
Innovative Strategies for Combating Multidrug-Resistant Tuberculosis: Advances in Drug Delivery Systems and Treatment


Abstract
:1. Introduction
2. Peculiarities of TB and Mechanisms of MDR-TB
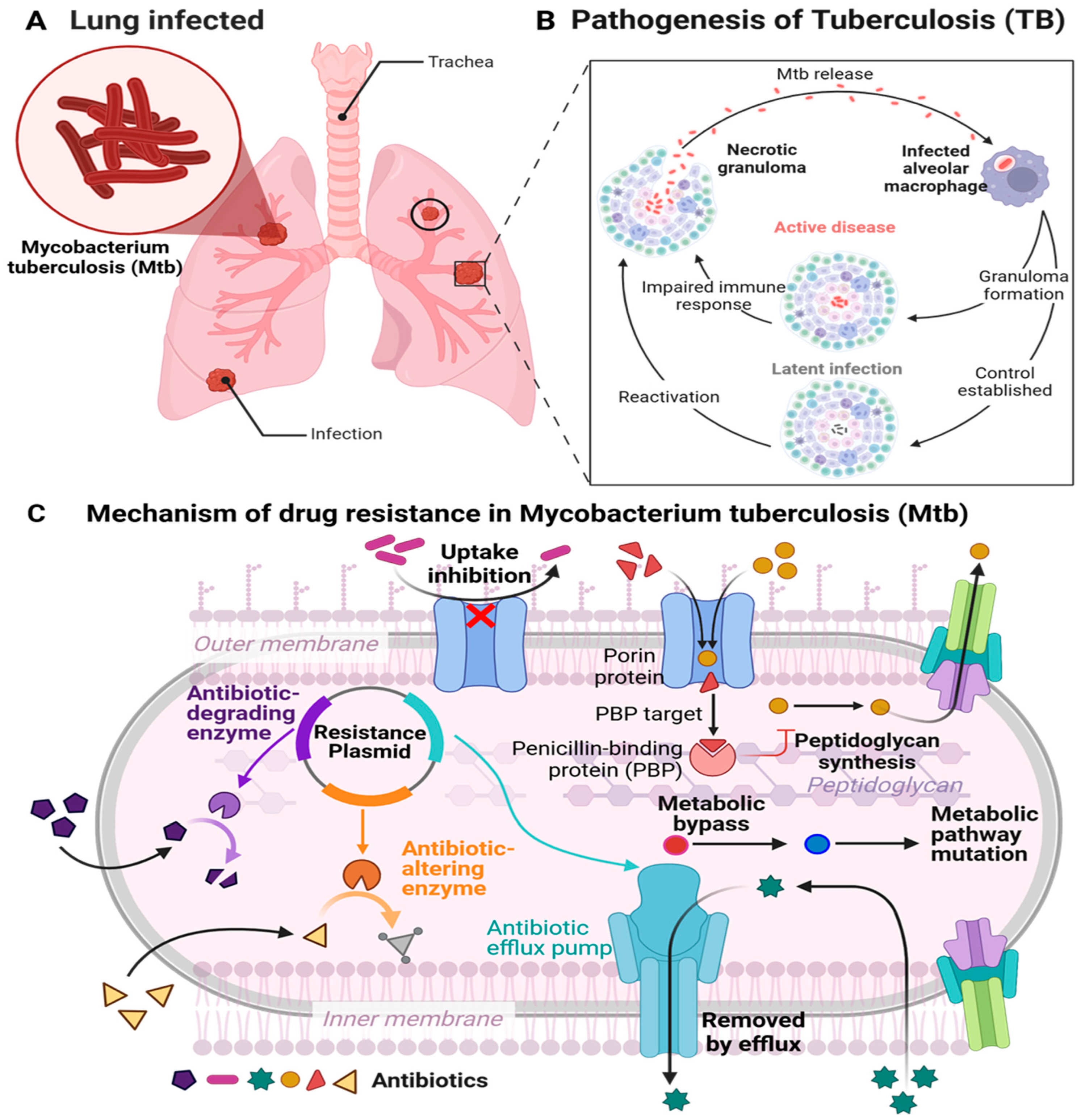
2.1. Tuberculosis Pathophysiology and Drug Resistance Mechanisms
2.1.1. Pathophysiology of TB
2.1.2. Conventional Anti-Tubercular Pharmaceuticals
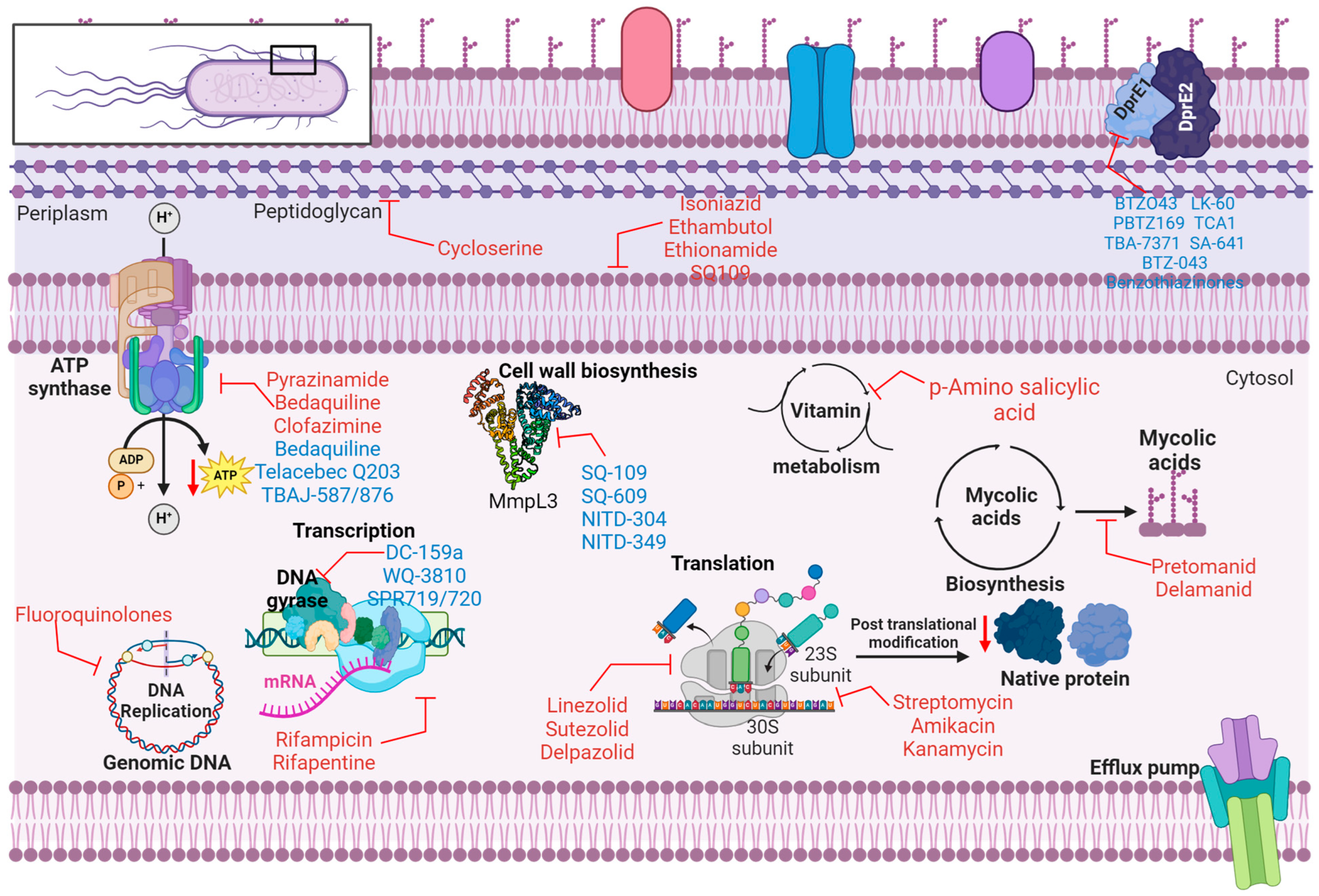
2.2. Mechanism of Action of Anti-Tubercular Agents
2.3. Mechanism of Drug Resistance (MDR-TB)
2.3.1. Genetic Mechanisms of Resistance
- Isoniazid Resistance
- Rifampicin Resistance
2.3.2. Non-Genetic Mechanisms
- Efflux Pump Mechanisms
- Phenotypic Adaptation
- Oxidative Stress and Adaptation
3. Advances in MDR-TB Treatment Strategies
3.1. Emerging Drug Regimens
Bedaquiline-Based Regimens
3.2. Precision Medicine in MDR-TB
3.2.1. Tailor-Made Treatment Regimens
3.2.2. Therapeutic Drug Monitoring (TDM)
3.2.3. Biomarker-Guided Therapy
3.3. Novel Drug Candidates
3.3.1. Delamanid
3.3.2. Pretomanid
3.4. Repurposed Drugs for MDR-TB
3.4.1. Linezolid
3.4.2. Clofazimine
3.4.3. Cycloserine
3.5. Host-Directed Therapies (HDT)
3.5.1. Metformin
3.5.2. Non-Steroidal Anti-Inflammatory Drugs (NSAID)
3.5.3. Vitamin D3
3.6. Enhancing Autophagy in Therapy
Autophagy Inducers
4. Innovative Drug Delivery Systems
4.1. Nanoparticle-Based Drug Delivery
4.1.1. Liposomal Systems
4.1.2. Metallic Nanoparticles
4.1.3. Polymeric Nanoparticles (PNP)
4.2. Gene Therapy and RNA-Based Therapy in the Treatment of MDR-TB
4.2.1. Gene Therapy
4.2.2. RNA-Based Therapy for Treatment of MDR-TB
4.3. Pulmonary Delivery Systems
Dry Powder Inhalers (DPI)
4.4. Combination Delivery Systems
Fixed-Dose Combination (FDC) Formulations
5. Challenges in the Implementation of Novel Delivery Strategies
5.1. Manufacture and Scale-Up
5.2. Safety and Biocompatibility
5.3. Regulatory Approval
5.4. Patient Adherence
5.5. Cost-Effectiveness
5.6. Stability Issues
6. Future Directions
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Raviglione, M.; Sulis, G. Tuberculosis 2015: Burden, Challenges and Strategy for Control and Elimination. Infect. Dis. Rep. 2016, 8, 6570. [Google Scholar] [CrossRef]
- WHO. Global Tuberculosis Report 2021; World Health Organization: Geneva, Switzerland, 2021. [Google Scholar]
- Song, H.-W.; Tian, J.-H.; Song, H.-P.; Guo, S.-J.; Lin, Y.-H.; Pan, J.-S. Tracking Multidrug Resistant Tuberculosis: A 30-Year Analysis of Global, Regional, and National Trends. Front. Public Health 2024, 12, 1408316. [Google Scholar] [CrossRef]
- Gygli, S.M.; Borrell, S.; Trauner, A.; Gagneux, S. Antimicrobial Resistance in Mycobacterium tuberculosis: Mechanistic and Evolutionary Perspectives. FEMS Microbiol. Rev. 2017, 41, 354–373. [Google Scholar] [CrossRef]
- Gupta, S.; Kakkar, V. Recent Technological Advancements in Tuberculosis Diagnostics—A Review. Biosens. Bioelectron. 2018, 115, 14–29. [Google Scholar] [CrossRef]
- Nurkanto, A.; Masrukhin; Tampubolon, J.C.E.; Ewaldo, M.F.; Putri, A.L.; Ratnakomala, S.; Setiawan, R.; Fathoni, A.; Palupi, K.D.; Rahmawati, Y.; et al. Exploring Indonesian Actinomycete Extracts for Anti-Tubercular Compounds: Integrating Inhibition Assessment, Genomic Analysis, and Prediction of Its Target by Molecular Docking. Heliyon 2024, 10, e35648. [Google Scholar] [CrossRef]
- Parida, K.K.; Lahiri, M.; Ghosh, M.; Dalal, A.; Kalia, N.P. P-Glycoprotein Inhibitors as an Adjunct Therapy for TB. Drug Discov. Today 2024, 29, 104108. [Google Scholar] [CrossRef]
- van Staden, D. Development of a Topical Self-Emulsifying Drug Delivery System for Optimised Delivery. Ph.D. Thesis, North West University, Potchefstroom, South Africa, 2020. [Google Scholar]
- Singh, V.; Chibale, K. Strategies to Combat Multi-Drug Resistance in Tuberculosis. Acc. Chem. Res. 2021, 54, 2361–2376. [Google Scholar] [CrossRef]
- Dheda, K.; Gumbo, T.; Maartens, G.; Dooley, K.E.; McNerney, R.; Murray, M.; Furin, J.; Nardell, E.A.; London, L.; Lessem, E.; et al. The Epidemiology, Pathogenesis, Transmission, Diagnosis, and Management of Multidrug-Resistant, Extensively Drug-Resistant, and Incurable Tuberculosis. Lancet Respir. Med. 2017, 5, 291–360. [Google Scholar] [CrossRef]
- Shah, N.S.; Auld, S.C.; Brust, J.C.M.; Mathema, B.; Ismail, N.; Moodley, P.; Mlisana, K.; Allana, S.; Campbell, A.; Mthiyane, T.; et al. Transmission of Extensively Drug-Resistant Tuberculosis in South Africa. N. Engl. J. Med. 2017, 376, 243–253. [Google Scholar] [CrossRef]
- Chandila, S.; Kaushik, H. Current trends and challenges in tuberculosis: A systemic review. World J. Pharm. Res. 2023, 12, 355–367. [Google Scholar]
- Conradie, F.; Diacon, A.H.; Ngubane, N.; Howell, P.; Everitt, D.; Crook, A.M.; Mendel, C.M.; Egizi, E.; Moreira, J.; Timm, J.; et al. Treatment of Highly Drug-Resistant Pulmonary Tuberculosis. N. Engl. J. Med. 2020, 382, 893–902. [Google Scholar] [CrossRef]
- Dartois, V.A.; Rubin, E.J. Anti-Tuberculosis Treatment Strategies and Drug Development: Challenges and Priorities. Nat. Rev. Microbiol. 2022, 20, 685–701. [Google Scholar] [CrossRef]
- Koul, A.; Arnoult, E.; Lounis, N.; Guillemont, J.; Andries, K. The Challenge of New Drug Discovery for Tuberculosis. Nature 2011, 469, 483–490. [Google Scholar] [CrossRef]
- Borah, P.; Deb, P.K.; Venugopala, K.N.; Al-Shar’i, N.A.; Singh, V.; Deka, S.; Srivastava, A.; Tiwari, V.; Mailavaram, R.P. Tuberculosis: An Update on Pathophysiology, Molecular Mechanisms of Drug Resistance, Newer Anti-TB Drugs, Treatment Regimens and Host- Directed Therapies. Curr. Top. Med. Chem. 2021, 21, 547–570. [Google Scholar] [CrossRef]
- Gordon, S.B.; Bruce, N.G.; Grigg, J.; Hibberd, P.L.; Kurmi, O.P.; Lam, K.H.; Mortimer, K.; Asante, K.P.; Balakrishnan, K.; Balmes, J.; et al. Respiratory Risks from Household Air Pollution in Low and Middle Income Countries. Lancet Respir. Med. 2014, 2, 823–860. [Google Scholar] [CrossRef]
- Suri, S.S.; Fenniri, H.; Singh, B. Nanotechnology-Based Drug Delivery Systems. J. Occup. Med. Toxicol. 2007, 2, 16. [Google Scholar] [CrossRef]
- Kia, P.; Ruman, U.; Pratiwi, A.R.; Hussein, M.Z. Innovative Therapeutic Approaches Based on Nanotechnology for the Treatment and Management of Tuberculosis. Int. J. Nanomed. 2023, 18, 1159–1191. [Google Scholar] [CrossRef]
- Lawn, S.D.; Mwaba, P.; Bates, M.; Piatek, A.; Alexander, H.; Marais, B.J.; Cuevas, L.E.; McHugh, T.D.; Zijenah, L.; Kapata, N.; et al. Advances in Tuberculosis Diagnostics: The Xpert MTB/RIF Assay and Future Prospects for a Point-of-Care Test. Lancet Infect. Dis. 2013, 13, 349–361. [Google Scholar] [CrossRef]
- Sunny, P.S. The Lessons Learned from An Active Tuberculosis Genotyping Cluster Investigation in Allegheny County. Ph.D. Thesis, University of Pittsburgh, Pittsburgh, MA, USA, 2024. [Google Scholar]
- Feldmann-Jensen, S.; O’Sullivan, T.M. Informing Adaptation with Lessons Learned from Key 21st Century Infectious Disease Outbreaks. In Current and Emerging Trends in the Management of International Disasters; Mavs Open Press: Arlington, TX, USA, 2024. [Google Scholar]
- Sparrow, A.; Smith-Torino, M.; Shamamba, S.; Chirakarhula, B.; Lwaboshi, M.; Benn, C.; Chumakov, K. A Risk Management Approach to Global Pandemics of Infectious Disease and Anti-Microbial Resistance. Trop. Med. Infect. Dis. 2024, 9, 280. [Google Scholar] [CrossRef]
- WHO. 2024a Tuberculosis (TB). Available online: https://www.who.int/news-room/fact-sheets/detail/tuberculosis (accessed on 30 January 2025).
- CDC. 2024a Clinical Overview of Latent Tuberculosis Infection. Available online: https://www.cdc.gov/tb/hcp/clinical-overview/latent-tuberculosis-infection.html (accessed on 30 January 2025).
- Gairola, A.; Benjamin, A.; Weatherston, J.D.; Cirillo, J.D.; Wu, H. Recent Developments in Drug Delivery for Treatment of Tuberculosis by Targeting Macrophages. Adv. Ther. 2022, 5, 2100193. [Google Scholar] [CrossRef] [PubMed]
- Maison, D.P. Tuberculosis Pathophysiology and Anti-VEGF Intervention. J. Clin. Tuberc. Other Mycobact. Dis. 2022, 27, 100300. [Google Scholar] [CrossRef]
- Bekraki, I.A. Liposomes-and Niosomes-Based Drug Delivery Systems for Tuberculosis Treatment. In Nanotechnology Based Approaches for Tuberculosis Treatment; Elsevier: Amsterdam, The Netherlands, 2020; pp. 107–122. ISBN 9780128198117. [Google Scholar]
- De Chastellier, C. The Many Niches and Strategies Used by Pathogenic Mycobacteria for Survival within Host Macrophages. Immunobiology 2009, 214, 526–542. [Google Scholar] [CrossRef]
- Jang, J.G.; Chung, J.H. Diagnosis and Treatment of Multidrug-Resistant Tuberculosis. Yeungnam Univ. J. Med. 2020, 37, 277–285. [Google Scholar] [CrossRef]
- Singh, A.; Gupta, A.K.; Singh, S. Molecular Mechanisms of Drug Resistance in Mycobacterium tuberculosis: Role of Nanoparticles Against Multi-Drug-Resistant Tuberculosis (MDR-TB). In NanoBioMedicine; Saxena, S.K., Khurana, S.M.P., Eds.; Springer: Singapore, 2020; pp. 285–314. ISBN 9789813298989. [Google Scholar]
- Mamatha Bhanu, L.S. Anti-Tuberculosis Drugs and Mechanisms of Action: Review. Int. J. Infect. Dis. Res. 2023, 4, 1–7. [Google Scholar]
- Khawbung, J.L.; Nath, D.; Chakraborty, S. Drug Resistant Tuberculosis: A Review. Comp. Immunol. Microbiol. Infect. Dis. 2021, 74, 101574. [Google Scholar] [CrossRef]
- Batt, J.; Khan, K. Responsible Use of Rifampin for the Treatment of Latent Tuberculosis Infection. Can. Med. Assoc. J. 2019, 191, E678–E679. [Google Scholar] [CrossRef]
- Santucci, P.; Greenwood, D.J.; Fearns, A.; Chen, K.; Jiang, H.; Gutierrez, M.G. Intracellular Localisation of Mycobacterium tuberculosis Affects Efficacy of the Antibiotic Pyrazinamide. Nat. Commun. 2021, 12, 3816. [Google Scholar] [CrossRef]
- Lee, N.; Patel, P.; Nguyen, H. Ethambutol. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Campbell, R.E.; Chen, C.H.; Edelstein, C.L. Overview of Antibiotic-Induced Nephrotoxicity. Kidney Int. Rep. 2023, 8, 2211–2225. [Google Scholar] [CrossRef]
- Sabur, N.F.; Brar, M.S.; Wu, L.; Brode, S.K. Low-Dose Amikacin in the Treatment of Multidrug-Resistant Tuberculosis (MDR-TB). BMC Infect. Dis. 2021, 21, 254. [Google Scholar] [CrossRef]
- Shibeshi, W.; Sheth, A.N.; Admasu, A.; Berha, A.B.; Negash, Z.; Yimer, G. Nephrotoxicity and Ototoxic Symptoms of Injectable Second-Line Anti-Tubercular Drugs among Patients Treated for MDR-TB in Ethiopia: A Retrospective Cohort Study. BMC Pharmacol. Toxicol. 2019, 20, 31. [Google Scholar] [CrossRef]
- Espinosa-Pereiro, J.; Sánchez-Montalvá, A.; Aznar, M.L.; Espiau, M. MDR Tuberculosis Treatment. Medicina 2022, 58, 188. [Google Scholar] [CrossRef]
- Täubel, J.; Prasad, K.; Rosano, G.; Ferber, G.; Wibberley, H.; Cole, S.T.; Van Langenhoven, L.; Fernandes, S.; Djumanov, D.; Sugiyama, A. Effects of the Fluoroquinolones Moxifloxacin and Levofloxacin on the QT Subintervals: Sex Differences in Ventricular Repolarization. J. Clin. Pharma 2020, 60, 400–408. [Google Scholar] [CrossRef]
- Chiang, C.-Y.; Van Deun, A.; Rieder, H.L. Gatifloxacin for Short, Effective Treatment of Multidrug-Resistant Tuberculosis. Int. J. Tuberc. Lung Dis. 2016, 20, 1143–1147. [Google Scholar] [CrossRef]
- Fox, G.J.; Nhung, N.V.; Cam Binh, N.; Hoa, N.B.; Garden, F.L.; Benedetti, A.; Ngoc Yen, P.; Cuong, N.K.; MacLean, E.L.; Yapa, H.M.; et al. Levofloxacin for the Prevention of Multidrug-Resistant Tuberculosis in Vietnam. N. Engl. J. Med. 2024, 391, 2304–2314. [Google Scholar] [CrossRef]
- Angula, K.T.; Legoabe, L.J.; Beteck, R.M. Chemical Classes Presenting Novel Antituberculosis Agents Currently in Different Phases of Drug Development: A 2010–2020 Review. Pharmaceuticals 2021, 14, 461. [Google Scholar] [CrossRef]
- Imran, M.; Abida; Alotaibi, N.M.; Thabet, H.K.; Alruwaili, J.A.; Asdaq, S.M.B.; Eltaib, L.; Alshehri, A.; Alsaiari, A.A.; Almehmadi, M.; et al. QcrB Inhibition as a Potential Approach for the Treatment of Tuberculosis: A Review of Recent Developments, Patents, and Future Directions. J. Infect. Public Health 2023, 16, 928–937. [Google Scholar] [CrossRef]
- Wallis, R.S.; Dawson, R.; Friedrich, S.O.; Venter, A.; Paige, D.; Zhu, T.; Silvia, A.; Gobey, J.; Ellery, C.; Zhang, Y.; et al. Mycobactericidal Activity of Sutezolid (PNU-100480) in Sputum (EBA) and Blood (WBA) of Patients with Pulmonary Tuberculosis. PLoS ONE 2014, 9, e94462. [Google Scholar] [CrossRef]
- Gao, C.; Peng, C.; Shi, Y.; You, X.; Ran, K.; Xiong, L.; Ye, T.; Zhang, L.; Wang, N.; Zhu, Y.; et al. Benzothiazinethione Is a Potent Preclinical Candidate for the Treatment of Drug-Resistant Tuberculosis. Sci. Rep. 2016, 6, 29717. [Google Scholar] [CrossRef]
- Lu, X.; Williams, Z.; Hards, K.; Tang, J.; Cheung, C.-Y.; Aung, H.L.; Wang, B.; Liu, Z.; Hu, X.; Lenaerts, A.; et al. Pyrazolo[1,5-a]Pyridine Inhibitor of the Respiratory Cytochrome Bcc Complex for the Treatment of Drug-Resistant Tuberculosis. ACS Infect. Dis. 2019, 5, 239–249. [Google Scholar] [CrossRef]
- Hoelscher, M.; Barros-Aguirre, D.; Dara, M.; Heinrich, N.; Sun, E.; Lange, C.; Tiberi, S.; Wells, C. Candidate Anti-Tuberculosis Medicines and Regimens under Clinical Evaluation. Clin. Microbiol. Infect. 2024, 30, 1131–1138. [Google Scholar] [CrossRef]
- Heinrich, N.; De Jager, V.; Dreisbach, J.; Gross-Demel, P.; Schultz, S.; Gerbach, S.; Kloss, F.; Dawson, R.; Narunsky, K.; Leonie, M.; et al. BTZ-043 Shows Good Safety and Strong Bactericidal Activity in a Combined Phase1b/2a Study in Tuberculosis Patients 2023. Available online: https://papers.ssrn.com/sol3/papers.cfm?abstract_id=4601314 (accessed on 30 January 2025).
- Miotto, P.; Zhang, Y.; Cirillo, D.M.; Yam, W.C. Drug Resistance Mechanisms and Drug Susceptibility Testing for Tuberculosis. Respirology 2018, 23, 1098–1113. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yew, W.-W. Mechanisms of Drug Resistance in Mycobacterium tuberculosis: Update 2015. Int. J. Tuberc. Lung Dis. 2015, 19, 1276–1289. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, S.; Musser, J.M. Molecular Genetic Basis of Antimicrobial Agent Resistance in Mycobacterium tuberculosis: 1998 Update. Tuber. Lung Dis. 1998, 79, 3–29. [Google Scholar] [CrossRef]
- Ali, A.; Hasan, Z.; McNerney, R.; Mallard, K.; Hill-Cawthorne, G.; Coll, F.; Nair, M.; Pain, A.; Clark, T.G.; Hasan, R. Whole Genome Sequencing Based Characterization of Extensively Drug-Resistant Mycobacterium tuberculosis Isolates from Pakistan. PLoS ONE 2015, 10, e0117771. [Google Scholar] [CrossRef]
- Kanji, A.; Hasan, R.; Ali, A.; Zaver, A.; Zhang, Y.; Imtiaz, K.; Shi, W.; Clark, T.G.; McNerney, R.; Phelan, J.; et al. Single Nucleotide Polymorphisms in Efflux Pumps Genes in Extensively Drug Resistant Mycobacterium tuberculosis Isolates from Pakistan. Tuberculosis 2017, 107, 20–30. [Google Scholar] [CrossRef]
- Louw, G.E.; Warren, R.M.; Van Helden, P.D.; Victor, T.C. Rv2629 191A/C Nucleotide Change Is Not Associated with Rifampicin Resistance in Mycobacterium tuberculosis. Clin. Chem. Lab. Med. 2009, 47, 111. [Google Scholar] [CrossRef] [PubMed]
- Ghajavand, H.; Kargarpour Kamakoli, M.; Khanipour, S.; Pourazar Dizaji, S.; Masoumi, M.; Rahimi Jamnani, F.; Fateh, A.; Yaseri, M.; Siadat, S.D.; Vaziri, F. Scrutinizing the Drug Resistance Mechanism of Multi- and Extensively-Drug Resistant Mycobacterium tuberculosis: Mutations versus Efflux Pumps. Antimicrob. Resist. Infect. Control 2019, 8, 70. [Google Scholar] [CrossRef]
- Ramón-García, S.; Martín, C.; Thompson, C.J.; Aínsa, J.A. Role of the Mycobacterium tuberculosis P55 Efflux Pump in Intrinsic Drug Resistance, Oxidative Stress Responses, and Growth. Antimicrob. Agents Chemother. 2009, 53, 3675–3682. [Google Scholar] [CrossRef]
- Kanji, A.; Hasan, R.; Hasan, Z. Efflux Pump as Alternate Mechanism for Drug Resistance in Mycobacterium tuberculosis. Indian J. Tuberc. 2019, 66, 20–25. [Google Scholar] [CrossRef]
- Machado, D.; Couto, I.; Perdigão, J.; Rodrigues, L.; Portugal, I.; Baptista, P.; Veigas, B.; Amaral, L.; Viveiros, M. Contribution of Efflux to the Emergence of Isoniazid and Multidrug Resistance in Mycobacterium tuberculosis. PLoS ONE 2012, 7, e34538. [Google Scholar] [CrossRef]
- Gengenbacher, M.; Kaufmann, S.H.E. Mycobacterium tuberculosis: Success through Dormancy. FEMS Microbiol. Rev. 2012, 36, 514–532. [Google Scholar] [CrossRef]
- Dua, K.; Rapalli, V.K.; Shukla, S.D.; Singhvi, G.; Shastri, M.D.; Chellappan, D.K.; Satija, S.; Mehta, M.; Gulati, M.; Pinto, T.D.J.A.; et al. Multi-Drug Resistant Mycobacterium tuberculosis & Oxidative Stress Complexity: Emerging Need for Novel Drug Delivery Approaches. Biomed. Pharmacother. 2018, 107, 1218–1229. [Google Scholar] [CrossRef]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive Oxygen Species in Inflammation and Tissue Injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef]
- Portevin, D.; Sukumar, S.; Coscolla, M.; Shui, G.; Li, B.; Guan, X.L.; Bendt, A.K.; Young, D.; Gagneux, S.; Wenk, M.R. Lipidomics and Genomics of Mycobacterium tuberculosis Reveal Lineage-specific Trends in Mycolic Acid Biosynthesis. MicrobiologyOpen 2014, 3, 823–835. [Google Scholar] [CrossRef]
- Vilchèze, C.; Weisbrod, T.R.; Chen, B.; Kremer, L.; Hazbón, M.H.; Wang, F.; Alland, D.; Sacchettini, J.C.; Jacobs, W.R. Altered NADH/NAD+ Ratio Mediates Coresistance to Isoniazid and Ethionamide in Mycobacteria. Antimicrob. Agents Chemother. 2005, 49, 708–720. [Google Scholar] [CrossRef]
- Awuh, J.A.; Flo, T.H. Molecular Basis of Mycobacterial Survival in Macrophages. Cell. Mol. Life Sci. 2017, 74, 1625–1648. [Google Scholar] [CrossRef]
- Jaeger, T. Peroxiredoxin Systems in Mycobacteria. In Peroxiredoxin Systems; Flohé, L., Harris, J.R., Eds.; Subcellular Biochemistry; Springer Netherlands: Dordrecht, The Netherlands, 2007; Volume 44, pp. 207–217. ISBN 9781402060502. [Google Scholar]
- Lu, J.; Holmgren, A. The Thioredoxin Antioxidant System. Free Radic. Bio. Med. 2014, 66, 75–87. [Google Scholar] [CrossRef]
- Sivaramakrishnan, S.; Ortiz De Montellano, P. The DosS-DosT/DosR Mycobacterial Sensor System. Biosensors 2013, 3, 259–282. [Google Scholar] [CrossRef]
- Wadhwa, R.; Sehgal, N.; Aggarwal, T.; Satija, S.; Mehta, M.; Gupta, G.; Chellappan, D.K.; Tambuwala, M.M.; Oliver, B.; Collet, T.; et al. Oxidative Stress and Immunological Complexities in Multidrug-Resistant Tuberculosis. In Role of Oxidative Stress in Pathophysiology of Diseases; Maurya, P.K., Dua, K., Eds.; Springer: Singapore, 2020; pp. 107–124. ISBN 9789811515675. [Google Scholar]
- Tiwari, D.; Martineau, A.R. Inflammation-Mediated Tissue Damage in Pulmonary Tuberculosis and Host-Directed Therapeutic Strategies. Semin. Immunol. 2023, 65, 101672. [Google Scholar] [CrossRef] [PubMed]
- Mackieh, R.; Al-Bakkar, N.; Kfoury, M.; Roufayel, R.; Sabatier, J.-M.; Fajloun, Z. Inhibitors of ATP Synthase as New Antibacterial Candidates. Antibiotics 2023, 12, 650. [Google Scholar] [CrossRef] [PubMed]
- Hatami, H.; Sotgiu, G.; Bostanghadiri, N.; Shafiee Dolat Abadi, S.; Mesgarpour, B.; Goudarzi, H.; Battista Migliori, G.; Javad Nasiri, M. Bedaquiline-Containing Regimens and Multidrug-Resistant Tuberculosis: A Systematic Review and Meta-Analysis. J. Bras. Pneumol. 2022, 48, e20210384. [Google Scholar] [CrossRef] [PubMed]
- Ur Rehman, O.; Fatima, E.; Ali, A.; Akram, U.; Nashwan, A.; Yunus, F. Efficacy and Safety of Bedaquiline Containing Regimens in Patients of Drug-Resistant Tuberculosis: An Updated Systematic Review and Meta-Analysis. J. Clin. Tuberc. Other Mycobact. Dis. 2024, 34, 100405. [Google Scholar] [CrossRef] [PubMed]
- Thariqulhaq, M.F.; Wahyono, T.Y.M. The effectiveness and safety of bedaquiline-containing regimens in the treatment of patients with multi-drug resistant tuberculosis (MDR-TB): A Systematic Literature Review. J. EduHealth 2023, 14, 1382–1392. [Google Scholar] [CrossRef]
- Trevisi, L.; Hernán, M.A.; Mitnick, C.D.; Khan, U.; Seung, K.J.; Rich, M.L.; Bastard, M.; Huerga, H.; Melikyan, N.; Atwood, S.A.; et al. Effectiveness of Bedaquiline Use beyond Six Months in Patients with Multidrug-Resistant Tuberculosis. Am. J. Respir. Crit. Care Med. 2023, 207, 1525–1532. [Google Scholar] [CrossRef]
- Haagsma, A.C.; Podasca, I.; Koul, A.; Andries, K.; Guillemont, J.; Lill, H.; Bald, D. Probing the Interaction of the Diarylquinoline TMC207 with Its Target Mycobacterial ATP Synthase. PLoS ONE 2011, 6, e23575. [Google Scholar] [CrossRef]
- Guo, H.; Courbon, G.M.; Bueler, S.A.; Mai, J.; Liu, J.; Rubinstein, J.L. Structure of Mycobacterial ATP Synthase Bound to the Tuberculosis Drug Bedaquiline. Nature 2021, 589, 143–147. [Google Scholar] [CrossRef]
- Krah, A.; Grüber, G.; Bond, P.J. Binding Properties of the Anti-TB Drugs Bedaquiline and TBAJ-876 to a Mycobacterial F-ATP Synthase. Curr. Res. Struct. Biol. 2022, 4, 278–284. [Google Scholar] [CrossRef]
- Giraud-Gatineau, A.; Coya, J.M.; Maure, A.; Biton, A.; Thomson, M.; Bernard, E.M.; Marrec, J.; Gutierrez, M.G.; Larrouy-Maumus, G.; Brosch, R.; et al. The Antibiotic Bedaquiline Activates Host Macrophage Innate Immune Resistance to Bacterial Infection. eLife 2020, 9, e55692. [Google Scholar] [CrossRef]
- Mbuagbaw, L.; Guglielmetti, L.; Hewison, C.; Bakare, N.; Bastard, M.; Caumes, E.; Fréchet-Jachym, M.; Robert, J.; Veziris, N.; Khachatryan, N.; et al. Outcomes of Bedaquiline Treatment in Patients with Multidrug-Resistant Tuberculosis. Emerg. Infect. Dis. 2019, 25, 936–943. [Google Scholar] [CrossRef]
- Maranchick, N.F.; Peloquin, C.A. Role of Therapeutic Drug Monitoring in the Treatment of Multi-Drug Resistant Tuberculosis. J. Clin. Tuberc. Other Mycobact. Dis. 2024, 36, 100444. [Google Scholar] [CrossRef]
- WHO. 2023b WHO Publishes Information Notes on the Use of Bedaquiline and Delamanid in Children and Adolescents with Drug-Resistant Tuberculosis. Available online: https://www.who.int/news/item/28-06-2023-who-publishes-information-notes-on-the-use-of-bedaquiline-and-delamanid-in-children-and-adolescents-with-drug-resistant-tuberculosis (accessed on 30 January 2025).
- Vanino, E.; Granozzi, B.; Akkerman, O.W.; Munoz-Torrico, M.; Palmieri, F.; Seaworth, B.; Tiberi, S.; Tadolini, M. Update of Drug-Resistant Tuberculosis Treatment Guidelines: A Turning Point. Int. J. Infect. Dis. 2023, 130, S12–S15. [Google Scholar] [CrossRef]
- Van Heeswijk, R.P.G.; Dannemann, B.; Hoetelmans, R.M.W. Bedaquiline: A Review of Human Pharmacokinetics and Drug-Drug Interactions. J. Antimicrob. Chemother. 2014, 69, 2310–2318. [Google Scholar] [CrossRef]
- Matteelli, A.; Carvalho, A.C.; Dooley, K.E.; Kritski, A. Tmc207: The First Compound of A New Class of Potent Anti-Tuberculosis Drugs. Future Microbiol. 2010, 5, 849–858. [Google Scholar] [CrossRef] [PubMed]
- Worley, M.V.; Estrada, S.J. Bedaquiline: A Novel Antitubercular Agent for the Treatment of Multidrug-Resistant Tuberculosis. Pharmacotherapy 2014, 34, 1187–1197. [Google Scholar] [CrossRef] [PubMed]
- Lange, C.; Aarnoutse, R.; Chesov, D.; Van Crevel, R.; Gillespie, S.H.; Grobbel, H.-P.; Kalsdorf, B.; Kontsevaya, I.; Van Laarhoven, A.; Nishiguchi, T.; et al. Perspective for Precision Medicine for Tuberculosis. Front. Immunol. 2020, 11, 566608. [Google Scholar] [CrossRef]
- Comas, I.; López, M.G.; Chiner-Oms, Á.; Farhat, M.R.; Ngabonziza, J.C.S.; Campos, J.; Moreno-Molina, M. Genomic Approaches for Tuberculosis Management and Control: The Challenge of Tuberculosis in the 21st Century, 3rd ed.; ERS Monograph Series; European Respiratory Society: Sheffield, UK, 2023; ISBN 9781849841696. [Google Scholar]
- Nahid, P.; Mase, S.R.; Migliori, G.B.; Sotgiu, G.; Bothamley, G.H.; Brozek, J.L.; Cattamanchi, A.; Cegielski, J.P.; Chen, L.; Daley, C.L.; et al. Treatment of Drug-Resistant Tuberculosis. An Official ATS/CDC/ERS/IDSA Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2019, 200, e93–e142. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.-S.; Lee, M.-H. Overview of Therapeutic Drug Monitoring. Korean J. Intern. Med. 2009, 24, 1. [Google Scholar] [CrossRef]
- Carranza, C.; Herrera, M.T.; Guzmán-Beltrán, S.; Salgado-Cantú, M.G.; Salido-Guadarrama, I.; Santiago, E.; Chávez-Galán, L.; Gutiérrez-González, L.H.; González, Y. A Dual Marker for Monitoring MDR-TB Treatment: Host-Derived miRNAs and M. Tuberculosis-Derived RNA Sequences in Serum. Front. Immunol. 2021, 12, 760468. [Google Scholar] [CrossRef]
- Goletti, D.; Lee, M.; Wang, J.; Walter, N.; Ottenhoff, T.H.M. Update on Tuberculosis Biomarkers: From Correlates of Risk, to Correlates of Active Disease and of Cure from Disease. Respirology 2018, 23, 455–466. [Google Scholar] [CrossRef]
- Lynch, J.; Szumowski, J. Profile of Delamanid for the Treatment of Multidrug-Resistant Tuberculosis. DDDT 2015, 677, S60923. [Google Scholar] [CrossRef]
- Nguyen, T.V.A.; Nguyen, Q.H.; Nguyen, T.N.T.; Anthony, R.M.; Vu, D.H.; Alffenaar, J.-W.C. Pretomanid Resistance: An Update on Emergence, Mechanisms and Relevance for Clinical Practice. Int. J. Antimicrob. Agents 2023, 62, 106953. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.F.K.; Laughon, B.E.; McHugh, T.D.; Lipman, M. New Drugs to Treat Difficult Tuberculous and Nontuberculous Mycobacterial Pulmonary Disease. Curr. Opin. Pulm. Med. 2019, 25, 271–280. [Google Scholar] [CrossRef]
- Keam, S.J. Pretomanid: First Approval. Drugs 2019, 79, 1797–1803. [Google Scholar] [CrossRef]
- Stover, C.K.; Warrener, P.; VanDevanter, D.R.; Sherman, D.R.; Arain, T.M.; Langhorne, M.H.; Anderson, S.W.; Towell, J.A.; Yuan, Y.; McMurray, D.N.; et al. A Small-Molecule Nitroimidazopyran Drug Candidate for the Treatment of Tuberculosis. Nature 2000, 405, 962–966. [Google Scholar] [CrossRef] [PubMed]
- Haver, H.L.; Chua, A.; Ghode, P.; Lakshminarayana, S.B.; Singhal, A.; Mathema, B.; Wintjens, R.; Bifani, P. Mutations in Genes for the F420 Biosynthetic Pathway and a Nitroreductase Enzyme Are the Primary Resistance Determinants in Spontaneous In Vitro -Selected PA-824-Resistant Mutants of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2015, 59, 5316–5323. [Google Scholar] [CrossRef]
- Mudde, S.E.; Upton, A.M.; Lenaerts, A.; Bax, H.I.; De Steenwinkel, J.E.M. Delamanid or Pretomanid? A Solomonic Judgement! J. Antimicrob. Chemother. 2022, 77, 880–902. [Google Scholar] [CrossRef] [PubMed]
- Peloquin, C.A.; Davies, G.R. The Treatment of Tuberculosis. Clin. Pharma. Ther. 2021, 110, 1455–1466. [Google Scholar] [CrossRef]
- Ginsberg, A.M.; Laurenzi, M.W.; Rouse, D.J.; Whitney, K.D.; Spigelman, M.K. Safety, Tolerability, and Pharmacokinetics of PA-824 in Healthy Subjects. Antimicrob. Agents Chemother. 2009, 53, 3720–3725. [Google Scholar] [CrossRef]
- Winter, H.; Ginsberg, A.; Egizi, E.; Erondu, N.; Whitney, K.; Pauli, E.; Everitt, D. Effect of a High-Calorie, High-Fat Meal on the Bioavailability and Pharmacokinetics of PA-824 in Healthy Adult Subjects. Antimicrob. Agents Chemother. 2013, 57, 5516–5520. [Google Scholar] [CrossRef]
- Ayodele, S.; Kumar, P.; Van Eyk, A.; Choonara, Y.E. Advances in Immunomodulatory Strategies for Host-Directed Therapies in Combating Tuberculosis. Biomed. Pharmacother. 2023, 162, 114588. [Google Scholar] [CrossRef]
- Fatima, S.; Bhaskar, A.; Dwivedi, V.P. Repurposing Immunomodulatory Drugs to Combat Tuberculosis. Front. Immunol. 2021, 12, 645485. [Google Scholar] [CrossRef]
- Kadura, S.; King, N.; Nakhoul, M.; Zhu, H.; Theron, G.; Köser, C.U.; Farhat, M. Systematic Review of Mutations Associated with Resistance to the New and Repurposed Mycobacterium tuberculosis Drugs Bedaquiline, Clofazimine, Linezolid, Delamanid and Pretomanid. J. Antimicrob. Chemother. 2020, 75, 2031–2043. [Google Scholar] [CrossRef]
- Lee, M.; Lee, J.; Carroll, M.W.; Choi, H.; Min, S.; Song, T.; Via, L.E.; Goldfeder, L.C.; Kang, E.; Jin, B.; et al. Linezolid for Treatment of Chronic Extensively Drug-Resistant Tuberculosis. N. Engl. J. Med. 2012, 367, 1508–1518. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Cocker, D.; Ryan, H.; Sloan, D.J. Linezolid for Drug-Resistant Pulmonary Tuberculosis. Cochrane Database Syst. Rev. 2019, 3, CD012836. [Google Scholar] [CrossRef] [PubMed]
- Agyeman, A.A.; Ofori-Asenso, R. Efficacy and Safety Profile of Linezolid in the Treatment of Multidrug-Resistant (MDR) and Extensively Drug-Resistant (XDR) Tuberculosis: A Systematic Review and Meta-Analysis. Ann. Clin. Microbiol. Antimicrob. 2016, 15, 41. [Google Scholar] [CrossRef]
- Ramírez-Lapausa, M.; Pascual Pareja, J.F.; Carrillo Gómez, R.; Martínez-Prieto, M.; González-Ruano Pérez, P.; Noguerado Asensio, A. Retrospective Study of Tolerability and Efficacy of Linezolid in Patients with Multidrug-Resistant Tuberculosis (1998–2014). Enferm. Infecc. Microbiol. Clín. 2016, 34, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Lange, C.; Chesov, D.; Heyckendorf, J. Clofazimine for the Treatment of Multidrug-Resistant Tuberculosis. Clin. Microbiol. Infect. 2019, 25, 128–130. [Google Scholar] [CrossRef]
- Browne, S.G.; Hogerzeil, L.M. “B 663” in the Treatment of Leprosy. Preliminary Report of a Pilot Trial. Lepr. Rev. 1962, 33, 6–10. [Google Scholar]
- Naranjo, M.F.; Kumar, A.; Ratrey, P.; Hudson, S.P. Pre-Formulation of an Additive Combination of Two Antimicrobial Agents, Clofazimine and Nisin A, to Boost Antimicrobial Activity. J. Mater. Chem. B 2024, 12, 1558–1568. [Google Scholar] [CrossRef]
- Lamprecht, D.A.; Finin, P.M.; Rahman, M.A.; Cumming, B.M.; Russell, S.L.; Jonnala, S.R.; Adamson, J.H.; Steyn, A.J.C. Turning the Respiratory Flexibility of Mycobacterium tuberculosis against Itself. Nat. Commun. 2016, 7, 12393. [Google Scholar] [CrossRef]
- Epstein, I.G.; Nair, K.G.S.; Boyd, L.J. Cycloserine, a New Antibiotic, in the Treatment of Human Pulmonary Tuberculosis: A Preliminary Report. Antibiot. Med. 1955, 1, 80–93. [Google Scholar] [PubMed]
- Li, Y.; Wang, F.; Wu, L.; Zhu, M.; He, G.; Chen, X.; Sun, F.; Liu, Q.; Wang, X.; Zhang, W. Cycloserine for Treatment of Multidrug-Resistant Tuberculosis: A Retrospective Cohort Study in China. IDR 2019, 12, 721–731. [Google Scholar] [CrossRef]
- Wang, J.; Pang, Y.; Jing, W.; Chen, W.; Guo, R.; Han, X.; Wu, L.; Yang, G.; Yang, K.; Chen, C.; et al. Efficacy and Safety of Cycloserine-Containing Regimens in the Treatment of Multidrug-Resistant Tuberculosis: A Nationwide Retrospective Cohort Study in China. Infect. Drug Resist. 2019, 12, 763–770. [Google Scholar] [CrossRef]
- Young, C.; Walzl, G.; Du Plessis, N. Therapeutic Host-Directed Strategies to Improve Outcome in Tuberculosis. Mucosal Immunol. 2020, 13, 190–204. [Google Scholar] [CrossRef] [PubMed]
- Saini, S.; Gangwar, A.; Sharma, R. Harnessing Host-Pathogen Interactions for Innovative Drug Discovery and Host-Directed Therapeutics to Tackle Tuberculosis. Microbiol. Res. 2023, 275, 127466. [Google Scholar] [CrossRef]
- Torfs, E.; Piller, T.; Cos, P.; Cappoen, D. Opportunities for Overcoming Mycobacterium tuberculosis Drug Resistance: Emerging Mycobacterial Targets and Host-Directed Therapy. Int. J. Mol. Sci. 2019, 20, 2868. [Google Scholar] [CrossRef] [PubMed]
- Khoza, L.J.; Kumar, P.; Dube, A.; Demana, P.H.; Choonara, Y.E. Insights into Innovative Therapeutics for Drug-Resistant Tuberculosis: Host-Directed Therapy and Autophagy Inducing Modified Nanoparticles. Int. J. Pharm. 2022, 622, 121893. [Google Scholar] [CrossRef] [PubMed]
- Kilinç, G.; Saris, A.; Ottenhoff, T.H.M.; Haks, M.C. Host-directed Therapy to Combat Mycobacterial Infections*. Immunol. Rev. 2021, 301, 62–83. [Google Scholar] [CrossRef]
- Singh, P.; Subbian, S. Harnessing the mTOR Pathway for Tuberculosis Treatment. Front. Microbiol. 2018, 9, 70. [Google Scholar] [CrossRef]
- Kolloli, A.; Subbian, S. Host-Directed Therapeutic Strategies for Tuberculosis. Front. Med. 2017, 4, 171. [Google Scholar] [CrossRef]
- Lee, C.; Bhakta, S. The Prospect of Repurposing Immunomodulatory Drugs for Adjunctive Chemotherapy against Tuberculosis: A Critical Review. Antibiotics 2021, 10, 91. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.; Ippolito, G.; Mfinanga, S.; Ntoumi, F.; Yeboah-Manu, D.; Vilaplana, C.; Zumla, A.; Maeurer, M. Improving Treatment Outcomes for MDR-TB—Novel Host-Directed Therapies and Personalised Medicine of the Future. Int. J. Infect. Dis. 2019, 80, S62–S67. [Google Scholar] [CrossRef]
- Naicker, N.; Sigal, A.; Naidoo, K. Metformin as Host-Directed Therapy for TB Treatment: Scoping Review. Front. Microbiol. 2020, 11, 435. [Google Scholar] [CrossRef]
- Bahlool, A.Z.; Grant, C.; Cryan, S.-A.; Keane, J.; O’Sullivan, M.P. All Trans Retinoic Acid as a Host-Directed Immunotherapy for Tuberculosis. Curr. Res. Immunol. 2022, 3, 54–72. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-J.; Han, S.K.; Park, J.H.; Lee, J.K.; Kim, D.K.; Chung, H.S.; Heo, E.Y. The Effect of Metformin on Culture Conversion in Tuberculosis Patients with Diabetes Mellitus. Korean J. Intern. Med. 2018, 33, 933–940. [Google Scholar] [CrossRef]
- Singhal, A.; Jie, L.; Kumar, P.; Hong, G.S.; Leow, M.K.-S.; Paleja, B.; Tsenova, L.; Kurepina, N.; Chen, J.; Zolezzi, F.; et al. Metformin as Adjunct Antituberculosis Therapy. Sci. Transl. Med. 2014, 6, 263ra159–3009885. [Google Scholar] [CrossRef] [PubMed]
- Yew, W.W.; Chang, K.C.; Chan, D.P.; Zhang, Y. Metformin as a Host-Directed Therapeutic in Tuberculosis: Is There a Promise? Tuberculosis 2019, 115, 76–80. [Google Scholar] [CrossRef]
- Chun, R.F.; Adams, J.S.; Hewison, M. Immunomodulation by Vitamin D: Implications for TB. Expert Rev. Clin. Pharmacol. 2011, 4, 583–591. [Google Scholar] [CrossRef]
- Deretic, V.; Saitoh, T.; Akira, S. Autophagy in Infection, Inflammation and Immunity. Nat. Rev. Immunol. 2013, 13, 722–737. [Google Scholar] [CrossRef]
- Paik, S.; Kim, J.K.; Chung, C.; Jo, E.-K. Autophagy: A New Strategy for Host-Directed Therapy of Tuberculosis. Virulence 2019, 10, 448–459. [Google Scholar] [CrossRef]
- Chai, Q.; Wang, L.; Liu, C.H.; Ge, B. New Insights into the Evasion of Host Innate Immunity by Mycobacterium tuberculosis. Cell Mol. Immunol. 2020, 17, 901–913. [Google Scholar] [CrossRef] [PubMed]
- Kimmey, J.M.; Stallings, C.L. Bacterial Pathogens versus Autophagy: Implications for Therapeutic Interventions. Trends Mol. Med. 2016, 22, 1060–1076. [Google Scholar] [CrossRef]
- Maphasa, R.E.; Meyer, M.; Dube, A. The Macrophage Response to Mycobacterium tuberculosis and Opportunities for Autophagy Inducing Nanomedicines for Tuberculosis Therapy. Front. Cell. Infect. Microbiol. 2021, 10, 618414. [Google Scholar] [CrossRef]
- Sarkar, K.; Kumar, M.; Jha, A.; Bharti, K.; Das, M.; Mishra, B. Nanocarriers for Tuberculosis Therapy: Design of Safe and Effective Drug Delivery Strategies to Overcome the Therapeutic Challenges. J. Drug Deliv. Sci. Technol. 2022, 67, 102850. [Google Scholar] [CrossRef]
- Nair, A.; Greeny, A.; Nandan, A.; Sah, R.K.; Jose, A.; Dyawanapelly, S.; Junnuthula, V.K.V.A.; Sadanandan, P. Advanced Drug Delivery and Therapeutic Strategies for Tuberculosis Treatment. J. Nanobiotechnol. 2023, 21, 414. [Google Scholar] [CrossRef]
- Miretti, M.; Juri, L.; Cosiansi, M.C.; Tempesti, T.C.; Baumgartner, M.T. Antimicrobial Effects of ZnPc Delivered into Liposomes on Multidrug Resistant (MDR)- Mycobacterium tuberculosis. ChemistrySelect 2019, 4, 9726–9730. [Google Scholar] [CrossRef]
- Sadhu, P.K.; Saisivam, S.; Debnath, S.K. Design and characterization of niosomes of ethionamide for multi drug resistance tuberculosis. World J. Pharm. Res. 2019, 8, 921–933. [Google Scholar]
- El-Ridy, M.S.; Yehia, S.A.; Kassem, M.A.-E.-M.; Mostafa, D.M.; Nasr, E.A.; Asfour, M.H. Niosomal Encapsulation of Ethambutol Hydrochloride for Increasing Its Efficacy and Safety. Drug Deliv. 2015, 22, 21–36. [Google Scholar] [CrossRef]
- Kulkarni, P.; Rawtani, D.; Barot, T. Formulation and Optimization of Long Acting Dual Niosomes Using Box-Behnken Experimental Design Method for Combinative Delivery of Ethionamide and D-Cycloserine in Tuberculosis Treatment. Colloids Surf. A Physicochem. Eng. Asp. 2019, 565, 131–142. [Google Scholar] [CrossRef]
- Yu, D.; Xu, J.; Li, R.; Zhao, J.; Li, F.; Zhai, Y.; Xue, J.; Song, H.; Yang, F.; Xu, P.; et al. Synergetic Effect of Rifampin Loaded Mussel-Inspired Silver Nanoparticles for Enhanced Antibacterial Activity Against Multidrug-Resistant Strain of Mycobacterium tuberculosis. ChemistrySelect 2021, 6, 10682–10687. [Google Scholar] [CrossRef]
- Kreytsberg, G.N.; Gracheva, I.E.; Kibrik, B.S.; Golikov, I.V. Antituberculous Effect of Silver Nanoparticles. J. Phys.Conf. Ser. 2011, 291, 012030. [Google Scholar] [CrossRef]
- Montelongo-Peralta, L.Z.; León-Buitimea, A.; Palma-Nicolás, J.P.; Gonzalez-Christen, J.; Morones-Ramírez, J.R. Antibacterial Activity of Combinatorial Treatments Composed of Transition-Metal/Antibiotics against Mycobacterium tuberculosis. Sci. Rep. 2019, 9, 5471. [Google Scholar] [CrossRef]
- Punjabi, K.; Mehta, S.; Chavan, R.; Chitalia, V.; Deogharkar, D.; Deshpande, S. Efficiency of Biosynthesized Silver and Zinc Nanoparticles Against Multi-Drug Resistant Pathogens. Front. Microbiol. 2018, 9, 2207. [Google Scholar] [CrossRef]
- Selim, A.; Elhaig, M.M.; Taha, S.A.; Nasr, E.A. Antibacterial Activity of Silver Nanoparticles against Field and Reference Strains of Mycobacterium tuberculosis, Mycobacterium bovis and Multiple-Drug-Resistant Tuberculosis Strains: -EN- -FR- Activité Antibactérienne Des Nanoparticules d’argent Contre Des Souches de Terrain et de Référence de Mycobacterium tuberculosis et Mycobacterium bovis et Des Souches Multirésistantes Aux Médicaments Contre La Tuberculose -ES- Actividad Antibacteriana de Las Nanopartículas de Plata Contra Cepas Salvajes y de Referencia de Mycobacterium tuberculosis y Mycobacterium bovis y Cepas de Tuberculosis Multirresistente. Rev. Sci. Tech. OIE 2018, 37, 823–830. [Google Scholar] [CrossRef]
- Yaghubi Kalurazi, T.; Jafari, A. Evaluation of Magnesium Oxide and Zinc Oxide Nanoparticles against Multi-Drug-Resistance Mycobacterium tuberculosis. Indian J. Tuberc. 2021, 68, 195–200. [Google Scholar] [CrossRef]
- Heidary, M.; Zaker Bostanabad, S.; Amini, S.M.; Jafari, A.; Ghalami Nobar, M.; Ghodousi, A.; Kamalzadeh, M.; Darban-Sarokhalil, D. The Anti-Mycobacterial Activity Of Ag, ZnO, And Ag- ZnO Nanoparticles Against MDR- And XDR-Mycobacterium tuberculosis. IDR 2019, 12, 3425–3435. [Google Scholar] [CrossRef]
- Vemuri, N.; Khuller, G.K.; Prabhakar, T.; Pal, N.; Gupta, P.; Gupta, U. Nanoformulations of Moxifloxacin, Econozole and Ethionamide as Novel Treatment Regimens Against MDR TB—An Experimental Study. Curr. Nanosci. 2016, 12, 110–117. [Google Scholar]
- Abdelghany, S.; Parumasivam, T.; Pang, A.; Roediger, B.; Tang, P.; Jahn, K.; Britton, W.J.; Chan, H.-K. Alginate Modified-PLGA Nanoparticles Entrapping Amikacin and Moxifloxacin as a Novel Host-Directed Therapy for Multidrug-Resistant Tuberculosis. J. Drug Deliv. Sci. Technol. 2019, 52, 642–651. [Google Scholar] [CrossRef]
- Li, G.; Li, J.; Hou, Y.; Xie, S.; Xu, J.; Yang, M.; Li, D.; Du, Y. Levofloxacin-Loaded Nanosonosensitizer as a Highly Efficient Therapy for Bacillus Calmette-Guérin Infections Based on Bacteria-Specific Labeling and Sonotheranostic Strategy. Int. J. Nanomed. 2021, 16, 6553–6573. [Google Scholar] [CrossRef]
- D’Souza, S.; Du Plessis, S.; Egieyeh, S.; Bekale, R.; Maphasa, R.; Irabin, A.; Sampson, S.; Dube, A. Physicochemical and Biological Evaluation of Curdlan-Poly(Lactic-Co-Glycolic Acid) Nanoparticles as a Host-Directed Therapy Against Mycobacterium tuberculosis. J. Pharm. Sci. 2022, 111, 469–478. [Google Scholar] [CrossRef]
- Pawde, D.M.; Viswanadh, M.K.; Mehata, A.K.; Sonkar, R.; Narendra; Poddar, S.; Burande, A.S.; Jha, A.; Vajanthri, K.Y.; Mahto, S.K.; et al. Mannose Receptor Targeted Bioadhesive Chitosan Nanoparticles of Clofazimine for Effective Therapy of Tuberculosis. Saudi Pharm. J. 2020, 28, 1616–1625. [Google Scholar] [CrossRef] [PubMed]
- Sheikhpour, M.; Delorme, V.; Kasaeian, A.; Amiri, V.; Masoumi, M.; Sadeghinia, M.; Ebrahimzadeh, N.; Maleki, M.; Pourazar, S. An Effective Nano Drug Delivery and Combination Therapy for the Treatment of Tuberculosis. Sci. Rep. 2022, 12, 9591. [Google Scholar] [CrossRef]
- Akinnawo, C.A.; Dube, A. Clinically Relevant Metallic Nanoparticles in Tuberculosis Diagnosis and Therapy. Adv. Ther. 2024, 2400189. [Google Scholar] [CrossRef]
- Hussain, A.; Singh, S.; Das, S.S.; Anjireddy, K.; Karpagam, S.; Shakeel, F. Nanomedicines as Drug Delivery Carriers of Anti-Tubercular Drugs: From Pathogenesis to Infection Control. Curr. Drug Deliv. 2019, 16, 400–429. [Google Scholar] [CrossRef] [PubMed]
- Caminero, J.A.; Lasserra, P.; Piubello, A.; Singla, R. Adverse Anti-Tuberculosis Drug Events and Their Management. Eur. Respir. Monogr. 2018, 82, 205–227. [Google Scholar]
- Kumar, M.; Virmani, T.; Kumar, G.; Deshmukh, R.; Sharma, A.; Duarte, S.; Brandão, P.; Fonte, P. Nanocarriers in Tuberculosis Treatment: Challenges and Delivery Strategies. Pharmaceuticals 2023, 16, 1360. [Google Scholar] [CrossRef]
- Li, M.; Liu, Y.; Gong, Y.; Yan, X.; Wang, L.; Zheng, W.; Ai, H.; Zhao, Y. Recent Advances in Nanoantibiotics against Multidrug-Resistant Bacteria. Nanoscale Adv. 2023, 5, 6278–6317. [Google Scholar] [CrossRef]
- Spirescu, V.A.; Chircov, C.; Grumezescu, A.M.; Andronescu, E. Polymeric Nanoparticles for Antimicrobial Therapies: An up-to-Date Overview. Polymers 2021, 13, 724. [Google Scholar] [CrossRef]
- Kamaruzzaman, N.F.; Tan, L.P.; Hamdan, R.H.; Choong, S.S.; Wong, W.K.; Gibson, A.J.; Chivu, A.; Pina, M.D.F. Antimicrobial Polymers: The Potential Replacement of Existing Antibiotics? Int. J. Mol. Sci. 2019, 20, 2747. [Google Scholar] [CrossRef]
- Cano, A.; Ettcheto, M.; Espina, M.; López-Machado, A.; Cajal, Y.; Rabanal, F.; Sánchez-López, E.; Camins, A.; García, M.L.; Souto, E.B. State-of-the-Art Polymeric Nanoparticles as Promising Therapeutic Tools against Human Bacterial Infections. J. Nanobiotechnol. 2020, 18, 156. [Google Scholar] [CrossRef]
- Pang, X.; Xiao, Q.; Cheng, Y.; Ren, E.; Lian, L.; Zhang, Y.; Gao, H.; Wang, X.; Leung, W.; Chen, X.; et al. Bacteria-Responsive Nanoliposomes as Smart Sonotheranostics for Multidrug Resistant Bacterial Infections. ACS Nano 2019, 13, acsnano.8b09336. [Google Scholar] [CrossRef]
- Sun, D.; Pang, X.; Cheng, Y.; Ming, J.; Xiang, S.; Zhang, C.; Lv, P.; Chu, C.; Chen, X.; Liu, G.; et al. Ultrasound-Switchable Nanozyme Augments Sonodynamic Therapy against Multidrug-Resistant Bacterial Infection. ACS Nano 2020, 14, 2063–2076. [Google Scholar] [CrossRef] [PubMed]
- Goodridge, H.S.; Wolf, A.J.; Underhill, D.M. Β-glucan Recognition by the Innate Immune System. Immunol. Rev. 2009, 230, 38–50. [Google Scholar] [CrossRef]
- Quesniaux, V.F.J.; Jacobs, M.; Allie, N.; Grivennikov, S.; Nedospasov, S.A.; Garcia, I.; Olleros, M.L.; Shebzukhov, Y.; Kuprash, D.; Vasseur, V.; et al. TNF in Host Resistance to Tuberculosis Infection. In Current Directions in Autoimmunity; Kollias, G., Sfikakis, P.P., Eds.; KARGER: Basel, Switzerland, 2010; Volume 11, pp. 157–179. ISBN 9783805593830. [Google Scholar]
- Kumar, P.V.; Asthana, A.; Dutta, T.; Jain, N.K. Intracellular Macrophage Uptake of Rifampicin Loaded Mannosylated Dendrimers. J. Drug Target. 2006, 14, 546–556. [Google Scholar] [CrossRef] [PubMed]
- Praphakar, R.A.; Shakila, H.; Azger Dusthackeer, V.N.; Munusamy, M.A.; Kumar, S.; Rajan, M. A Mannose-Conjugated Multi-Layered Polymeric Nanocarrier System for Controlled and Targeted Release on Alveolar Macrophages. Polym. Chem. 2018, 9, 656–667. [Google Scholar] [CrossRef]
- Karine De Sousa, A.; Rocha, J.E.; Gonçalves De Souza, T.; Sampaio De Freitas, T.; Ribeiro-Filho, J.; Melo Coutinho, H.D. New Roles of Fluoxetine in Pharmacology: Antibacterial Effect and Modulation of Antibiotic Activity. Microb. Pathog. 2018, 123, 368–371. [Google Scholar] [CrossRef]
- Munoz-Bellido, J.L.; Munoz-Criado, S.; Garcı̀a-Rodrı̀guez, J.A. Antimicrobial Activity of Psychotropic Drugs. Int. J. Antimicrob. Agents 2000, 14, 177–180. [Google Scholar] [CrossRef]
- Shao, L.; Shen, S.; Liu, H. Recent Advances in PLGA Micro/Nanoparticle Delivery Systems as Novel Therapeutic Approach for Drug-Resistant Tuberculosis. Front. Bioeng. Biotechnol. 2022, 10, 941077. [Google Scholar] [CrossRef]
- Shetty, A.; Kwas, H.; Rajhi, H.; Rangareddy, H.; Fryer, J. Revolutionizing Tuberculosis Management With Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-Cas Technology: A Comprehensive Literature Review. Cureus 2024, 16, 71697. [Google Scholar] [CrossRef]
- Rahman, K.; Jamal, M.; Chen, X.; Zhou, W.; Yang, B.; Zou, Y.; Xu, W.; Lei, Y.; Wu, C.; Cao, X.; et al. Reprogramming Mycobacterium tuberculosis CRISPR System for Gene Editing and Genome-Wide RNA Interference Screening. Genom. Proteom. Bioinf. 2022, 20, 1180–1196. [Google Scholar] [CrossRef]
- Feng, S.; Liang, L.; Shen, C.; Lin, D.; Lyu, L.; Liang, W.; Zhong, L.L.; Cook, G.M.; Doi, Y.; Chen, C. A CRISPR-Guided Mutagenic DNA Polymerase Strategy for the Detection of Antibiotic-Resistant Mutations in M. Tuberculosis: Molecular Therapy Nucleic Acids. Available online: https://www.cell.com/molecular-therapy-family/nucleic-acids/fulltext/S2162-2531(22)00173-1 (accessed on 31 January 2025).
- Tram, T.T.B.; Ha, V.T.N.; Trieu, L.P.T.; Ashton, P.M.; Crawford, E.D.; Thu, D.D.A.; Quang, N.L.; Thwaites, G.E.; Walker, T.M.; Anscombe, C.; et al. FLASH-TB: An Application of Next-Generation CRISPR to Detect Drug Resistant Tuberculosis from Direct Sputum. J. Clin. Microbiol. 2023, 61, e01634-22. [Google Scholar] [CrossRef] [PubMed]
- Bharti, R.; Roy, T.; Verma, S.; Reddy, D.S.; Shafi, H.; Verma, K.; Raman, S.K.; Pal, S.; Azmi, L.; Singh, A.K.; et al. Transient, inhaled gene therapy with gamma interferon mitigates pathology induced by host response in a mouse model of tuberculosis. Tuberculosis 2022, 1, 102198. [Google Scholar] [CrossRef]
- Yan, M.Y.; Li, S.S.; Ding, X.Y.; Guo, X.P.; Jin, Q.; Sun, Y.C.A. CRISPR-assisted nonhomologous end-joining strategy for efficient genome editing in Mycobacterium tuberculosis. MBio 2020, 11, 10–128. [Google Scholar] [CrossRef]
- Hussen, B.M.; Najmadden, Z.B.; Abdullah, S.R.; Rasul, M.F.; Mustafa, S.A.; Ghafouri-Fard, S.; Taheri, M. CRISPR/Cas9 Gene Editing: A Novel Strategy for Fighting Drug Resistance in Respiratory Disorders. Cell Commun. Signal. 2024, 22, 329. [Google Scholar] [CrossRef]
- Yan, M.-Y.; Zheng, D.; Li, S.-S.; Ding, X.-Y.; Wang, C.-L.; Guo, X.-P.; Zhan, L.; Jin, Q.; Yang, J.; Sun, Y.-C. Application of Combined CRISPR Screening for Genetic and Chemical-Genetic Interaction Profiling in Mycobacterium tuberculosis. Sci. Adv. 2022, 8, eadd5907. [Google Scholar] [CrossRef]
- Hernández-Bazán, S.; Mata-Espinosa, D.; Lozano-Ordaz, V.; Ramos-Espinosa, O.; Barrios-Payán, J.; López-Casillas, F.; Hernández Pando, R. Immune Regulatory Effect of Osteopontin Gene Therapy in a Murine Model of Multidrug Resistant Pulmonary Tuberculosis. Hum. Gene Ther. 2022, 33, 1037–1051. [Google Scholar] [CrossRef]
- Ahmed, S.; Raqib, R.; Guðmundsson, G.H.; Bergman, P.; Agerberth, B.; Rekha, R.S. Host-Directed Therapy as a Novel Treatment Strategy to Overcome Tuberculosis: Targeting Immune Modulation. Antibiotics 2020, 9, 21. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Chen, J.; Wang, P.; Li, H.; Zhou, Y.; Liu, H.; Liu, Z.; Zheng, R.; Wang, L.; Yang, H.; et al. MicroRNA-27a Controls the Intracellular Survival of Mycobacterium tuberculosis by Regulating Calcium-Associated Autophagy. Nat. Commun. 2018, 9, 4295. [Google Scholar] [CrossRef]
- Kumar, R.; Halder, P.; Sahu, S.K.; Kumar, M.; Kumari, M.; Jana, K.; Ghosh, Z.; Sharma, P.; Kundu, M.; Basu, J. Identification of a Novel Role of ESAT-6-Dependent miR-155 Induction during Infection of Macrophages with Mycobacterium tuberculosis: M. tuberculosis Induces miR-155 in Macrophages. Cell Microbiol. 2012, 14, 1620–1631. [Google Scholar] [CrossRef]
- Yan, H.; Xu, R.; Zhang, X.; Wang, Q.; Pang, J.; Zhang, X.; Chang, X.; Zhang, Y. Identifying Differentially Expressed Long Non-Coding RNAs in PBMCs in Response to the Infection of Multidrug-Resistant Tuberculosis. Infect. Drug Resist. 2018, 11, 945–959. [Google Scholar] [CrossRef]
- Li, G.; Feng, Z.; Song, H.; Wang, Y.; Zhu, L.; Li, Y. Long Non-Coding RNA Expression in PBMCs of Patients with Active Pulmonary Tuberculosis. Front. Microbiol. 2023, 14, 1257267. [Google Scholar] [CrossRef]
- Yang, X.; Bui, T.A.; Mei, H.; Aksoy, Y.A.; Deng, F.; Hutvagner, G.; Deng, W. Exploring the Potential and Challenges of CRISPR Delivery and Therapeutics for Genetic Disease Treatment. Adv. Funct. Mater. 2024, 34, 2402630. [Google Scholar] [CrossRef]
- Chen, S.; Yao, Y.; Zhang, Y.; Fan, G. CRISPR system: Discovery, development and off-target detection. Cell. Signal 2020, 70, 109577. [Google Scholar] [CrossRef]
- Winkle, M.; El-Daly, S.M.; Fabbri, M.; Calin, G.A. Noncoding RNA therapeutics—Challenges and potential solutions. Nat. Rev. Drug Discov. 2021, 20, 629–651. [Google Scholar] [CrossRef] [PubMed]
- Appaiahgari, M.B.; Vrati, S. Adenoviruses as gene/vaccine delivery vectors: Promises and pitfalls. Expert Opin. Biol. Ther. 2015, 15, 337–351. [Google Scholar] [CrossRef]
- Khare, G.; Nangpal, P.; Tyagi, A.K. Challenges and advances in TB drug discovery. In Mycobacterium tuberculosis: Molecular Infection Biology, Pathogenesis, Diagnostics and New Interventions; Hasnain, S., Ehtesham, N., Grover, S., Eds.; Springer: Singapore, 2019; pp. 463–495. [Google Scholar] [CrossRef]
- Ramachandran, S.; Prakash, P.; Mohtar, N.; Kumar, K.S.; Parumasivam, T. Review of Inhalable Nanoparticles for the Pulmonary Delivery of Anti-Tuberculosis Drugs. Pharm. Dev. Technol. 2023, 28, 978–991. [Google Scholar] [CrossRef]
- Mehta, P.; Bothiraja, C.; Kadam, S.; Pawar, A. Potential of Dry Powder Inhalers for Tuberculosis Therapy: Facts, Fidelity and Future. Artif. Cells Nanomed. Biotechnol. 2018, 46, S791–S806. [Google Scholar] [CrossRef]
- Braunstein, M.; Hickey, A.J.; Ekins, S. Why Wait? The Case for Treating Tuberculosis with Inhaled Drugs. Pharm. Res. 2019, 36, 166. [Google Scholar] [CrossRef]
- Nainwal, N.; Sharma, Y.; Jakhmola, V. Dry Powder Inhalers of Antitubercular Drugs. Tuberculosis 2022, 135, 102228. [Google Scholar] [CrossRef]
- Ranjan, R.; Devireddy, V.S.R. Prospects of Inhalable Formulations of Conventionally Administered Repurposed Drugs for Adjunctive Treatment of Drug-Resistant Tuberculosis: Supporting Evidence from Clinical Trials and Cohort Studies. J. Aerosol Med. Pulm. Drug Deliv. 2024. [Google Scholar] [CrossRef]
- Dharmadhikari, A.S.; Kabadi, M.; Gerety, B.; Hickey, A.J.; Fourie, P.B.; Nardell, E. Phase I, Single-Dose, Dose-Escalating Study of Inhaled Dry Powder Capreomycin: A New Approach to Therapy of Drug-Resistant Tuberculosis. Antimicrob. Agents Chemother. 2013, 57, 2613–2619. [Google Scholar] [CrossRef]
- McGee, B.; Dietze, R.; Hadad, D.J.; Molino, L.P.D.; Maciel, E.L.N.; Boom, W.H.; Palaci, M.; Johnson, J.L.; Peloquin, C.A. Population Pharmacokinetics of Linezolid in Adults with Pulmonary Tuberculosis. Antimicrob. Agents Chemother. 2009, 53, 3981–3984. [Google Scholar] [CrossRef]
- Rudolph, D.; Redinger, N.; Schwarz, K.; Li, F.; Hädrich, G.; Cohrs, M.; Dailey, L.A.; Schaible, U.E.; Feldmann, C. Amorphous Drug Nanoparticles for Inhalation Therapy of Multidrug-Resistant Tuberculosis. ACS Nano 2023, 17, 9478–9486. [Google Scholar] [CrossRef]
- Marwitz, F.; Hädrich, G.; Redinger, N.; Besecke, K.F.W.; Li, F.; Aboutara, N.; Thomsen, S.; Cohrs, M.; Neumann, P.R.; Lucas, H.; et al. Intranasal Administration of Bedaquiline-Loaded Fucosylated Liposomes Provides Anti-Tubercular Activity While Reducing the Potential for Systemic Side Effects. ACS Infect. Dis. 2024, 10, 3222–3232. [Google Scholar] [CrossRef]
- Paliwal, H.; Nakpheng, T.; Kumar Paul, P.; Prem Ananth, K.; Srichana, T. Development of a Self-Microemulsifying Drug Delivery System to Deliver Delamanid via a Pressurized Metered Dose Inhaler for Treatment of Multi-Drug Resistant Pulmonary Tuberculosis. Int. J. Pharm. 2024, 655, 124031. [Google Scholar] [CrossRef]
- Bahlool, A.Z.; Fattah, S.; O’Sullivan, A.; Cavanagh, B.; MacLoughlin, R.; Keane, J.; O’Sullivan, M.P.; Cryan, S.-A. Development of Inhalable ATRA-Loaded PLGA Nanoparticles as Host-Directed Immunotherapy against Tuberculosis. Pharmaceutics 2022, 14, 1745. [Google Scholar] [CrossRef]
- Mehta, P.P.; Ghoshal, D.; Pawar, A.P.; Kadam, S.S.; Dhapte-Pawar, V.S. Recent Advances in Inhalable Liposomes for Treatment of Pulmonary Diseases: Concept to Clinical Stance. J. Drug Deliv. Sci. Technol. 2020, 56, 101509. [Google Scholar] [CrossRef]
- Verma, S.; Dal, N.-J.K.; Srivastava, A.; Bharti, R.; Siva Reddy, D.V.; Sofi, H.S.; Roy, T.; Verma, K.; Raman, S.K.; Azmi, L.; et al. Inhaled Adjunct Therapy with Second-Line Drug Candidates for Dose Reduction in Chemotherapeutic Regimens for Multi-Drug-Resistant Tuberculosis. AAPS PharmSciTech 2023, 24, 130. [Google Scholar] [CrossRef]
- Makled, S.; Boraie, N.; Nafee, N. Nanoparticle-Mediated Macrophage Targeting—A New Inhalation Therapy Tackling Tuberculosis. Drug Deliv. Transl. Res. 2021, 11, 1037–1055. [Google Scholar] [CrossRef]
- Eedara, B.B.; Fan, C.; Sinha, S.; Khadka, P.; Das, S.C. Inhalable Combination Powder Formulations for Treating Latent and Multidrug-Resistant Tuberculosis: Formulation and In Vitro Characterization. Pharmaceutics 2023, 15, 2354. [Google Scholar] [CrossRef]
- Mitchell, S.L.; Carlson, E.E. Tiny Things with Enormous Impact: Nanotechnology in the Fight Against Infectious Disease. ACS Infect. Dis. 2018, 4, 1432–1435. [Google Scholar] [CrossRef]
- Sheard, D.E.; O’Brien-Simpson, N.M.; Wade, J.D.; Separovic, F. Combating Bacterial Resistance by Combination of Antibiotics with Antimicrobial Peptides. Pure Appl. Chem. 2019, 91, 199–209. [Google Scholar] [CrossRef]
- Alyami, M.H.; Dahmash, E.Z.; Ali, D.K.; Alyami, H.S.; AbdulKarim, H.; Alsudir, S.A. Novel Fluticasone Propionate and Salmeterol Fixed-Dose Combination Nano-Encapsulated Particles Using Polyamide Based on L-Lysine. Pharmaceuticals 2022, 15, 321. [Google Scholar] [CrossRef]
- Malik, M.A.; Wani, M.Y.; Hashmi, A.A. Chapter 1—Combination Therapy: Current Status and Future Perspectives. In Combination Therapy Against Multidrug Resistance; Wani, M.Y., Ahmad, A., Eds.; Academic Press: New York, NY, USA, 2020; pp. 1–38. ISBN 9780128205761. [Google Scholar]
- Wu, L.-P.; Wang, D.; Li, Z. Grand Challenges in Nanomedicine. Mater. Sci. Eng. C 2020, 106, 110302. [Google Scholar] [CrossRef]
- Buya, A.B.; Witika, B.A.; Bapolisi, A.M.; Mwila, C.; Mukubwa, G.K.; Memvanga, P.B.; Makoni, P.A.; Nkanga, C.I. Application of Lipid-Based Nanocarriers for Antitubercular Drug Delivery: A Review. Pharmaceutics 2021, 13, 2041. [Google Scholar] [CrossRef]
- Madkhali, O.A. Drug Delivery of Gelatin Nanoparticles as a Biodegradable Polymer for the Treatment of Infectious Diseases: Perspectives and Challenges. Polymers 2023, 15, 4327. [Google Scholar] [CrossRef]
- Sharma, S.; Parveen, R.; Chatterji, B.P. Toxicology of Nanoparticles in Drug Delivery. Curr. Pathobiol. Rep. 2021, 9, 133–144. [Google Scholar] [CrossRef]
- Chan, Y.; Ng, S.W.; Mehta, M.; Anand, K.; Kumar Singh, S.; Gupta, G.; Chellappan, D.K.; Dua, K. Advanced Drug Delivery Systems Can Assist in Managing Influenza Virus Infection: A Hypothesis. Med. Hypotheses 2020, 144, 110298. [Google Scholar] [CrossRef]
- Verma, N.; Arora, V.; Awasthi, R.; Chan, Y.; Jha, N.K.; Thapa, K.; Jawaid, T.; Kamal, M.; Gupta, G.; Liu, G.; et al. Recent Developments, Challenges and Future Prospects in Advanced Drug Delivery Systems in the Management of Tuberculosis. J. Drug Deliv. Sci. Technol. 2022, 75, 103690. [Google Scholar] [CrossRef]
- Kirtane, A.R.; Verma, M.; Karandikar, P.; Furin, J.; Langer, R.; Traverso, G. Nanotechnology Approaches for Global Infectious Diseases. Nat. Nanotechnol. 2021, 16, 369–384. [Google Scholar] [CrossRef]
- Satalkar, P.; Elger, B.S.; Hunziker, P.; Shaw, D. Challenges of Clinical Translation in Nanomedicine: A Qualitative Study. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 893–900. [Google Scholar] [CrossRef]
- Babel, A.; Taneja, R.; Mondello Malvestiti, F.; Monaco, A.; Donde, S. Artificial Intelligence Solutions to Increase Medication Adherence in Patients With Non-Communicable Diseases. Front. Digit. Health 2021, 3, 669869. [Google Scholar] [CrossRef]
- Sundar, S.; Chakravarty, J. Liposomal Amphotericin B and Leishmaniasis: Dose and Response. J. Glob. Infect. Dis. 2010, 2, 159. [Google Scholar] [CrossRef]
- Geethalakshmi, S.; Yadav, S. Advancements in Artificial Intelligence for the Diagnosis of Multidrug Resistance and Extensively Drug-Resistant Tuberculosis: A Comprehensive Review. Cureus 2024, 16, 60280. [Google Scholar] [CrossRef]
- Zhang, F.; Zhang, F.; Li, L.; Pang, Y. Clinical Utilization of Artificial Intelligence in Predicting Therapeutic Efficacy in Pulmonary Tuberculosis. J. Infect. Public Health 2024, 17, 632–641. [Google Scholar] [CrossRef]
- Singh, M.; Pujar, G.V.; Kumar, S.A.; Bhagyalalitha, M.; Akshatha, H.S.; Abuhaija, B.; Alsoud, A.R.; Abualigah, L.; Beeraka, N.M.; Gandomi, A.H. Evolution of Machine Learning in Tuberculosis Diagnosis: A Review of Deep Learning-Based Medical Applications. Electronics 2022, 11, 2634. [Google Scholar] [CrossRef]
- Olawade, D.B.; Eberhardt, J.; David-Olawade, A.C.; Balogun, M.A.; Bolarinwa, O.A.; Esan, D.T. Transforming Multidrug-Resistant Tuberculosis Care: The Potentials of Telemedicine in Resource-Limited Settings. Health Sci. Rev. 2024, 12, 100185. [Google Scholar] [CrossRef]
- Yadav, S.; Jeyaraman, N.; Jeyaraman, M.; Rawal, G. Artificial Intelligence in Tuberculosis Diagnosis: Revolutionizing Detection and Treatment. Indian J. Immunol. Respir. Med. 2024, 9, 85–87. [Google Scholar] [CrossRef]
- Patel, M.N.; Patel, A.J.; Nandpal, M.N.; Raval, M.A.; Patel, R.J.; Patel, A.A.; Paudel, K.R.; Hansbro, P.M.; Singh, S.K.; Gupta, G.; et al. Advancing against Drug-Resistant Tuberculosis: An Extensive Review, Novel Strategies and Patent Landscape. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2024, 398, 2127–2150. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Drug Name | Mechanism of Action | Target | Mode of Administration | Key Notes | Refs |
|---|---|---|---|---|---|
| Isoniazid (INH) | Inhibits mycolic acid synthesis | Cell wall | Oral | Effective for active TB; resistance due to KatG mutations | [32,33] |
| Rifampin (RIF) | Inhibits RNA polymerase | RNA synthesis | Oral | Active against latent and active TB | [32,34] |
| Pyrazinamide (PZA) | Disrupts mycobacterial membrane metabolism | Fatty acid synthase pathway | Oral | Effective in acidic environments (e.g., phagolysosomes) | [27,35] |
| Ethambutol (EMB) | Inhibits arabinosyl transferase | Cell wall | Oral | Used to prevent resistance to other drugs | [31,36] |
| Streptomycin (STREP) | Inhibits protein synthesis (30S ribosome) | Ribosome | Intramuscular | Aminoglycoside requires injection; nephrotoxicity risk | [31,37] |
| Amikacin (AMI) | Inhibits protein synthesis (30S ribosome) | Ribosome | Intravenous | Used for MDR-TB; nephrotoxicity and ototoxicity risks | [32,38] |
| Capreomycin (CAP) | Inhibits protein synthesis | Ribosome | Intramuscular | Effective for MDR-TB; injectable; toxicity concerns | [31,39,40] |
| Kanamycin (KAN) | Inhibits protein synthesis (30S ribosome) | Ribosome | Intravenous | Aminoglycoside: alternative to amikacin | [30,31] |
| Moxifloxacin (MOX) | Inhibits DNA gyrase | DNA replication | Oral/Intravenous | More potent fluoroquinolone; risk of electrocardiographic QT interval prolongation | [31,40,41] |
| Gatifloxacin (GAT) | Inhibits DNA gyrase | DNA replication | Oral | Less commonly used; associated with glycemic changes | [31,42] |
| Levofloxacin (LEV) | Inhibits DNA gyrase | DNA replication | Oral | Fluoroquinolone is effective in resistance settings | [31,43] |
| Ofloxacin (OFL) | Inhibits DNA gyrase | DNA replication | Oral | Older fluoroquinolone; declining use | [31] |
| P-aminosalicylic acid (PAS) | Inhibits folate metabolism | Metabolism | Oral | Gastrointestinal side effects limit usage | [31] |
| Prothionamide (PTA) | Inhibits mycolic acid synthesis | Cell wall | Oral | Like Ethionamide, used for MDR-TB | [40] |
| Terizidone (TZD) | Inhibits cell wall synthesis | Cell wall | Oral | Alternative to cycloserine; less neurotoxic | [40] |
| Cycloserine (CYS) | Inhibits cell wall synthesis | Cell wall | Oral | Central nervous system toxicity limits the use | [32,40] |
| Ethionamide (ETH) | Inhibits mycolic acid synthesis | Cell wall | Oral | Used for MDR-TB; gastrointestinal side effects | [32,40] |
| Bedaquiline (BDQ) | Inhibits ATP synthase | Cell wall | Oral | Reserved for MDR/XDR-TB; QT prolongation risk | [27,30,40] |
| Delamanid (DLM) | Inhibits mycolic acid synthesis | Cell wall | Oral | Used for MDR/XDR-TB; well-tolerated; alternative to bedaquiline | [30,40] |
| Pretomanid (PA-824) | Generates reactive nitrogen species | Cell respiration | Oral | Effective in combination therapy | [40,44] |
| Linezolid (LZD) | Inhibits protein synthesis (50S ribosome) | Ribosome | Oral/Intravenous | Significant adverse effects; used in refractory TB | [30,40] |
| Clofazimine (CFZ) | Generates reactive oxygen species | DNA | Oral | Also used for leprosy, lipophilic compound | [32,40] |
| Rifapentine (RFT) | Inhibits RNA polymerase | RNA synthesis | Oral | Longer half-life; used in shorter-course treatments | [27] |
| Drug Name | Mechanism of Action | Target | Route | Stage | Key Notes | Refs |
|---|---|---|---|---|---|---|
| Telacebec (Q203) | Inhibits cytochrome bc1 complex | Respiration | Oral | Phase II | Promising for MDR-TB: reduces bacterial burden | [45] |
| Sutezolid (PNU-100480) | Inhibits protein synthesis (50S ribosome) | Ribosome | Oral | Phase II | Improved safety profile compared with Linezolid | [44,46] |
| Benzothiazinones | Inhibits DprE1 enzyme | Cell wall | Oral | Preclinical | Effective in drug-resistant TB strains | [47] |
| TBA-7371 | Inhibits decaprenylphosphoryl-beta-D-ribose 2-epimerase | Cell wall | Oral | Phase I | Novel mechanism; active against MDR-TB | [48,49] |
| BTZ-043 | Inhibits DprE1 enzyme | Cell wall | Oral | Phase II | Promising preclinical results | [50] |
| SQ109 | Inhibits mycobacterial cell wall biosynthesis | Cell wall | Oral | Phase II | Synergistic with other TB drugs | [44,50] |
| Drug Delivery System | Active Agent | Target | Refs |
|---|---|---|---|
| Liposomes | Zn(II) phthalocyanine (ZnPc) | (ATCC 27294) Mtb and MDR-TB (9037R) | [140] |
| Niosomes | Ethionamide | MDR-TB | [141] |
| Niosomes | Ethionamide | Mtb (H37RV) | [142] |
| Niosomes | Lipophilic ETH and hydrophilic D-Cycloserine | Mycobacterium Smegmatis | [143] |
| Polydopamine-coated silver nanoparticles | Rifampin | Multidrug-resistant strain of Mtb | [144] |
| Silver nanoparticles | Isoniazid | Resistant TB | [145] |
| Silver nanoparticles | Isoniazid | Clinical strain of resistant Mtb | [146] |
| Silver nanoparticles (AgNP) and zinc nanoparticles (ZnNP) | AgNP and ZnNP | Mtb and an MDR strain | [147] |
| Silver nanoparticles | AgNP | Mycobacterium bovis and Mtb H37Rv | [148] |
| Magnesium oxide nanoparticles (MnONP) and zinc oxide nanoparticles (ZnONP) | MnONP and ZnONP | H37Rv Mtb and MDR-Mtb | [149] |
| Silver nanoparticles and zinc oxide nanoparticles | AgNP and ZnONP | MDR and XDR-Mtb | [150] |
| PLGA nanoparticles | Moxifloxacin, econazole, and ethionamide | MDR-TB-infected mice | [151] |
| Alginate modified-PLGA nanoparticles | Amikacin and moxifloxacin | MDR-TB-Mtb-infected macrophages | [152] |
| PLGA-PEG Nanoparticle conjugated with the BM2 aptamer | Levofloxacin | Mtb model (Bacillus Calmette-Guérin bacteria (BCG)) | [153] |
| Curdlan-functionalized PLGA Nanoparticles | Curdlan | Viable Mtb and MDR-TB | [154] |
| Mannose receptor-targeted bioadhesive chitosan Nanoparticles | Clofazimine | H37Rv Mtb strain | [155] |
| Multi-walled carbon nanotube nanofluid | Isoniazid and fluoxetine | Clinical strains of Mtb | [156]. |
| Active Agent | Inhalation Formulation | Dosage form or Inhalation Device | Target | Refs |
|---|---|---|---|---|
| Bedaquiline and 1,3-benzothiazin-4-one 043 (BTZ) | Amorphous nanoparticles | Nebulization/nebulizer | Granulomas in Mtb-infected lungs | [200] |
| Bedaquiline | Fucosylated and nonfucosylated liposomes | Nebulization/nebulizer | Mtb-burden lung | [201] |
| Delamanid | Self-microemulsifying drug delivery system (SMEDDS) | Pressurized metered dose inhalation/inhaler | MDR-pulmonary tuberculosis | [202] |
| All trans-retinoic acid | PLGA nanoparticles | Nebulization/nebulizer | H37Ra avirulent Mtb | [203] |
| Sutezolid | Biodegradable polymer poly(L-lactide)-polymeric particles | Dry Powder inhalation/Inhalers | Mtb-burden lung and spleen | [205] |
| Linezolid | Non-structured lipid carriers (NLC) and microparticles | Dry Powder inhalation/Inhalers | Mtb-infected macrophages | [206] |
| Pretomanid, pyrazinamide and moxifloxacin | Spray-dried L-leucine powder | Dry Powder inhalation/Inhalers | Active, latent, and resistant Mtb-infected alveolar regions of the lung | [207] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Omoteso, O.A.; Fadaka, A.O.; Walker, R.B.; Khamanga, S.M. Innovative Strategies for Combating Multidrug-Resistant Tuberculosis: Advances in Drug Delivery Systems and Treatment. Microorganisms 2025, 13, 722. https://doi.org/10.3390/microorganisms13040722
Omoteso OA, Fadaka AO, Walker RB, Khamanga SM. Innovative Strategies for Combating Multidrug-Resistant Tuberculosis: Advances in Drug Delivery Systems and Treatment. Microorganisms. 2025; 13(4):722. https://doi.org/10.3390/microorganisms13040722
Chicago/Turabian StyleOmoteso, Omobolanle A., Adewale O. Fadaka, Roderick B. Walker, and Sandile M. Khamanga. 2025. "Innovative Strategies for Combating Multidrug-Resistant Tuberculosis: Advances in Drug Delivery Systems and Treatment" Microorganisms 13, no. 4: 722. https://doi.org/10.3390/microorganisms13040722
APA StyleOmoteso, O. A., Fadaka, A. O., Walker, R. B., & Khamanga, S. M. (2025). Innovative Strategies for Combating Multidrug-Resistant Tuberculosis: Advances in Drug Delivery Systems and Treatment. Microorganisms, 13(4), 722. https://doi.org/10.3390/microorganisms13040722