
Mechanisms Driving Seasonal Succession and Community Assembly in Sediment Microbial Communities Across the Dali River Basin, the Loess Plateau, China

Abstract
:1. Introduction
2. Materials and Methods
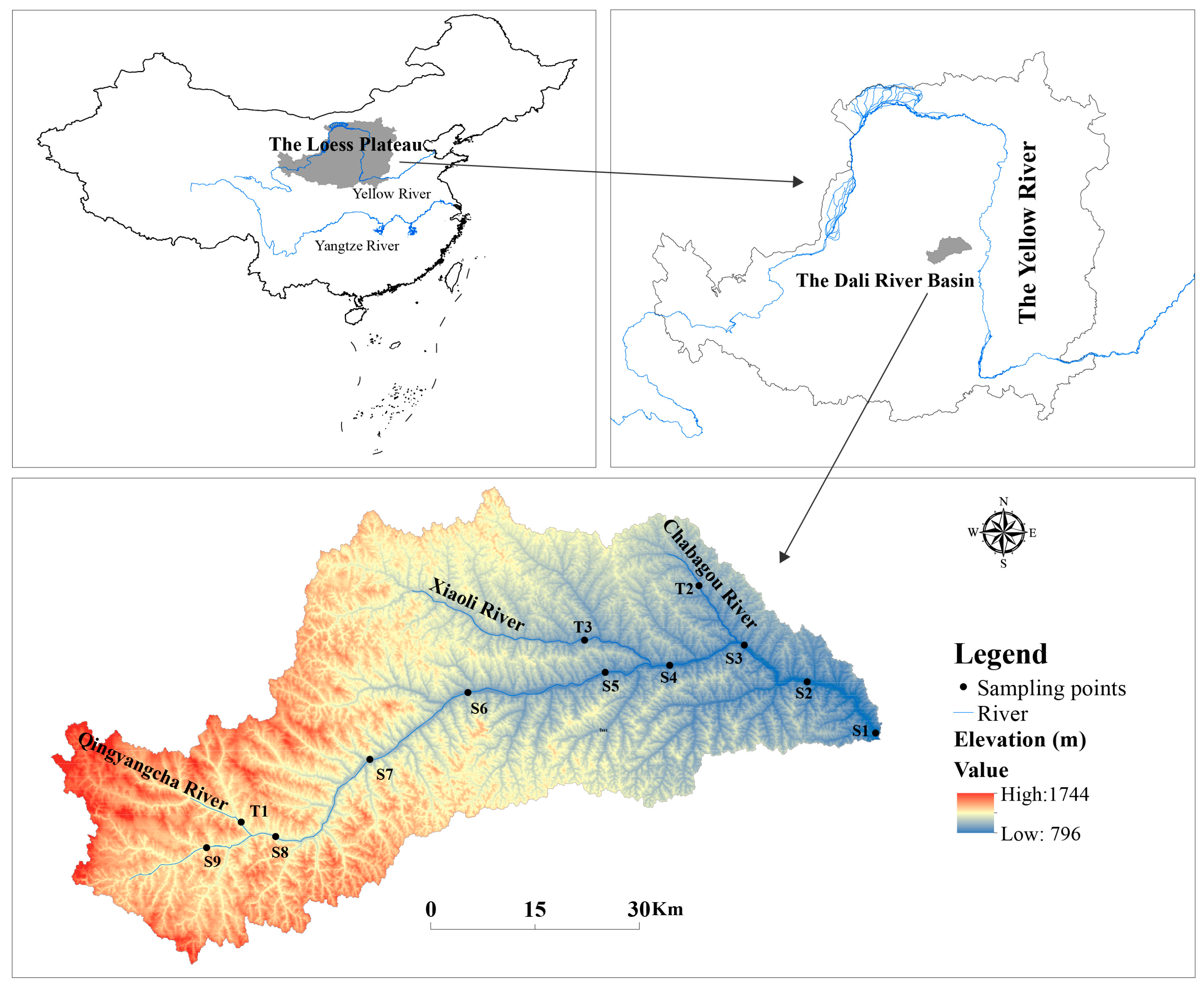
2.1. Study Area
2.2. Sampling Process and Measurement of Physicochemical Properties
2.3. DNA Extraction and PCR Amplification
2.4. Statistical Analyses
3. Results
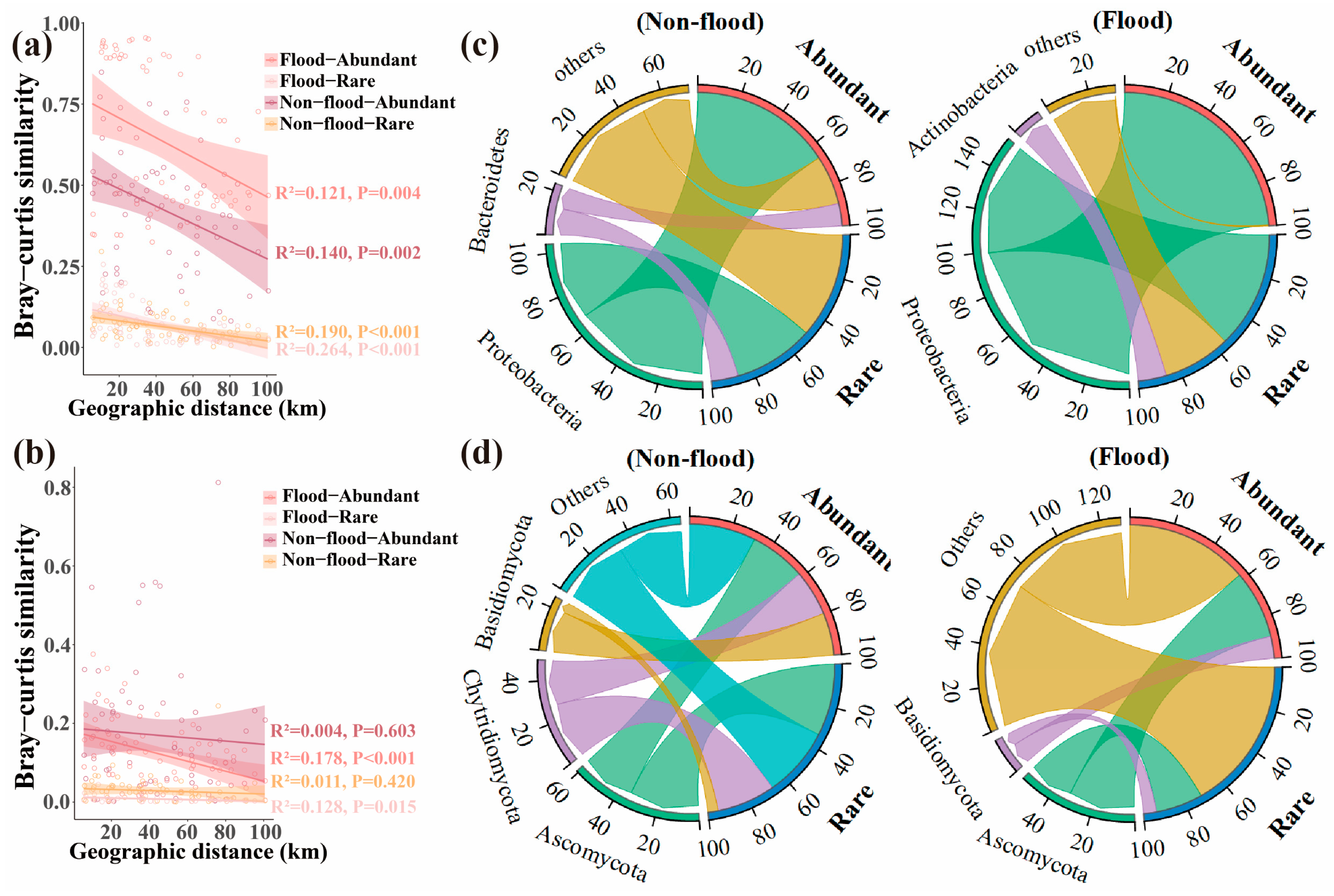
3.1. Diversity and Distance-Decay Patterns of the Sediment Microbial Communities
3.2. Structure of the Sediment Microbial Community
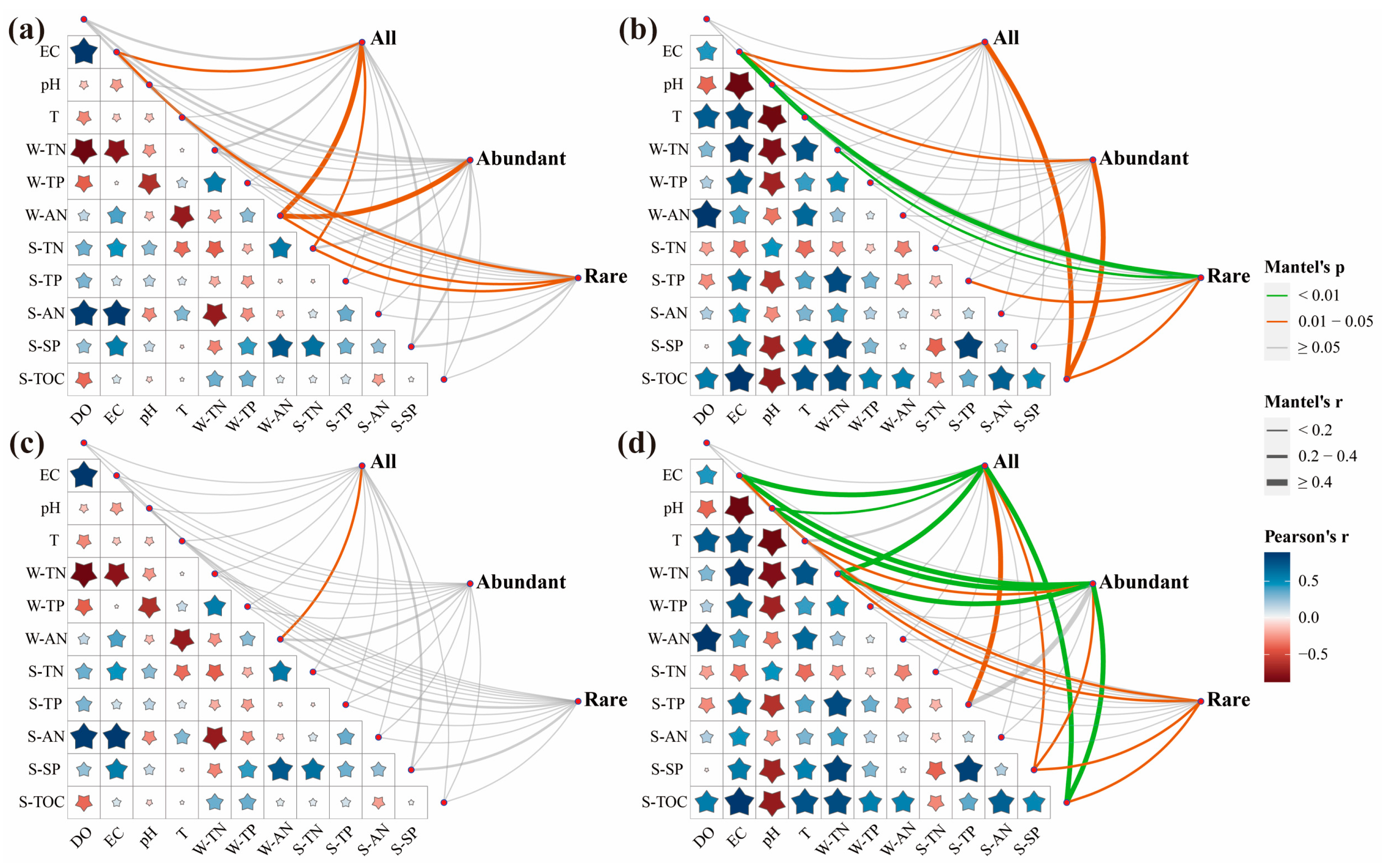
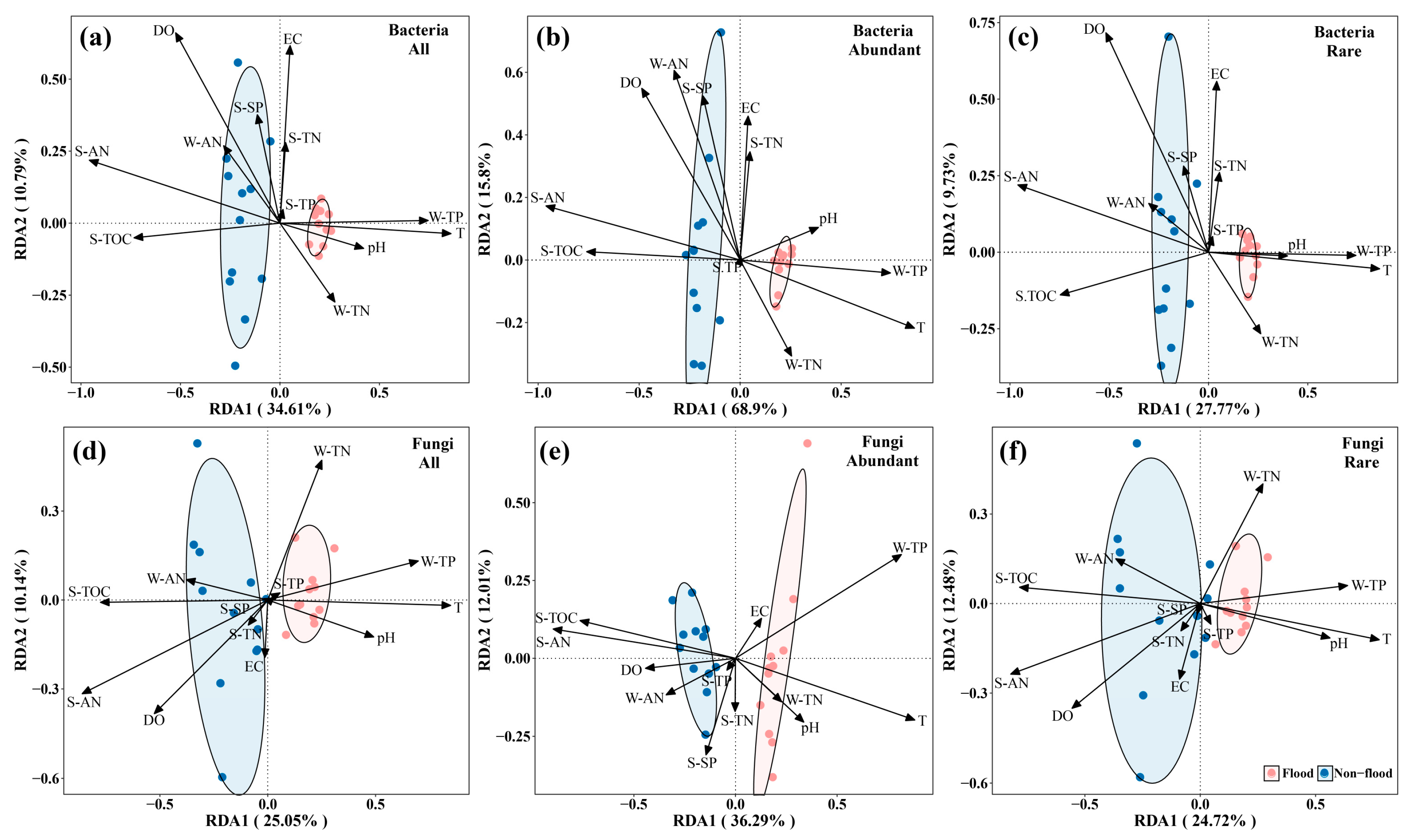
3.3. Environmental Factors Affecting Microbial Communities
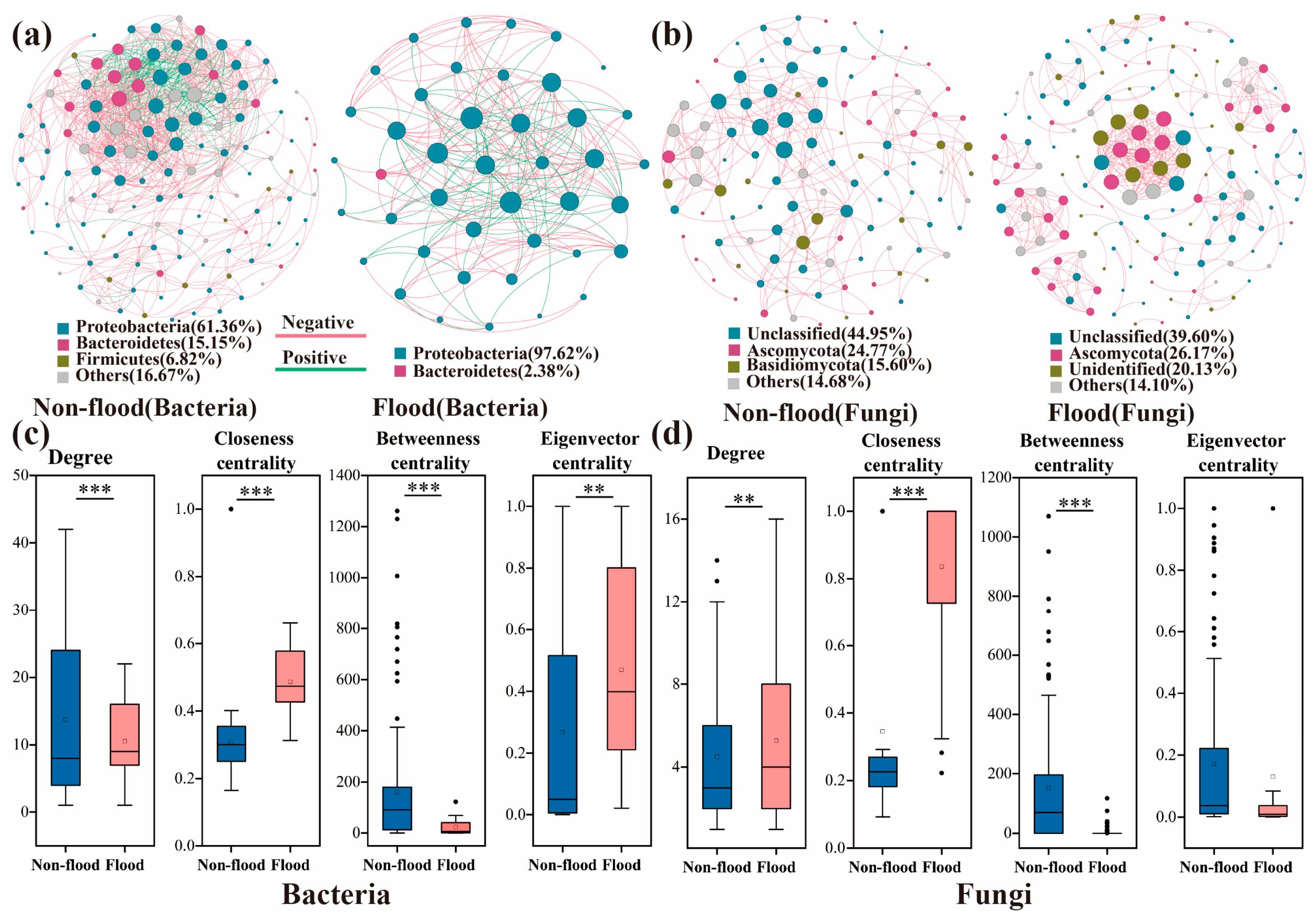
3.4. Symbiotic Networks and Functional Prediction of Microbial Communities
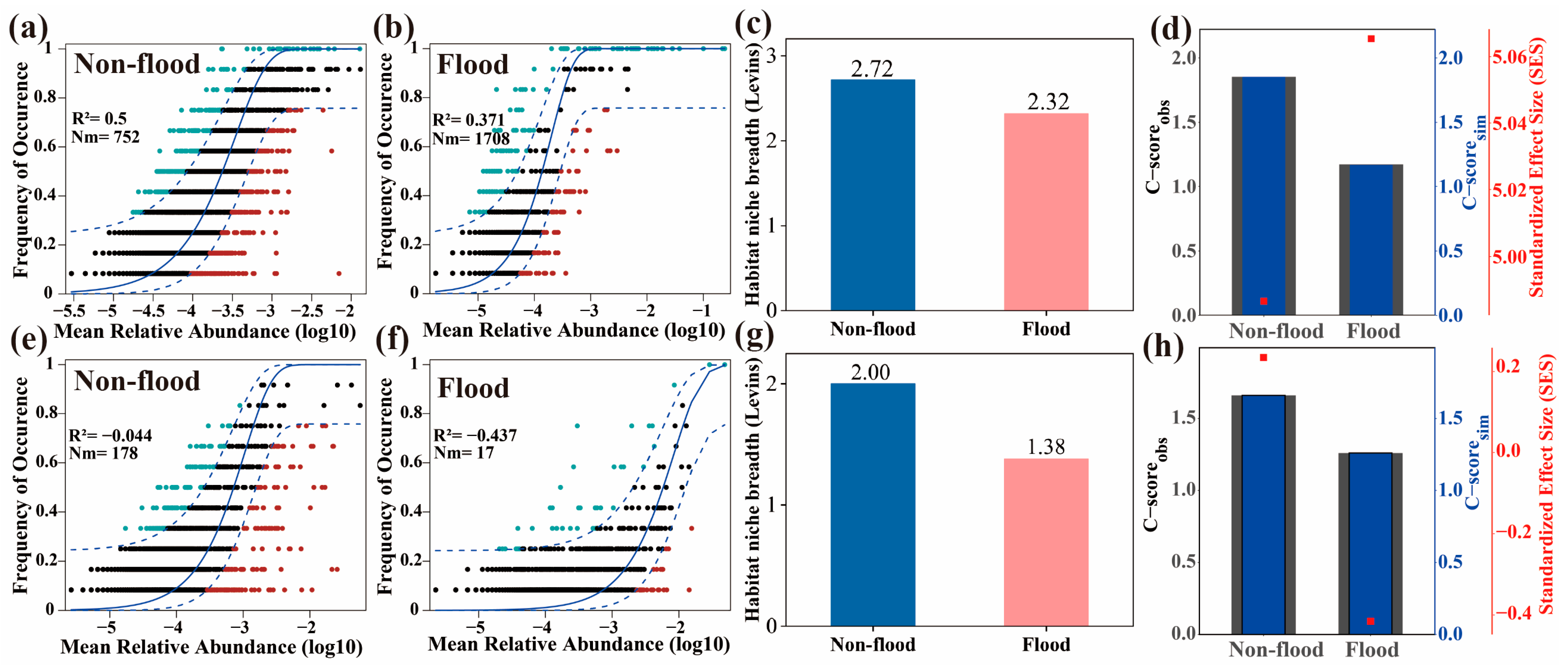
3.5. Sediment Microbial Community Assembly and Ecological Niche
4. Discussion
4.1. Seasonal Dynamics Affect Sediment Microbial Community Diversity
4.2. Seasonal Dynamics of the Dominant Phyla in the Microbial Community
4.3. Influencing Factors of Microbial Community
4.4. Assembly of Sediment Microbial Communities
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Armstrong, J.B.; Schindler, D.E.; Cunningham, C.J.; Deacy, W.; Walsh, P. Watershed complexity increases the capacity for salmon-wildlife interactions in coastal ecosystems. Conserv. Lett. 2020, 13, e12689. [Google Scholar] [CrossRef]
- Ge, Y.; Lou, Y.H.; Xu, M.M.; Wu, C.; Meng, J.; Shi, L.; Xia, F.; Xu, Y. Spatial distribution and influencing factors on the variation of bacterial communities in an urban river sediment. Environ. Pollut. 2020, 272, 115984. [Google Scholar] [CrossRef]
- Guo, S.Z.; Zhang, S.H.; Wang, S.P.; Lv, X.; Chen, H.Z.; Hu, X.R.; Ma, Y. Potamogeton crispus restoration increased the epiphytic microbial diversity and improved water quality in a micro-polluted urban river. Environ. Pollut. 2023, 326, 121485. [Google Scholar] [CrossRef]
- Damiani, M.; Lamouroux, N.; Pella, H.; Roux, P.; Loiseau, E.; Rosenbaum, R.K. Spatialized freshwater ecosystem life cycle impact assessment of water consumption based on instream habitat change modeling. Water Res. 2019, 163, 114884. [Google Scholar] [CrossRef] [PubMed]
- Shoemaker, W.R.; Locey, K.J.; Lennon, J.T. A macroecological theory of microbial biodiversity. Nat. Ecol. Evol. 2017, 1, 107. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.Y.; Cao, X.Y.; Huang, R.; Zeng, J.; Shen, F.; Xu, H.M.; Wang, S.C.; He, X.W.; Yu, Z.B. The heterogeneity of composition and assembly processes of the microbial community between different nutrient loading lake zones in Taihu Lake. Appl. Microbiol. Biotechnol. 2017, 101, 5913–5923. [Google Scholar] [CrossRef]
- Wu, K.; Liu, Y.; Liao, X.; Yang, X.; Chen, Z.; Mo, L.; Zhong, S.; Zhang, X. Fungal Diversity and Its Relationship with Environmental Factors in Coastal Sediments from Guangdong, China. J. Fungi 2023, 9, 101. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.Z.; Ning, D.L. Stochastic community assembly: Does it matter in microbial ecology? Microbiol. Mol. Biol. Rev. 2017, 81, e00002-17. [Google Scholar] [CrossRef]
- Goldford, J.E.; Lu, N.; Bajić, D.; Estrela, S.; Tikhonov, M.; Sanchez-Gorostiaga, A.; Segrè, D.; Mehta, P.; Sanchez, A. Emergent simplicity in microbial community assembly. Science 2018, 361, 469–474. [Google Scholar] [CrossRef]
- Guo, X.P.; Lu, D.P.; Niu, Z.S.; Feng, J.N.; Chen, Y.R.; Tou, F.Y.; Liu, M.; Yang, Y. Bacterial community structure in response to environmental impacts in the intertidal sediments along the Yangtze Estuary, China. Mar. Pollut. Bull. 2018, 126, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Xia, N.; Xia, X.H.; Liu, T.; Hu, L.J.; Zhu, B.T.; Zhang, X.T.; Dong, J.W. Characteristics of bacterial community in the water and surface sediment of the Yellow River, China, the largest turbid river in the world. J. Soils Sediments 2014, 14, 1894–1904. [Google Scholar] [CrossRef]
- Zhang, L.; Delgado-Baquerizo, M.; Shi, Y.; Liu, X.; Yang, Y.F.; Chu, H.Y. Co-existing water and sediment bacteria are driven by contrasting environmental factors across glacier-fed aquatic systems. Water Res. 2021, 198, 117139. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Yang, J.; Yu, Z.; Wilkinson, D.M. The biogeography of abundant and rare bacterioplankton in the lakes and reservoirs of China. ISME J. 2015, 9, 2068–2077. [Google Scholar] [CrossRef] [PubMed]
- Pester, M.; Bittner, N.; Deevong, P.; Wagner, M.; Loy, A. A ‘rare biosphere’ microorganism contributes to sulfate reduction in a peatland. ISME J. 2010, 4, 1591–1602. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.Q.; Cao, X.F.; Wang, J.; Zhao, L.; Sun, J.H.; Jiang, D.L.; Huang, Y. Similar community assembly mechanisms underlie similar biogeography of rare and abundant bacteria in lakes on Yungui Plateau, China. Limnol. Ocean. 2017, 62, 723–735. [Google Scholar] [CrossRef]
- Sun, H.; Pan, B.Z.; He, H.R.; Zhao, G.N.; Hou, Y.M.; Zhu, P.H. Assembly processes and co-occurrence relationships in the bacterioplankton communities of a large river system. Ecol. Indic. 2021, 126, 107643. [Google Scholar] [CrossRef]
- Obieze, C.C.; Wani, G.A.; Shah, M.A.; Reshi, Z.F.; Comeau, A.M.; Khasa, D.P. Anthropogenic activities and geographic locations regulate microbial diversity, community assembly and species sorting in Canadian and Indian freshwater lakes. Sci. Total Environ. 2022, 826, 154292. [Google Scholar] [CrossRef]
- Stegen, J.C.; Lin, X.J.; Konopka, A.E.; Fredrickson, J.K. Stochastic and deterministic assembly processes in subsurface microbial communities. ISME J. 2012, 6, 1653–1664. [Google Scholar] [CrossRef]
- Dini-Andreote, F.; Stegen, J.C.; van Elsas, J.D.; Salles, J.F. Disentangling mechanisms that mediate the balance between stochastic and deterministic processes in microbial succession. Proc. Natl. Acad. Sci. USA 2015, 112, E1326–E1332. [Google Scholar] [CrossRef]
- Jiao, C.C.; Zhao, D.Y.; Zeng, J.; Guo, L.; Yu, Z.B. Disentangling the seasonal co-occurrence patterns and ecological stochasticity of planktonic and benthic bacterial communities within multiple lakes. Sci. Total Environ. 2020, 740, 140010. [Google Scholar] [CrossRef]
- Zhang, Z.F.; Mao, J.; Cai, L. Dispersal Limitation Controlling the Assembly of the Fungal Community in Karst Caves. J. Fungi 2023, 9, 1013. [Google Scholar] [CrossRef]
- Ye, G.P.; Yang, N.; He, Z.Y.; Yang, P.; Ye, R.C.; Jiang, M.H.; Wang, D.; Cao, D.D.; Zhang, W.B.; Wei, X.Y.; et al. Seasonal patterns in diversity, complexity, and community assembly of soil microorganisms in a subtropical coastal wetland. Pedosphere 2024. [Google Scholar] [CrossRef]
- Chen, W.D.; Ren, K.X.; Isabwe, A.; Chen, H.H.; Liu, M.; Yang, J. Stochastic processes shape microeukaryotic community assembly in a subtropical river across wet and dry seasons. Microbiome 2019, 7, 138. [Google Scholar] [CrossRef]
- Chen, X.; Xu, G.C.; Xiong, P.; Peng, J.B.; Fang, K.; Wan, S.; Wang, B.; Gu, F.Y.; Li, J.; Xiong, H.J. Dry and wet seasonal variations of the sediment fungal community composition in the semi-arid region of the Dali River, Northwest China. Environ. Sci. Pollut. Res. 2023, 30, 123694–123709. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Xu, S.; Yan, R.M.; Wang, R.Y.; Gao, Y.X.; Kong, M.; Yi, Q.T.; Zhang, Y.M. Similar geographic patterns but distinct assembly processes of abundant and rare bacterioplankton communities in river networks of the Taihu Basin. Water Res. 2022, 211, 118057. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Zhang, P.; Li, X.D.; Yang, S.X.; Chao, X.; Liu, H.Q.; Ba, S. Distribution patterns and community assembly processes of eukaryotic microorganisms along an altitudinal gradient in the middle reaches of the Yarlung Zangbo River. Water Res. 2023, 239, 120047. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.Y.; Zhang, M.L.; Huang, S.Y.; Li, L.J.; Gao, Q.; Wang, Y. A highly conserved core bacterial microbiota with nitrogen-fixation capacity inhabits the xylem sap in maize plants. Nat. Commun. 2022, 13, 3361. [Google Scholar] [CrossRef] [PubMed]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol 2020, 38, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Mo, Y.Y.; Peng, F.; Gao, X.F.; Xiao, P.; Logares, R.; Jappesen, E.; Ren, K.X.; Xue, Y.Y.; Yang, J. Low shifts in salinity determined assembly processes and network stability of microeukaryotic plankton communities in a subtropical urban reservoir. Microbiome 2021, 9, 128. [Google Scholar] [CrossRef]
- López, R.P.; Valdivia, S.; Rivera, M.L.; Rios, R.S. Co-occurrence patterns along a regional aridity gradient of the subtropical Andes do not support stress gradient hypotheses. PLoS ONE 2013, 8, e58518. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Xie, G.J.; Shao, K.Q.; Hu, Y.; Cai, J.; Bai, C.R.; Gong, Y.; Gao, G. Contrast diversity patterns and processes of microbial community assembly in a river-lake continuum across a catchment scale in northwestern China. Environ. Microbiome 2020, 15, 10. [Google Scholar] [CrossRef]
- Liao, J.Q.; Cao, X.F.; Zhao, L.; Wang, J.; Gao, Z.; Wang, M.C.; Huang, Y. The importance of neutral and niche processes for bacterial community assembly differs between habitat generalists and specialists. FEMS Microbiol. Ecol. 2016, 11, fiw174. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Jiang, Y.; Huang, H.; Mou, L.; Ru, J.; Zhao, J.; Xiao, S. Long-term and high-concentration heavy-metal contamination strongly influences the microbiome and functional genes in Yellow River sediments. Sci. Total Environ. 2018, 637–638, 1400–1412. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.H.; Jiang, W.L.; Sun, N.; Shi, J.F.; Zhang, D.P.; Zhang, J.J.; Wang, Z.K.; Yang, J.S.; Yu, J.B.; Lv, Z.B. Responses of the structure and function of microbes in Yellow River Estuary sediments to different levels of mercury. Mar. Environ. Res. 2023, 190, 106097. [Google Scholar] [CrossRef]
- Wu, Y.; Mao, X.F.; Xia, L.; Yu, H.Y.; Yu, Y.; Tang, W.J.; Xiao, F.; Ji, H.C. Microbial Community Abundance Affects the Methane Ebullition Flux in Dahejia Reservoir of the Yellow River in the Warm Season. Diversity 2023, 15, 154. [Google Scholar] [CrossRef]
- Li, H.N.; Tian, S.M.; Shang, F.D.; Shi, X.Y.; Zhang, Y.; Cao, Y.T. Impacts of oxbow lake evolution on sediment microbial community structure in the Yellow River source region. Environ. Res. 2024, 252, 119042. [Google Scholar] [CrossRef]
- Crump, B.C.; Peterson, B.J.; Raymond, P.A.; Amon, R.M.W.; Rinehart, A.; McClelland, J.W.; Holmes, R.M. Circumpolar synchrony in big river bacterioplankton. Proc. Natl. Acad. Sci. USA 2009, 106, 21208–21212. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Wang, P.F.; Huang, S.H.; Li, Z.Y.; Gong, H.Z.; Huang, W.J.; Zhao, Z.L.; Yu, Z.H. Environmental filtering dominates bacterioplankton community assembly in a highly urbanized estuarine ecosystem. Water Res. 2021, 196, 110934. [Google Scholar] [CrossRef]
- Liu, R.; Zhou, X.D. Influence of sediment characteristics and overlying water quality on sediment bacterial communities in a seasonal sandy river. China Environ. Sci. 2017, 37, 4342–4352. (In Chinese) [Google Scholar] [CrossRef]
- Bai, Y.; Shi, Q.; Wen, D.; Li, Z.; Jefferson, W.A.; Feng, C.; Tang, X. Bacterial communities in the sediments of Dianchi Lake, a partitioned eutrophic waterbody in China. PLoS ONE 2012, 7, e37796. [Google Scholar] [CrossRef] [PubMed]
- Dash, H.R.; Mangwani, N.; Chakraborty, J.; Kumari, S.; Das, S. Marine bacteria: Potential candidates for enhanced bioremediation. Appl. Microbiol. Biotechnol. 2013, 97, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Raiyani, N.M.; Singh, S.P. Taxonomic and functional profiling of the microbial communities of Arabian Sea: A metagenomics approach. Genomics 2020, 112, 4361–4369. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Chen, X.; Jiang, X.; Zheng, B. Characterization of sediment bacterial communities in plain lakes with different trophic statuses. MicrobiologyOpen 2017, 6, e503. [Google Scholar] [CrossRef] [PubMed]
- Oni, O.E.; Schmidt, F.; Miyatake, T.; Kasten, S.; Witt, M.; Hinrichs, K.; Friedrich, M.W. Microbial Communities and Organic Matter Composition in Surface and Subsurface Sediments of the Helgoland Mud Area, North Sea. Front. Microbiol. 2015, 6, 635–644. [Google Scholar] [CrossRef]
- Zhang, J.W.; Ge, Z.H.; Ma, Z.H.; Huang, D.Y.; Zhang, J.B. Seasonal changes driving shifts of aquatic rhizosphere microbial community structure and the functional properties. J. Environ. Manag. 2022, 322, 116124. [Google Scholar] [CrossRef] [PubMed]
- Qi, H.Y.; Huang, D.; Wang, F.H.; Ye, M.; Jiang, X. Spatial dynamics of prokaryotic microbial communities in sediments of the Yellow Sea: Assembly process, co-occurrence relationships, and environmental implications. J. Environ. Manag. 2022, 319, 115645. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Dangi, P.; Choudhary, M. Actinomycetes: Source, identification, and their applications. Int. J. Curr. Microbiol. Appl. Sci. 2014, 3, 801–832. [Google Scholar]
- Catão, E.C.; Lopes, F.A.; Araújo, J.F.; de Castro, A.P.; Barreto, C.C.; Bustamante, M.M.C.; Quirino, B.F.; Krüger, R.H. Soil acidobacterial 16S rRNA gene sequences reveal subgroup level differences between Savanna-Like cerrado and atlantic forest Brazilian biomes. Int. J. Microbiol. 2014, 2014, 156341. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, J.N.; Gao, G.H.; Bartlam, M.G.; Wang, Y.Y. Distribution and diversity of fungi in freshwater sediments on a river catchment scale. Front. Microbiol. 2015, 6, 329. [Google Scholar] [CrossRef]
- Wan, W.J.; Gadd, G.M.; Gu, J.D.; Liu, W.Z.; Chen, P.; Zhang, Q.F.; Yang, Y.Y. Beyond biogeographic patterns: Processes shaping the microbial landscape in soils and sediments along the Yangtze River. mLife 2023, 2, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Caiafa, M.V.; Gómez-Hernández, M.; Williams-Linera, G.; Ramírez-Cruz, V. Functional diversity of macromycete communities along an environmental gradient in a Mexican seasonally dry tropical forest. Fungal Ecol. 2017, 28, 66–75. [Google Scholar] [CrossRef]
- Jia, T.; Guo, T.Y.; Yao, Y.S.; Wang, R.H.; Chai, B.F. Seasonal microbial community characteristic and its driving factors in a copper tailings dam in the Chinese Loess Plateau. Front. Microbiol. 2020, 11, 1574. [Google Scholar] [CrossRef]
- Shang, Y.Q.; Wu, X.Y.; Wang, X.B.; Dou, H.S.; Wei, Q.G.; Ma, S.C.; Sun, G.L.; Wang, L.D.; Sha, W.L.; Zhang, H.H. Environmental factors and stochasticity affect the fungal community structures in the water and sediments of Hulun Lake, China. Ecol. Evol. 2022, 12, e9510. [Google Scholar] [CrossRef]
- Kagami, M.; Miki, T.; Takimoto, G. Mycoloop: Chytrids in aquatic food webs. Front. Microbiol. 2014, 5, 166. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.D.M.; Forn, I.; Gadelha, C.; Egan, M.J.; Bass, D.; Massana, R.; Richards, T.A. Discovery of novel intermediate forms redefines the fungal tree of life. Nature 2011, 474, 200–203. [Google Scholar] [CrossRef] [PubMed]
- Cunliffe, M.; Hollingsworth, A.; Bain, C.; Sharma, V.; Taylor, J.D. Algal polysaccharide utilisation by saprotrophic planktonic marine fungi. Funct. Ecol. 2017, 30, 135–138. [Google Scholar] [CrossRef]
- Zhang, Z.M.; Deng, Q.H.; Cao, X.Y.; Zhou, Y.Y.; Song, C.L. Patterns of sediment fungal community dependent on farming practices in aquaculture ponds. Front. Microbiol. 2021, 12, 542064. [Google Scholar] [CrossRef] [PubMed]
- Stockdale, E.A.; Shepherd, M.A.; Fortune, S.; Cuttle, S.P. Soil fertility in organic farming systems-fundamentally different? Soil Use Manag. 2002, 18, 301–308. [Google Scholar] [CrossRef]
- Fang, W.; Fan, T.; Wang, S.; Yu, X.; Lu, A.; Wang, X.; Zhou, W.; Yuan, H.; Zhang, L. Seasonal changes driving shifts in microbial community assembly and species coexistence in an urban river. Sci. Total Environ. 2023, 905, 167027. [Google Scholar] [CrossRef] [PubMed]
- Shang, Y.Q.; Wu, X.Y.; Wang, X.B.; Wei, Q.G.; Ma, S.C.; Sun, G.L.; Zhang, H.X.; Wang, L.D.; Dou, H.S.; Zhang, H.H. Factors affecting seasonal variation of microbial community structure in Hulun Lake, China. Sci. Total Environ. 2022, 805, 150294. [Google Scholar] [CrossRef] [PubMed]
- Čačković, A.; Kajan, K.; Selak, L.; Marković, T.; Brozičević, A.; Pjevac, P.; Orlić, S. Hydrochemical and Seasonally Conditioned Changes of Microbial Communities in the Tufa-Forming Freshwater Network Ecosystem. mSphere 2023, 8, e00602-22. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.X.; Zhang, B.; Ning, D.L.; Zhang, Y.; Dai, T.J.; Wu, L.W.; Li, T.L.; Liu, W.; Zhou, J.Z.; Wen, X.H. Seasonal dynamics of the microbial community in two full-scale wastewater treatment plants: Diversity, composition, phylogenetic group based assembly and cooccurrence pattern. Water Res. 2021, 200, 117295. [Google Scholar] [CrossRef]
- Huang, Y.J.; Feng, J.C.; Kong, J.; Sun, L.; Zhang, M.; Huang, Y.; Tang, L.; Zhang, S.; Yang, Z. Community assemblages and species coexistence of prokaryotes controlled by local environmental heterogeneity in a cold seep water column. Sci. Total Environ. 2023, 868, 161725. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nodes | Links | Modularity | Clustering Coefficient | Average Path Length | Network Diameter | Average Degree | Graph Density | Positive Correlation | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Bacteria | Non-flood | 132 | 904 | 0.377 | 0.584 | 3.534 | 10 | 13.697 | 0.105 | 74.12% |
| Flood | 43 | 221 | 0.33 | 0.712 | 2.135 | 5 | 10.524 | 0.257 | 61.09% | |
| Fungi | Non-flood | 109 | 245 | 0.678 | 0.475 | 5.146 | 16 | 4.495 | 0.042 | 99.59% |
| Flood | 149 | 393 | 0.833 | 0.855 | 1.786 | 6 | 5.275 | 0.036 | 99.75% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, X.; Li, J.; Xu, G.; Fang, K.; Wan, S.; Wang, B.; Gu, F. Mechanisms Driving Seasonal Succession and Community Assembly in Sediment Microbial Communities Across the Dali River Basin, the Loess Plateau, China. Microorganisms 2025, 13, 319. https://doi.org/10.3390/microorganisms13020319
Chen X, Li J, Xu G, Fang K, Wan S, Wang B, Gu F. Mechanisms Driving Seasonal Succession and Community Assembly in Sediment Microbial Communities Across the Dali River Basin, the Loess Plateau, China. Microorganisms. 2025; 13(2):319. https://doi.org/10.3390/microorganisms13020319
Chicago/Turabian StyleChen, Xin, Jing Li, Guoce Xu, Kang Fang, Shun Wan, Bin Wang, and Fengyou Gu. 2025. "Mechanisms Driving Seasonal Succession and Community Assembly in Sediment Microbial Communities Across the Dali River Basin, the Loess Plateau, China" Microorganisms 13, no. 2: 319. https://doi.org/10.3390/microorganisms13020319
APA StyleChen, X., Li, J., Xu, G., Fang, K., Wan, S., Wang, B., & Gu, F. (2025). Mechanisms Driving Seasonal Succession and Community Assembly in Sediment Microbial Communities Across the Dali River Basin, the Loess Plateau, China. Microorganisms, 13(2), 319. https://doi.org/10.3390/microorganisms13020319