
Composition, Distribution and Mobility Potential of the Antibiotic Resistome in Sediments from the East China Sea Revealed by Metagenomic Analysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Determination of Environmental Factors
2.2. DNA Extraction and Metagenomic Sequencing Analysis
2.3. ARG and ARG Host Analysis
2.4. Mobility Potential of ARGs
2.5. Statistical Analysis
3. Results
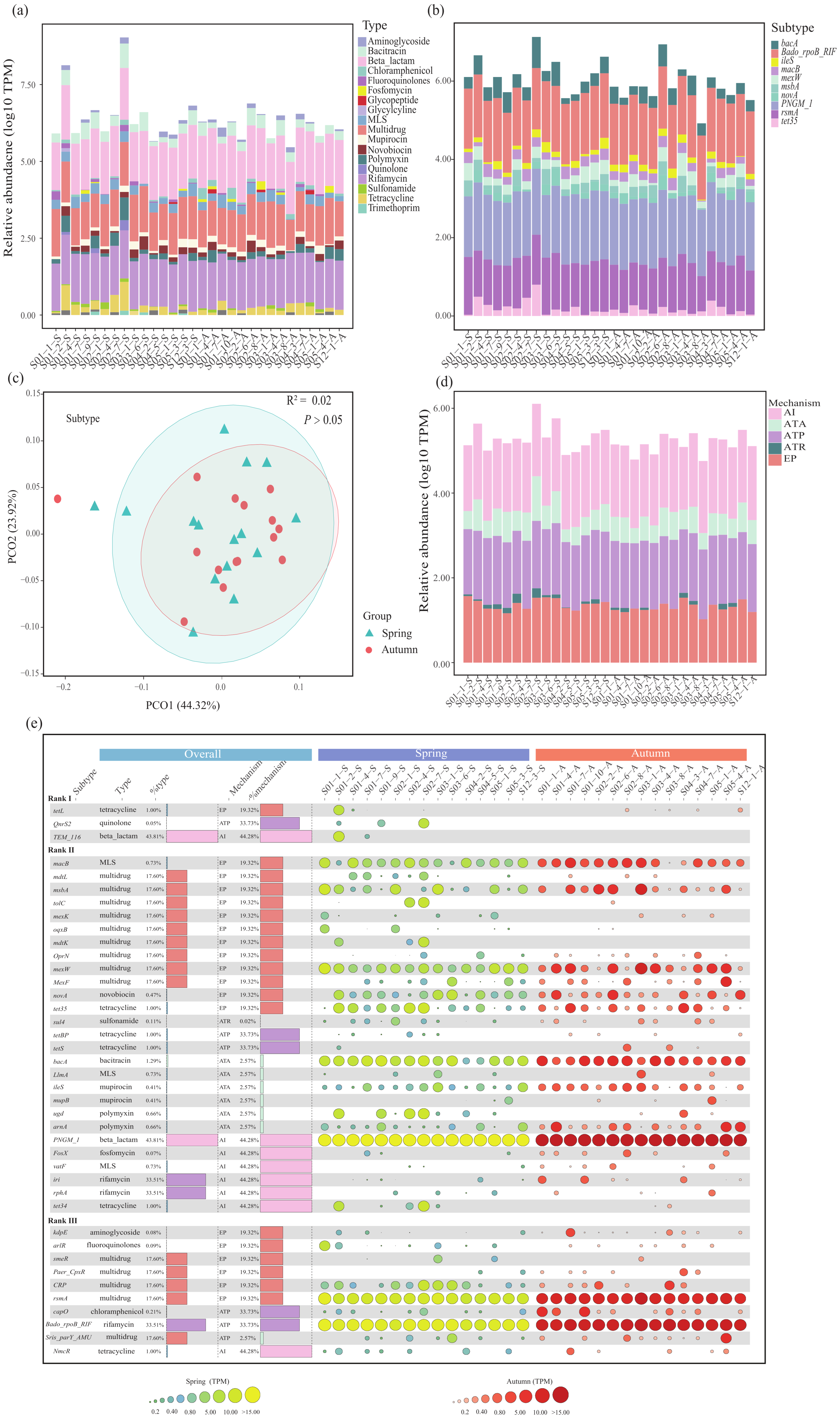
3.1. Composition and Distribution of ARGs
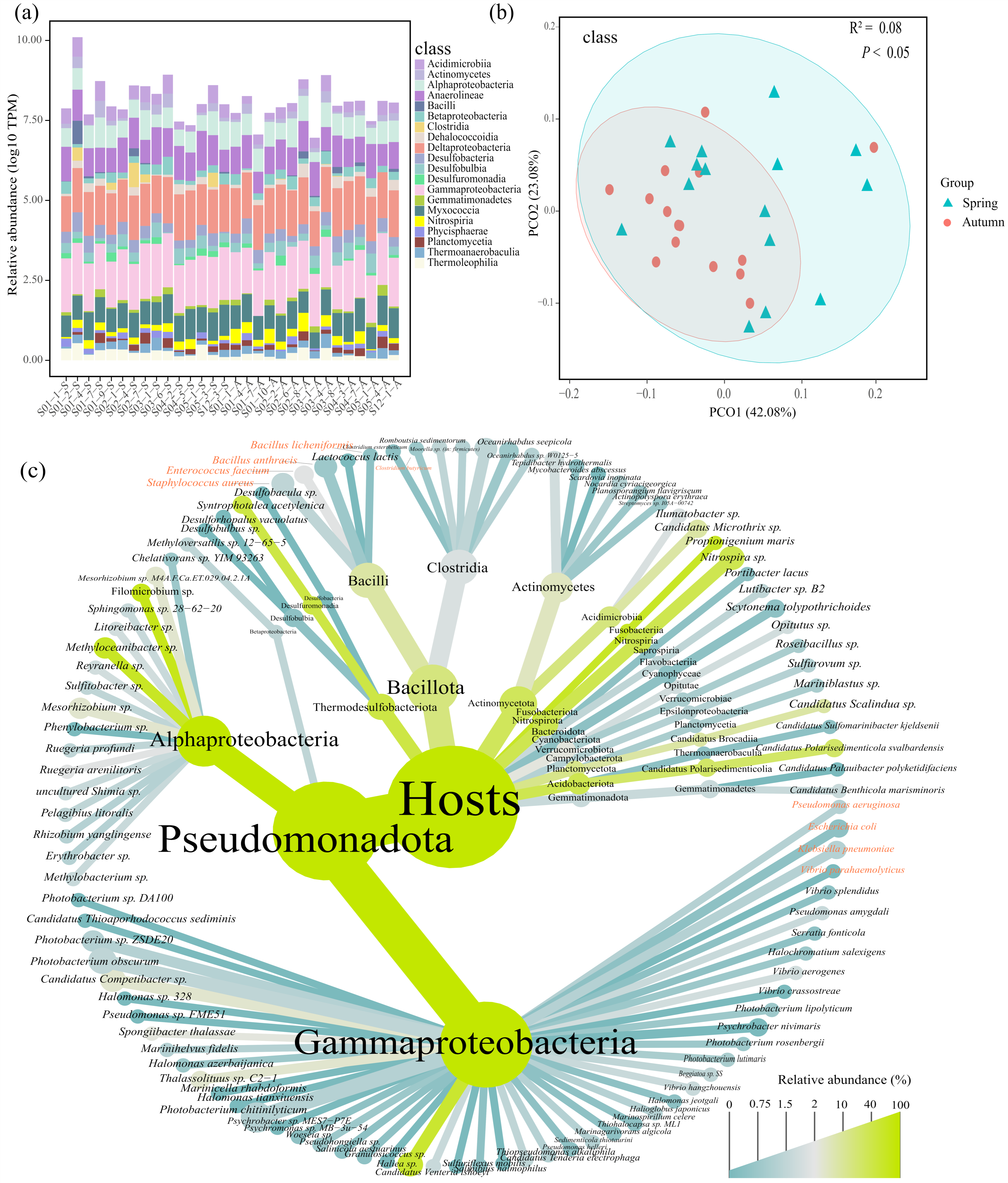
3.2. Community Composition of ARG Hosts
3.3. Assessment of ARG Mobility Potential
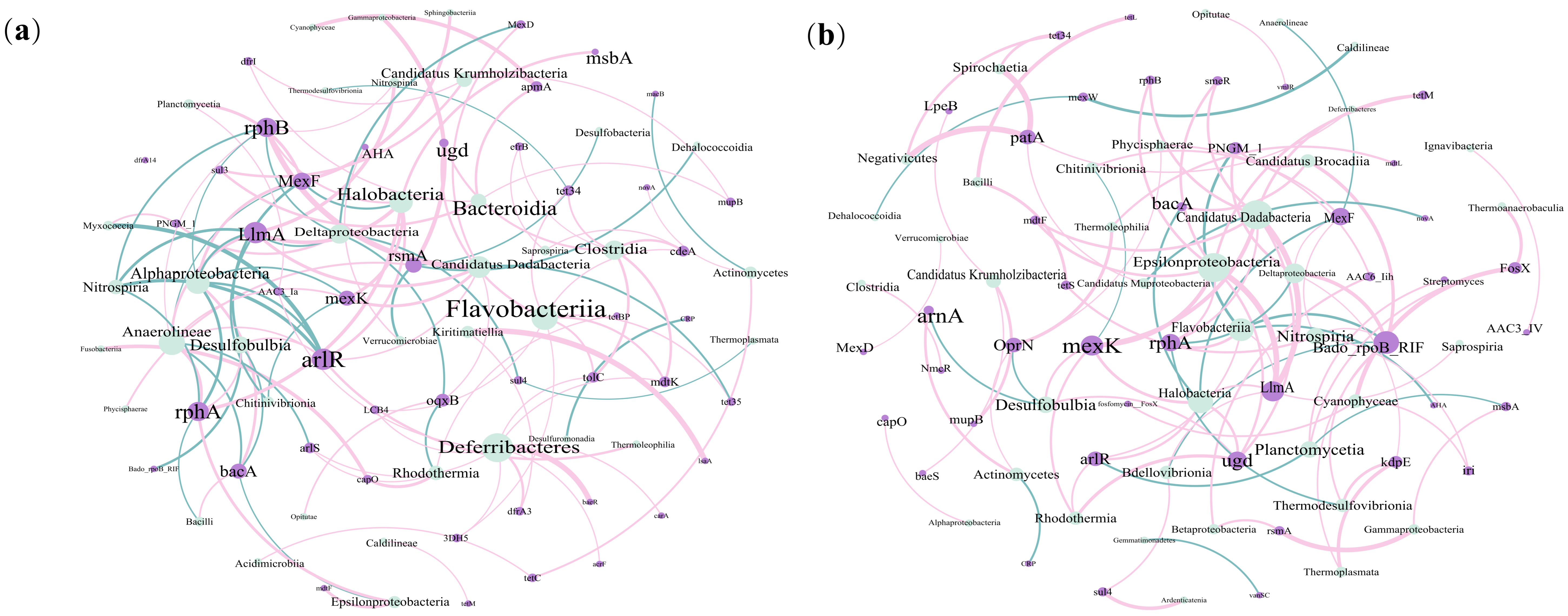
3.4. Relationships Among ARGs, MGEs and ARG Hosts
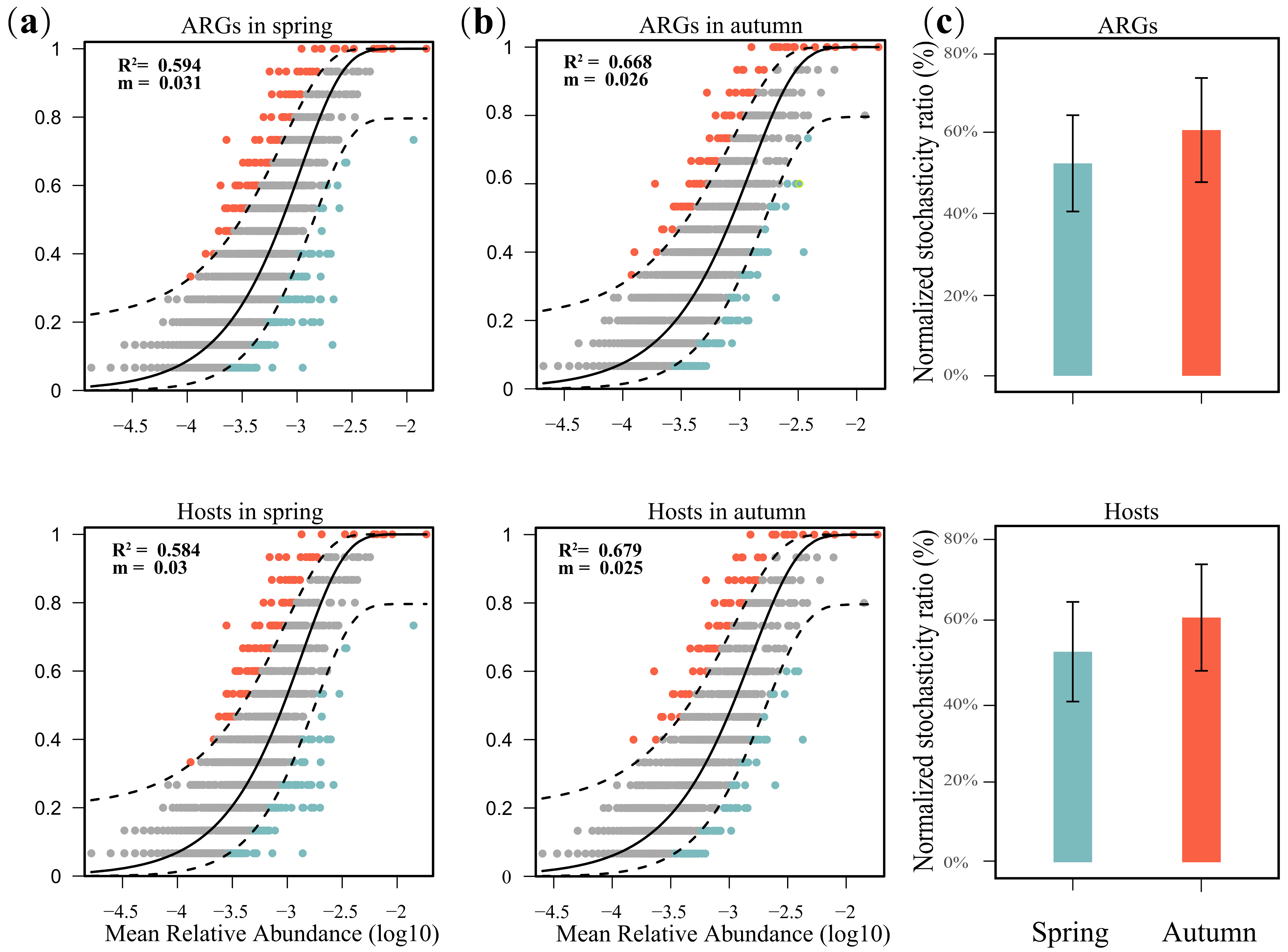
3.5. Driving Forces and Risk Assessment of ARGs
4. Discussion
4.1. Distribution Patterns of ARGs and ARG Hosts in Sediments of the East China Sea
4.2. Mobility Potential and Health Risk of ARGs in Sediments of the East China Sea
4.3. Factors That Regulate the Antibiotic Resistome in Sediments of the East China Sea
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hutchings, M.I.; Truman, A.W.; Wilkinson, B. Antibiotics: Past, Present and Future. Antimicrobials 2019, 51, 72–80. [Google Scholar] [CrossRef]
- Pruden, A.; Pei, R.; Storteboom, H.; Carlson, K.H. Antibiotic Resistance Genes as Emerging Contaminants: Studies in Northern Colorado. Environ. Sci. Technol. 2006, 40, 7445–7450. [Google Scholar] [CrossRef] [PubMed]
- Fischbach, M.A.; Walsh, C.T. Antibiotics for Emerging Pathogens. Science 2009, 325, 1089–1093. [Google Scholar] [CrossRef]
- Berendonk, T.U.; Manaia, C.M.; Merlin, C.; Fatta-Kassinos, D.; Cytryn, E.; Walsh, F.; Bürgmann, H.; Sørum, H.; Norström, M.; Pons, M.-N.; et al. Tackling Antibiotic Resistance: The Environmental Framework. Nat. Rev. Microbiol. 2015, 13, 310–317. [Google Scholar] [CrossRef]
- Fang, H.; Han, L.; Zhang, H.; Long, Z.; Cai, L.; Yu, Y. Dissemination of Antibiotic Resistance Genes and Human Pathogenic Bacteria from a Pig Feedlot to the Surrounding Stream and Agricultural Soils. J. Hazard. Mater. 2018, 357, 53–62. [Google Scholar] [CrossRef]
- Cassini, A.; Högberg, L.D.; Plachouras, D.; Quattrocchi, A.; Hoxha, A.; Simonsen, G.S.; Colomb-Cotinat, M.; Kretzschmar, M.E.; Devleesschauwer, B.; Cecchini, M.; et al. Attributable Deaths and Disability-Adjusted Life-Years Caused by Infections with Antibiotic-Resistant Bacteria in the EU and the European Economic Area in 2015: A Population-Level Modelling Analysis. Lancet Infect. Dis. 2019, 19, 56–66. [Google Scholar] [CrossRef]
- D’Costa, V.M.; King, C.E.; Kalan, L.; Morar, M.; Sung, W.W.L.; Schwarz, C.; Froese, D.; Zazula, G.; Calmels, F.; Debruyne, R.; et al. Antibiotic Resistance Is Ancient. Nature 2011, 477, 457–461. [Google Scholar] [CrossRef]
- Zhu, Y.-G.; Zhao, Y.; Li, B.; Huang, C.-L.; Zhang, S.-Y.; Yu, S.; Chen, Y.-S.; Zhang, T.; Gillings, M.R.; Su, J.-Q. Continental-Scale Pollution of Estuaries with Antibiotic Resistance Genes. Nat. Microbiol. 2017, 2, 16270. [Google Scholar] [CrossRef]
- Munoz-Lopez, M.; Garcia-Perez, J.L. DNA Transposons: Nature and Applications in Genomics. Curr. Genom. 2010, 11, 115–128. [Google Scholar] [CrossRef]
- Tennstedt, T.; Szczepanowski, R.; Krahn, I.; Pühler, A.; Schlüter, A. Sequence of the 68,869 bp IncP-1α Plasmid pTB11 from a Waste-Water Treatment Plant Reveals a Highly Conserved Backbone, a Tn402-like Integron and Other Transposable Elements. Plasmid 2005, 53, 218–238. [Google Scholar] [CrossRef]
- Ferreira da Silva, M.; Vaz-Moreira, I.; Gonzalez-Pajuelo, M.; Nunes, O.C.; Manaia, C.M. Antimicrobial Resistance Patterns in Enterobacteriaceae Isolated from an Urban Wastewater Treatment Plant. FEMS Microbiol. Ecol. 2007, 60, 166–176. [Google Scholar] [CrossRef]
- Taviani, E.; Ceccarelli, D.; Lazaro, N.; Bani, S.; Cappuccinelli, P.; Colwell, R.R.; Colombo, M.M. Environmental Vibrio Spp., Isolated in Mozambique, Contain a Polymorphic Group of Integrative Conjugative Elements and Class 1 Integrons. FEMS Microbiol. Ecol. 2008, 64, 45–54. [Google Scholar] [CrossRef]
- Szczepanowski, R.; Krahn, I.; Linke, B.; Goesmann, A.; Pühler, A.; Schlüter, A. Antibiotic Multiresistance Plasmid pRSB101 Isolated from a Wastewater Treatment Plant Is Related to Plasmids Residing in Phytopathogenic Bacteria and Carries Eight Different Resistance Determinants Including a Multidrug Transport System. Microbiology 2004, 150, 3613–3630. [Google Scholar]
- Chen, H.; Zheng, M.; Mu, H.; Kuang, S.; Zhang, S.; Liu, S. Spatial Distribution of Antibiotic Resistance Genes in Marine Sediments of East China Seas. J. Hazard. Mater. Adv. 2023, 11, 100363. [Google Scholar] [CrossRef]
- Guo, X.; Yang, Y.; Lu, D.; Niu, Z.; Feng, J.; Chen, Y.; Tou, F.; Garner, E.; Xu, J.; Liu, M.; et al. Biofilms as a Sink for Antibiotic Resistance Genes (ARGs) in the Yangtze Estuary. Water Res. 2018, 129, 277–286. [Google Scholar] [CrossRef]
- Lu, J.; Zhang, Y.; Wu, J.; Wang, J.; Zhang, C.; Lin, Y. Occurrence and Spatial Distribution of Antibiotic Resistance Genes in the Bohai Sea and Yellow Sea Areas, China. Environ. Pollut. 2019, 252, 450–460. [Google Scholar] [CrossRef] [PubMed]
- Gyraitė, G.; Kataržytė, M.; Espinosa, R.P.; Kalvaitienė, G.; Lastauskienė, E. Microbiome and Resistome Studies of the Lithuanian Baltic Sea Coast and the Curonian Lagoon Waters and Sediments. Antibiotics 2024, 13, 1013. [Google Scholar] [CrossRef]
- Habibi, N.; Uddin, S.; Lyons, B.; Al-Sarawi, H.A.; Behbehani, M.; Shajan, A.; Razzack, N.A.; Zakir, F.; Alam, F. Antibiotic Resistance Genes Associated with Marine Surface Sediments: A Baseline from the Shores of Kuwait. Sustainability 2022, 14, 8029. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, Y.; Li, J.; Tian, P.; Zhong, J.; Gong, H. Evaluation and Analysis of Coastal Complex Ecological Resilience Based on Multidimensional Data: A Case Study of East China Sea. Ecol. Indic. 2023, 155, 110981. [Google Scholar] [CrossRef]
- Li, F.; Chen, L.; Chen, W.; Bao, Y.; Zheng, Y.; Huang, B.; Mu, Q.; Wen, D.; Feng, C. Antibiotics in Coastal Water and Sediments of the East China Sea: Distribution, Ecological Risk Assessment and Indicators Screening. Mar. Pollut. Bull. 2020, 151, 110810. [Google Scholar] [CrossRef]
- Zhang, R.; Tang, J.; Li, J.; Zheng, Q.; Liu, D.; Chen, Y.; Zou, Y.; Chen, X.; Luo, C.; Zhang, G. Antibiotics in the Offshore Waters of the Bohai Sea and the Yellow Sea in China: Occurrence, Distribution and Ecological Risks. Environ. Pollut. 2013, 174, 71–77. [Google Scholar] [CrossRef]
- Fu, C.; Qin, Y.; Xiang, Q.; Qiao, M.; Zhu, Y. pH Drives the Spatial Variation of Antibiotic Resistance Gene Profiles in Riparian Soils at a Watershed Scale. Environ. Pollut. 2023, 326, 121486. [Google Scholar] [CrossRef]
- Chen, J.; Su, Z.; Dai, T.; Huang, B.; Mu, Q.; Zhang, Y.; Wen, D. Occurrence and Distribution of Antibiotic Resistance Genes in the Sediments of the East China Sea Bays. J. Environ. Sci. 2019, 81, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Yang, Y.; Liang, X.; Yu, K.; Zhang, T.; Li, X. Metagenomic Profiles of Antibiotic Resistance Genes (ARGs) between Human Impacted Estuary and Deep Ocean Sediments. Environ. Sci. Technol. 2013, 47, 12753–12760. [Google Scholar] [CrossRef]
- Cui, G.; Liu, Z.; Xu, W.; Gao, Y.; Yang, S.; Grossart, H.-P.; Li, M.; Luo, Z. Metagenomic Exploration of Antibiotic Resistance Genes and Their Hosts in Aquaculture Waters of the Semi-Closed Dongshan Bay (China). Sci. Total Environ. 2022, 838, 155784. [Google Scholar] [CrossRef]
- Ke, Y.; Sun, W.; Chen, X.; Zhu, Y.; Guo, X.; Yan, W.; Xie, S. Seasonality Determines the Variations of Biofilm Microbiome and Antibiotic Resistome in a Pilot-Scale Chlorinated Drinking Water Distribution System Deciphered by Metagenome Assembly. Environ. Sci. Technol. 2023, 57, 11430–11441. [Google Scholar] [CrossRef]
- Sun, F.; Xu, Z.; Fan, L. Response of Heavy Metal and Antibiotic Resistance Genes and Related Microorganisms to Different Heavy Metals in Activated Sludge. J. Environ. Manag. 2021, 300, 113754. [Google Scholar] [CrossRef]
- Yu, Q.; Han, Q.; Shi, S.; Sun, X.; Wang, X.; Wang, S.; Yang, J.; Su, W.; Nan, Z.; Li, H. Metagenomics Reveals the Response of Antibiotic Resistance Genes to Elevated Temperature in the Yellow River. Sci. Total Environ. 2023, 859, 160324. [Google Scholar] [CrossRef]
- Peng, J.; Wang, D.; He, P.; Wei, P.; Zhang, L.; Lan, W.; Zhang, X.; Guan, J.; Chen, Y.; Li, W.; et al. Seasonal Dynamics of Antibiotic Resistance Genes and Mobile Genetic Elements in a Subtropical Coastal Ecosystem: Implications for Environmental Health Risks. Environ. Res. 2024, 257, 119298. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Pan, R.; Dong, P.; Liu, S.; Chen, Q.; Borthwick, A.G.L.; Sun, L.; Xu, N.; Ni, J. Supercarriers of Antibiotic Resistome in a World’s Large River. Microbiome 2022, 10, 111. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.K.; Jain, M. NGS QC Toolkit: A Toolkit for Quality Control of Next Generation Sequencing Data. PLoS ONE 2012, 7, e30619. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.-M.; Luo, R.; Sadakane, K.; Lam, T.-W. MEGAHIT: An Ultra-Fast Single-Node Solution for Large and Complex Metagenomics Assembly via Succinct de Bruijn Graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef]
- Hyatt, D.; Chen, G.-L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic Gene Recognition and Translation Initiation Site Identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for Clustering the next-Generation Sequencing Data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Zheng, X.; Li, L.; Zhang, A.-N.; Jiang, X.-T.; Zhang, T. ARGs-OAP v3.0: Antibiotic-Resistance Gene Database Curation and Analysis Pipeline Optimization. Engineering 2022, 27, 234–241. [Google Scholar] [CrossRef]
- Shen, W.; Ren, H. TaxonKit: A Practical and Efficient NCBI Taxonomy Toolkit. Spec. Issue Microbiome 2021, 48, 844–850. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Ju, F.; Cai, L.; Zhang, T. Profile and Fate of Bacterial Pathogens in Sewage Treatment Plants Revealed by High-Throughput Metagenomic Approach. Environ. Sci. Technol. 2015, 49, 10492–10502. [Google Scholar] [CrossRef]
- Brown, C.L.; Mullet, J.; Hindi, F.; Stoll, J.E.; Gupta, S.; Choi, M.; Keenum, I.; Vikesland, P.; Pruden, A.; Zhang, L. mobileOG-Db: A Manually Curated Database of Protein Families Mediating the Life Cycle of Bacterial Mobile Genetic Elements. Appl. Environ. Microbiol. 2022, 88, e00991-22. [Google Scholar] [CrossRef]
- Forsberg, K.J.; Patel, S.; Gibson, M.K.; Lauber, C.L.; Knight, R.; Fierer, N.; Dantas, G. Bacterial Phylogeny Structures Soil Resistomes across Habitats. Nature 2014, 509, 612–616. [Google Scholar] [CrossRef]
- Zhao, R.; Yu, K.; Zhang, J.; Zhang, G.; Huang, J.; Ma, L.; Deng, C.; Li, X.; Li, B. Deciphering the Mobility and Bacterial Hosts of Antibiotic Resistance Genes under Antibiotic Selection Pressure by Metagenomic Assembly and Binning Approaches. Water Res. 2020, 186, 116318. [Google Scholar] [CrossRef]
- Ning, D.; Deng, Y.; Tiedje, J.M.; Zhou, J. A General Framework for Quantitatively Assessing Ecological Stochasticity. Proc. Natl. Acad. Sci. USA 2019, 116, 16892–16898. [Google Scholar] [CrossRef] [PubMed]
- Sloan, W.T.; Lunn, M.; Woodcock, S.; Head, I.M.; Nee, S.; Curtis, T.P. Quantifying the Roles of Immigration and Chance in Shaping Prokaryote Community Structure. Environ. Microbiol. 2006, 8, 732–740. [Google Scholar] [CrossRef] [PubMed]
- Jia, S.; Bian, K.; Shi, P.; Ye, L.; Liu, C. Metagenomic Profiling of Antibiotic Resistance Genes and Their Associations with Bacterial Community during Multiple Disinfection Regimes in a Full-Scale Drinking Water Treatment Plant. Water Res. 2020, 176, 115721. [Google Scholar] [CrossRef]
- Han, M.; Zhang, L.; Zhang, N.; Mao, Y.; Peng, Z.; Huang, B.; Zhang, Y.; Wang, Z. Antibiotic Resistome in a Large Urban-Lake Drinking Water Source in Middle China: Dissemination Mechanisms and Risk Assessment. J. Hazard. Mater. 2022, 424, 127745. [Google Scholar] [CrossRef]
- Yang, T.; Wang, X.; Hui, X.; Jiang, L.; Bi, X.; Ng, H.Y.; Zheng, X.; Huang, S.; Jiang, B.; Zhou, X. Antibiotic Resistome Associated with Inhalable Bioaerosols from Wastewater to Atmosphere: Mobility, Bacterial Hosts, Source Contributions and Resistome Risk. Water Res. 2023, 243, 120403. [Google Scholar] [CrossRef]
- Zhang, Q.; Ying, G.-G.; Pan, C.; Liu, Y.; Zhao, J. Comprehensive Evaluation of Antibiotics Emission and Fate in the River Basins of China: Source Analysis, Multimedia Modeling, and Linkage to Bacterial Resistance. Environ. Sci. Technol. 2015, 49, 6772–6782. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Zhang, G.; Zhang, K.; Sun, J.; Cui, Z.; Guo, Y.; Liu, H.; Xu, W. Strong Variation in Sedimental Antibiotic Resistomes among Urban Rivers, Estuaries and Coastal Oceans: Evidence from a River-Connected Coastal Water Ecosystem in Northern China. J. Environ. Manag. 2023, 342, 118132. [Google Scholar] [CrossRef]
- Ma, L.; Li, B.; Zhang, T. Abundant Rifampin Resistance Genes and Significant Correlations of Antibiotic Resistance Genes and Plasmids in Various Environments Revealed by Metagenomic Analysis. Appl. Microbiol. Biotechnol. 2014, 98, 5195–5204. [Google Scholar] [CrossRef]
- Li, S.; Zhu, Y.; Zhong, G.; Huang, Y.; Jones, K.C. Comprehensive Assessment of Environmental Emissions, Fate, and Risks of Veterinary Antibiotics in China: An Environmental Fate Modeling Approach. Environ. Sci. Technol. 2024, 58, 5534–5547. [Google Scholar] [CrossRef]
- Quillaguamán, J.; Guzmán, D.; Campero, M.; Hoepfner, C.; Relos, L.; Mendieta, D.; Higdon, S.M.; Eid, D.; Fernández, C.E. The Microbiome of a Polluted Urban Lake Harbors Pathogens with Diverse Antimicrobial Resistance and Virulence Genes. Environ. Pollut. 2021, 273, 116488. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Xie, S. Overview of Sulfonamide Biodegradation and the Relevant Pathways and Microorganisms. Sci. Total Environ. 2018, 640–641, 1465–1477. [Google Scholar] [CrossRef]
- Zhang, K.; Li, K.; Xin, R.; Han, Y.; Guo, Z.; Zou, W.; Wei, W.; Cui, X.; Zhang, Z.; Zhang, Y. Antibiotic Resistomes in Water Supply Reservoirs Sediments of Central China: Main Biotic Drivers and Distribution Pattern. Environ. Sci. Pollut. Res. 2022, 29, 37712–37721. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Yang, Y.; Ma, L.; Ju, F.; Guo, F.; Tiedje, J.M.; Zhang, T. Metagenomic and Network Analysis Reveal Wide Distribution and Co-Occurrence of Environmental Antibiotic Resistance Genes. ISME J. 2015, 9, 2490–2502. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.; George, A. The ABC Transporter Structure and Mechanism: Perspectives on Recent Research. Cell. Mol. Life Sci. 2004, 61, 682–699. [Google Scholar] [CrossRef]
- Wang, J.; Wang, J.; Zhao, Z.; Chen, J.; Lu, H.; Liu, G.; Zhou, J.; Guan, X. PAHs Accelerate the Propagation of Antibiotic Resistance Genes in Coastal Water Microbial Community. Environ. Pollut. 2017, 231, 1145–1152. [Google Scholar] [CrossRef]
- Von Wintersdorff, C.J.H.; Penders, J.; Van Niekerk, J.M.; Mills, N.D.; Majumder, S.; Van Alphen, L.B.; Savelkoul, P.H.M.; Wolffs, P.F.G. Dissemination of Antimicrobial Resistance in Microbial Ecosystems through Horizontal Gene Transfer. Front. Microbiol. 2016, 7, 173. [Google Scholar] [CrossRef]
- Sun, J.; Deng, Z.; Yan, A. Bacterial Multidrug Efflux Pumps: Mechanisms, Physiology and Pharmacological Exploitations. Integr. Glycobiol. Future Perspect. 2014, 453, 254–267. [Google Scholar] [CrossRef]
- Kong, M.; Zhang, Y.; Ma, Y.; Fang, H.; Wang, W.; Shi, G.; Yan, Y.; Zhang, S. Antibiotics and Antibiotic Resistance Change Bacterial Community Compositions in Marine Sediments. Environ. Res. 2024, 244, 118005. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhang, Z.; Lu, T.; Yu, Y.; Penuelas, J.; Zhu, Y.-G.; Qian, H. Gammaproteobacteria, a Core Taxon in the Guts of Soil Fauna, Are Potential Responders to Environmental Concentrations of Soil Pollutants. Microbiome 2021, 9, 196. [Google Scholar] [CrossRef]
- Hu, A.; Yanxia, N.; Yu, G.; Conghai, H.; He, J.; He, N.; Liu, S.; Deng, J.; Shen, W.; Zhangf, G. Diurnal Temperature Variation and Plants Drive Latitudinal Patterns in Seasonal Dynamics of Soil Microbial Community. Front. Microbiol. 2019, 10, 674. [Google Scholar] [CrossRef]
- Siguier, P.; Perochon, J.; Lestrade, L.; Mahillon, J.; Chandler, M. ISfinder: The Reference Centre for Bacterial Insertion Sequences. Nucleic Acids Res. 2006, 34, D32–D36. [Google Scholar] [CrossRef]
- Bengtsson-Palme, J.; Boulund, F.; Fick, J.; Kristiansson, E.; Larsson, D.G.J. Shotgun Metagenomics Reveals a Wide Array of Antibiotic Resistance Genes and Mobile Elements in a Polluted Lake in India. Front. Microbiol. 2014, 5, 648. [Google Scholar] [CrossRef]
- Kristiansson, E.; Fick, J.; Janzon, A.; Grabic, R.; Rutgersson, C.; Weijdegård, B.; Söderström, H.; Larsson, D.G.J. Pyrosequencing of Antibiotic-Contaminated River Sediments Reveals High Levels of Resistance and Gene Transfer Elements. PLoS ONE 2011, 6, e17038. [Google Scholar] [CrossRef]
- Li, W.; Zhang, G. Detection and Various Environmental Factors of Antibiotic Resistance Gene Horizontal Transfer. Environ. Res. 2022, 212, 113267. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Huang, K.; Yu, J.; Ding, C.; Wang, Z.; Zhao, C.; Yuan, H.; Wang, Z.; Wang, S.; Hu, J.; et al. Metagenomic Analysis of Bacterial Communities and Antibiotic Resistance Genes in the Eriocheir Sinensis Freshwater Aquaculture Environment. Chemosphere 2019, 224, 202–211. [Google Scholar] [CrossRef]
- Wei, Y.; Palacios Araya, D.; Palmer, K.L. Enterococcus Faecium: Evolution, Adaptation, Pathogenesis and Emerging Therapeutics. Nat. Rev. Microbiol. 2024, 22, 705–721. [Google Scholar] [CrossRef]
- Wozniak, R.A.F.; Waldor, M.K. Integrative and Conjugative Elements: Mosaic Mobile Genetic Elements Enabling Dynamic Lateral Gene Flow. Nat. Rev. Microbiol. 2010, 8, 552–563. [Google Scholar] [CrossRef]
- Allen, H.K.; Donato, J.; Wang, H.H.; Cloud-Hansen, K.A.; Davies, J.; Handelsman, J. Call of the Wild: Antibiotic Resistance Genes in Natural Environments. Nat. Rev. Microbiol. 2010, 8, 251–259. [Google Scholar] [CrossRef]
- Ji, X.; Shen, Q.; Liu, F.; Ma, J.; Xu, G.; Wang, Y.; Wu, M. Antibiotic Resistance Gene Abundances Associated with Antibiotics and Heavy Metals in Animal Manures and Agricultural Soils Adjacent to Feedlots in Shanghai; China. J. Hazard. Mater. 2012, 235–236, 178–185. [Google Scholar] [CrossRef]
- Xu, Y.; Gao, H.; Li, R.; Lou, Y.; Li, B.; Cheng, G.; Na, G. Occurrence and Distribution of Antibiotics and Antibiotic Resistance Genes from the Land to Ocean in Daliao River-Liaodong Bay, China. Mar. Environ. Res. 2024, 197, 106470. [Google Scholar] [CrossRef]
- Bearson, B.L.; Brunelle, B.W. Fluoroquinolone Induction of Phage-Mediated Gene Transfer in Multidrug-Resistant Salmonella. Int. J. Antimicrob. Agents 2015, 46, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Zhao, S.; Chen, Y.; Yang, J.; Hou, L.; Liu, M.; Yang, Y. Antibiotic Resistance Genes in Sediments of the Yangtze Estuary: From 2007 to 2019. Sci. Total Environ. 2020, 744, 140713. [Google Scholar] [CrossRef]
- Li, W.; Su, H.; Cao, Y.; Wang, L.; Hu, X.; Xu, W.; Xu, Y.; Li, Z.; Wen, G. Antibiotic Resistance Genes and Bacterial Community Dynamics in the Seawater Environment of Dapeng Cove, South China. Sci. Total Environ. 2020, 723, 138027. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, X.; Gao, L.; Kou, Y.; Wang, X.; Li, X.; He, H.; Wang, M. Composition, Distribution and Mobility Potential of the Antibiotic Resistome in Sediments from the East China Sea Revealed by Metagenomic Analysis. Microorganisms 2025, 13, 697. https://doi.org/10.3390/microorganisms13030697
Chen X, Gao L, Kou Y, Wang X, Li X, He H, Wang M. Composition, Distribution and Mobility Potential of the Antibiotic Resistome in Sediments from the East China Sea Revealed by Metagenomic Analysis. Microorganisms. 2025; 13(3):697. https://doi.org/10.3390/microorganisms13030697
Chicago/Turabian StyleChen, Xiaozhong, Long Gao, Yanxue Kou, Xiaoxuan Wang, Xintong Li, Hui He, and Min Wang. 2025. "Composition, Distribution and Mobility Potential of the Antibiotic Resistome in Sediments from the East China Sea Revealed by Metagenomic Analysis" Microorganisms 13, no. 3: 697. https://doi.org/10.3390/microorganisms13030697
APA StyleChen, X., Gao, L., Kou, Y., Wang, X., Li, X., He, H., & Wang, M. (2025). Composition, Distribution and Mobility Potential of the Antibiotic Resistome in Sediments from the East China Sea Revealed by Metagenomic Analysis. Microorganisms, 13(3), 697. https://doi.org/10.3390/microorganisms13030697