tikhe, C.V.; Barrera, R.; Dimopoulos, G. Microbial diversity of adult Aedes aegypti and water collected from different mosquito aquatic habitats in Puerto Rico. Microb. Ecol. 2022, 83, 182–201. [Google Scholar] [CrossRef] [PubMed]
- Aželytė, J.; Maitre, A.; Abuin-Denis, L.; Wu-Chuang, A.; Žiegytė, R.; Mateos-Hernandez, L.; Obregon, D.; Palinauskas, V.; Cabezas-Cruz, A. Nested patterns of commensals and endosymbionts in microbial communities of mosquito vectors. BMC Microbiol. 2024, 24, 434. [Google Scholar] [CrossRef] [PubMed]
- Mee, P.T.; Lynch, S.E.; Walker, P.J.; Melville, L.; Duchemin, J.B. Detection of Elizabethkingia spp. in Culicoides Biting Midges, Australia. Emerg. Infect. Dis. 2017, 23, 1409–1410. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gilbreath, T.M., 3rd; Kukutla, P.; Yan, G.; Xu, J. Dynamic gut microbiome across life history of the malaria mosquito Anopheles gambiae in Kenya. PLoS ONE 2011, 6, e24767. [Google Scholar]
- Ngwa, C.J.; Glöckner, V.; Abdelmohsen, U.R.; Scheuermayer, M.; Fischer, R.; Hentschel, U.; Pradel, G. 16S rRNA gene-based identification of Elizabethkingia meningoseptica (Flavobacteriales: Flavobacteriaceae) as a dominant midgut bacterium of the Asian malaria vector Anopheles stephensi (Dipteria: Culicidae) with antimicrobial activities. J. Med. Entomol. 2013, 50, 404–414. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.K.; Wu, A.K.; Teng, J.L.; Tse, H.; Curreem, S.O.; Tsui, S.K.; Huang, Y.; Chen, J.H.; Lee, R.A.; Yuen, K.Y.; et al. Evidence for Elizabethkingia anophelis transmission from mother to infant, Hong Kong. Emerg. Infect. Dis. 2015, 21, 232–241. [Google Scholar] [CrossRef]
- Kang, X.; Wang, Y.; Li, S.; Sun, X.; Lu, X.; Rajaofera, M.J.N.; Lu, Y.; Kang, L.; Zheng, A.; Zou, Z.; et al. Comparative analysis of the gut microbiota of adult mosquitoes from eight locations in Hainan, China. Front. Cell. Infect. Microbiol. 2020, 10, 596750. [Google Scholar] [CrossRef]
- Sonenshine, D.E.; Stewart, P.E. Microbiomes of blood-feeding arthropods: Genes coding for essential nutrients and relation to vector fitness and pathogenic infections. A review. Microorganisms 2021, 9, 2433. [Google Scholar] [CrossRef]
- Coutinho-Abreu, I.V.; Jamshidi, O.; Raban, R.; Atabakhsh, K.; Merriman, J.A.; Fischbach, M.A.; Akbari, O.S. Identification of human skin microbiome odorants that manipulate mosquito landing behavior. Sci. Rep. 2024, 14, 1631. [Google Scholar] [CrossRef]
- Kalappa, D.M.; Subramani, P.A.; Basavanna, S.K.; Ghosh, S.K.; Sundaramurthy, V.; Uragayala, S.; Tiwari, S.; Anvikar, A.R.; Valecha, N. Influence of midgut microbiota in Anopheles stephensi on Plasmodium berghei infections. Malar. J. 2018, 17, 385. [Google Scholar] [CrossRef]
- Moretó, M.; Pérez-Bosque, A. Dietary plasma proteins, the intestinal immune system, and the barrier functions of the intestinal mucosa. J. Anim. Sci. 2009, 87, E92–E100. [Google Scholar] [CrossRef]
- Zheng, Z.; Deng, W.; Bai, Y.; Miao, R.; Mei, S.; Zhang, Z.; Pan, Y.; Wang, Y.; Min, R.; Deng, F.; et al. The lysosomal rag-ragulator complex licenses RIPK1 and caspase-8-mediated pyroptosis by Yersinia. Science 2021, 372, eabg0269. [Google Scholar] [CrossRef] [PubMed]
- Chapelle, E.; Mendes, R.; Bakker, P.A.; Raaijmakers, J.M. Fungal invasion of the rhizosphere microbiome. ISME J. 2016, 10, 265–268. [Google Scholar] [CrossRef] [PubMed]
- Xie, R.; Dong, C.; Wang, S.; Danso, B.; Dar, M.A.; Pandit, R.S.; Pawar, K.D.; Geng, A.; Zhu, D.; Li, X.; et al. Host-specific diversity of culturable bacteria in the gut systems of fungus-growing Termites and their potential functions towards lignocellulose bioconversion. Insects 2023, 14, 403. [Google Scholar] [CrossRef]
- Zhou, F.; Gao, Y.; Liu, M.; Xu, L.; Wu, X.; Zhao, X.; Zhang, X. Bacterial inhibition on Beauveria bassiana contributes to microbiota stability in Delia antiqua. Front. Microbiol. 2021, 12, 710800. [Google Scholar] [CrossRef]
- Fritts, R.K.; McCully, A.L.; McKinlay, J.B. Extracellular Metabolism Sets the Table for Microbial Cross-Feeding. Microbiol. Mol. Biol. Rev. 2021, 85, e00135-20. [Google Scholar] [CrossRef]
- Gao, L.; Yang, W.; Wang, J. Implications of mosquito metabolism on vector competence. Insect Sci. 2024, 31, 674–682. [Google Scholar] [CrossRef] [PubMed]
- Pavlides, S.C.; Pavlides, S.A.; Tammariello, S.P. Proteomic and phosphoproteomic profiling during diapause entrance in the flesh fly, Sarcophaga crassipalpis. J. Insect Physiol. 2011, 57, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Ragland, G.J.; Denlinger, D.L.; Hahn, D.A. Mechanisms of suspended animation are revealed by transcript profiling of diapause in the flesh fly. Proc. Natl. Acad. Sci. USA 2010, 107, 14909–14914. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.H.; Ikeda, M.; Sato, Y.; Saito, H.; Imai, K.; Isobe, M.; Yamashita, O. Molecular characterization of ovary trehalase of the silkworm, Bombyx mori and its transcriptional activation by diapause hormone. Biochim. Biophys. Acta 1994, 1218, 366–374. [Google Scholar] [CrossRef]
- Kageyama, T.; Ohnishi, E. Carbohydrate metabolism in the eggs of the silkworm, Bombyx mori. I. Absence of phosphofructokinase in the diapausing egg. Dev. Growth Differ. 1971, 13, 97–106. [Google Scholar] [CrossRef]
- Klowden, M.J. Physiological Systems in Insects; Academic Press: San Diego, CA, USA, 2002. [Google Scholar]
- Kunieda, T.; Fujiyuki, T.; Kucharski, R.; Foret, S.; Ament, S.A.; Toth, A.L.; Ohashi, K.; Takeuchi, H.; Kamikouchi, A.; Kage, E.; et al. Carbohydrate metabolism genes and pathways in insects: Insights from the honey bee genome. Insect Mol. Biol. 2006, 15, 563–576. [Google Scholar] [CrossRef] [PubMed]
- Ratnayake, O.C.; Chotiwan, N.; Saavedra-Rodriguez, K.; Perera, R. The buzz in the field: The interaction between viruses, mosquitoes, and metabolism. Front. Cell. Infect. Microbiol. 2023, 13, 1128577. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | Raw Reads | Clean Reads | Raw Tags | Clean Tags | Chimera | Effective Tags |
|---|---|---|---|---|---|---|
| CK_4W1 | 125,826 | 125,725 | 124,117 | 123,665 | 7212 | 116,453 |
| CK_4W2 | 124,908 | 124,815 | 123,285 | 122,951 | 9406 | 113,545 |
| CK_4W3 | 68,129 | 68,079 | 67,193 | 66,989 | 5427 | 61,562 |
| CK_CW1 | 135,349 | 134,439 | 119,449 | 118,428 | 5363 | 113,065 |
| CK_CW2 | 128,135 | 127,512 | 117,013 | 116,482 | 5549 | 110,933 |
| CK_CW3 | 131,175 | 130,061 | 116,129 | 114,688 | 6126 | 108,562 |
| CO_4W1 | 123,780 | 123,682 | 122,163 | 121,860 | 5414 | 116,446 |
| CO_4W2 | 115,998 | 115,899 | 114,404 | 114,090 | 4035 | 110,055 |
| CO_4W3 | 128,115 | 128,017 | 126,494 | 126,156 | 6316 | 119,840 |
| CO_CW1 | 124,746 | 124,120 | 114,681 | 113,974 | 2609 | 111,365 |
| CO_CW2 | 121,009 | 120,447 | 112,761 | 111,903 | 4137 | 107,766 |
| CO_CW3 | 125,995 | 125,335 | 114,293 | 113,793 | 9011 | 104,782 |
| S_4W1 | 111,938 | 111,842 | 110,280 | 109,916 | 2352 | 107,564 |
| S_4W2 | 121,867 | 121,752 | 120,144 | 119,668 | 2995 | 116,673 |
| S_4W3 | 113,832 | 113,733 | 112,242 | 111,945 | 2697 | 109,248 |
| S_CW1 | 116,513 | 116,406 | 115,066 | 114,643 | 2828 | 111,815 |
| S_CW2 | 132,052 | 131,957 | 130,225 | 129,834 | 6147 | 123,687 |
| S_CW3 | 117,870 | 117,749 | 116,260 | 115,856 | 5566 | 110,290 |
| B_4W1 | 117,559 | 117,515 | 116,836 | 116,708 | 5001 | 111,707 |
| B_4W2 | 113,668 | 113,621 | 113,096 | 112,751 | 4267 | 108,484 |
| B_4W3 | 123,419 | 123,373 | 122,707 | 121,556 | 4283 | 117,273 |
| B_CW1 | 113,555 | 113,515 | 113,066 | 112,955 | 6063 | 106,892 |
| B_CW2 | 130,802 | 130,749 | 130,044 | 129,882 | 9759 | 120,123 |
| B_CW3 | 121,045 | 120,995 | 120,423 | 120,266 | 7759 | 112,507 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yin, Y.; Liu, Y.; Fan, J.; Yu, L.; Nie, M.; Zhang, Z.; Han, Q.; Liao, C. Analysis of Midgut Bacterial Communities in Larvae and Adult Mosquitoes of Aedes aegypti Invaded by Three Different Microorganisms. Microorganisms 2025, 13, 248. https://doi.org/10.3390/microorganisms13020248
Yin Y, Liu Y, Fan J, Yu L, Nie M, Zhang Z, Han Q, Liao C. Analysis of Midgut Bacterial Communities in Larvae and Adult Mosquitoes of Aedes aegypti Invaded by Three Different Microorganisms. Microorganisms. 2025; 13(2):248. https://doi.org/10.3390/microorganisms13020248
Chicago/Turabian StyleYin, Yanan, Yanhui Liu, Jieli Fan, Lingling Yu, Meng Nie, Zhiqi Zhang, Qian Han, and Chenghong Liao. 2025. "Analysis of Midgut Bacterial Communities in Larvae and Adult Mosquitoes of Aedes aegypti Invaded by Three Different Microorganisms" Microorganisms 13, no. 2: 248. https://doi.org/10.3390/microorganisms13020248
APA StyleYin, Y., Liu, Y., Fan, J., Yu, L., Nie, M., Zhang, Z., Han, Q., & Liao, C. (2025). Analysis of Midgut Bacterial Communities in Larvae and Adult Mosquitoes of Aedes aegypti Invaded by Three Different Microorganisms. Microorganisms, 13(2), 248. https://doi.org/10.3390/microorganisms13020248