
Automation of RNA-Seq Sample Preparation and Miniaturized Parallel Bioreactors Enable High-Throughput Differential Gene Expression Studies


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Strain
2.2. Media
2.3. Parallel Stirred-Tank Bioreactors
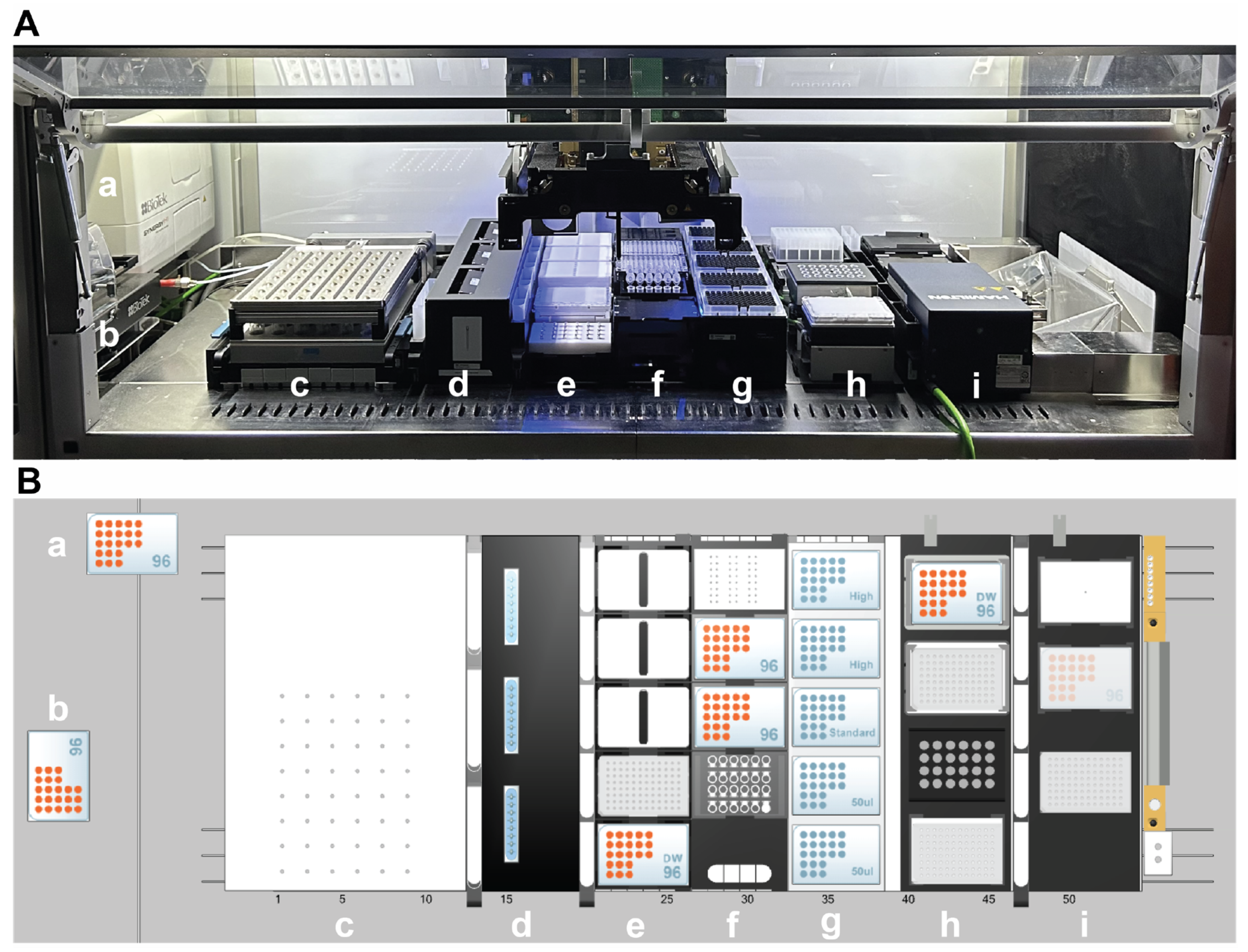
2.4. Automated RNA-Seq Sample Preparation Workflow
2.5. RNA-Seq Data Acquisition and Analysis
2.6. Data Availability
3. Results
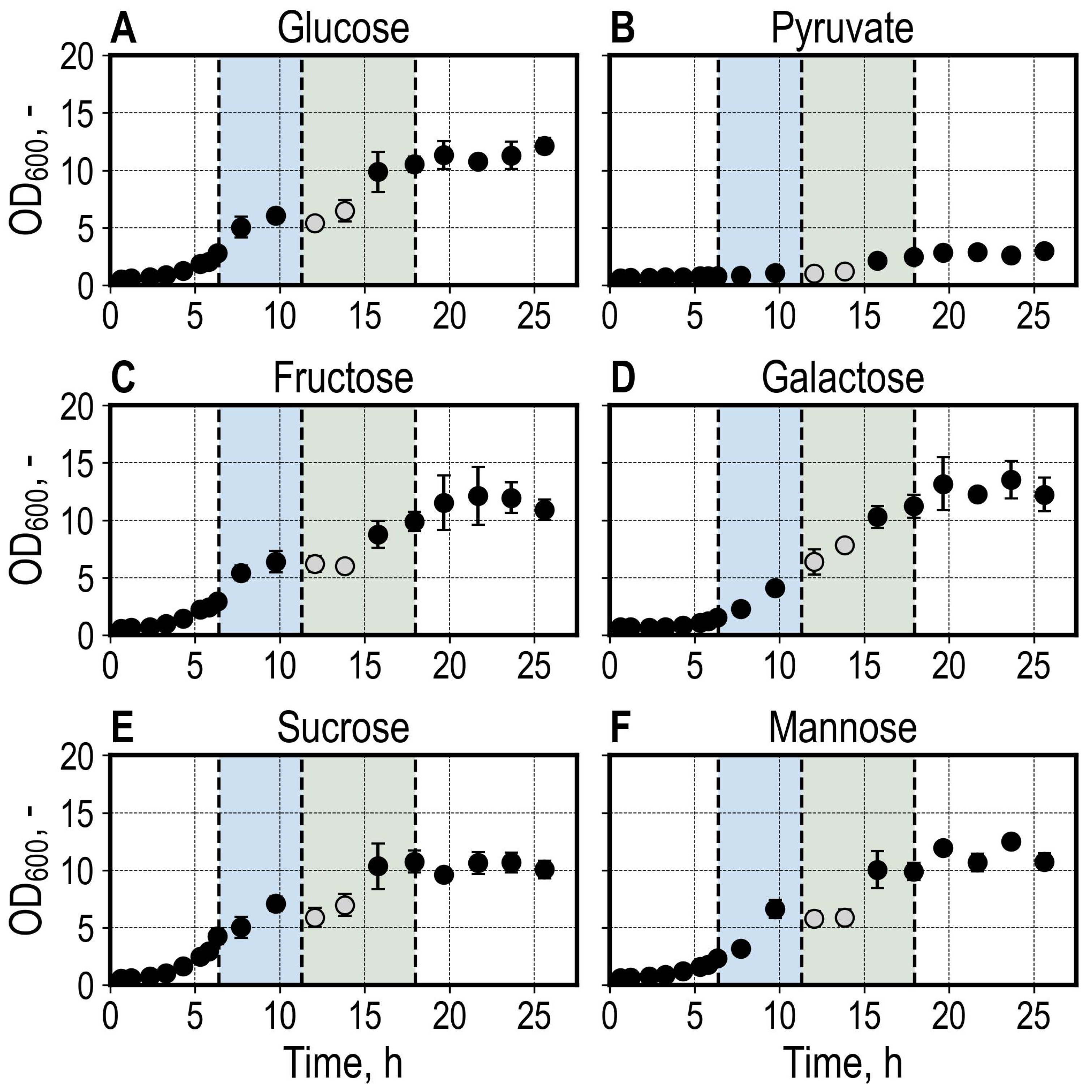
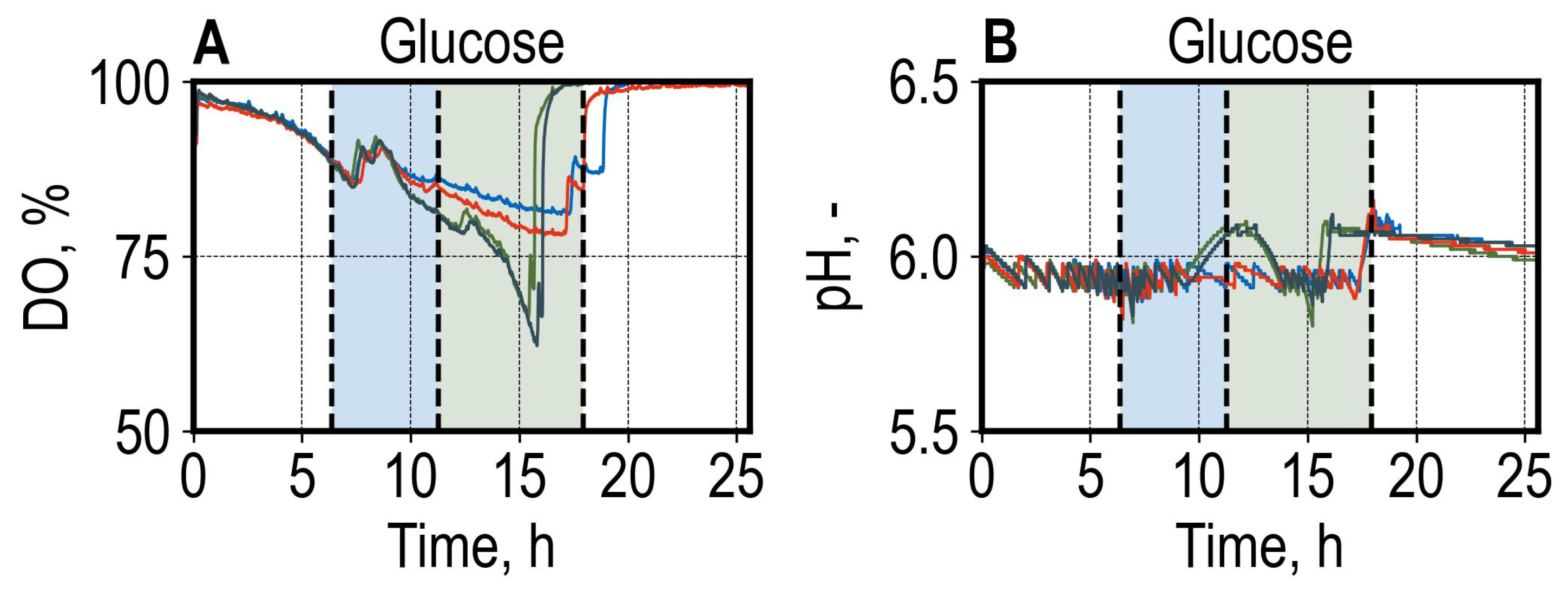
3.1. Batch Growth of Saccharomyces cerevisiae Utilizing Different Carbon Sources
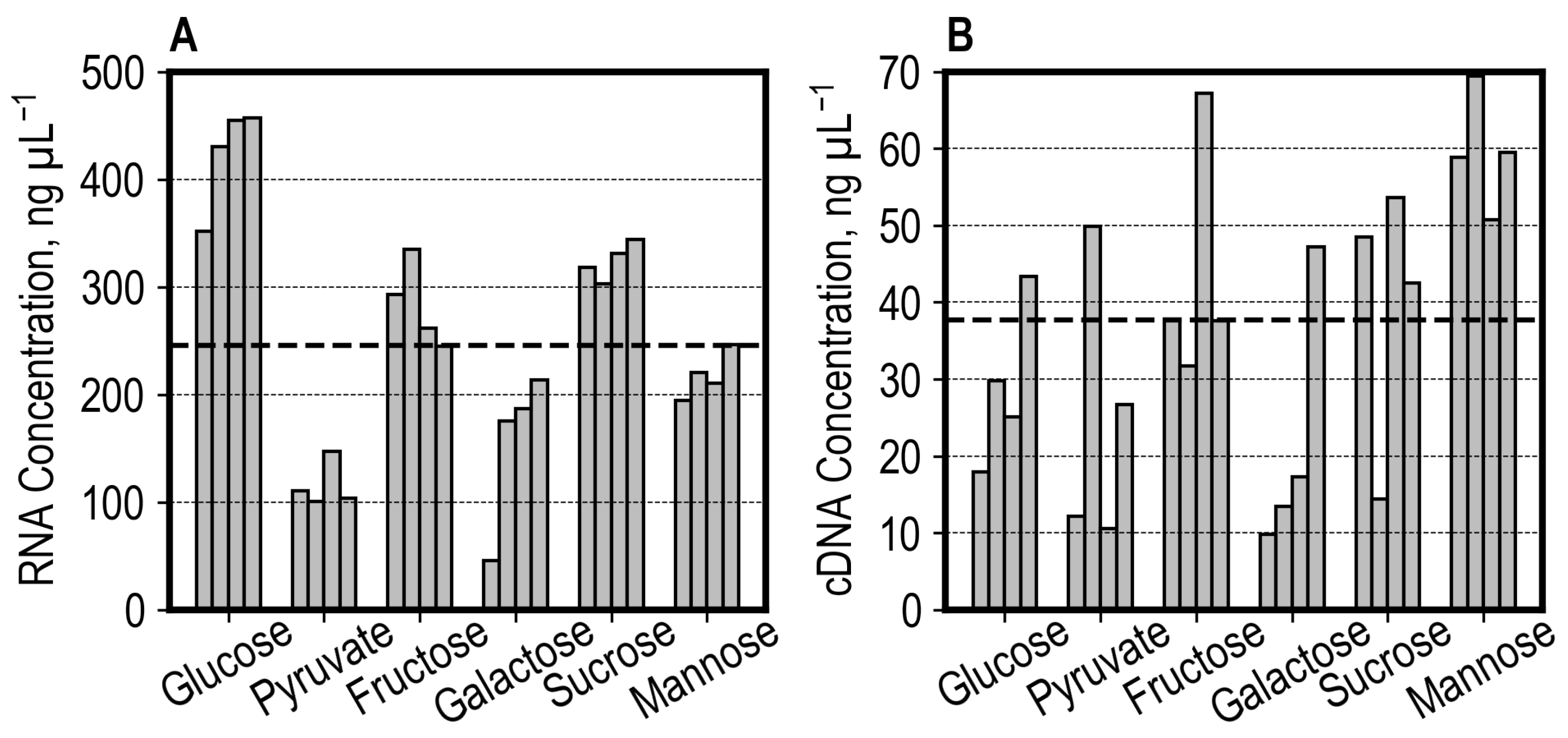
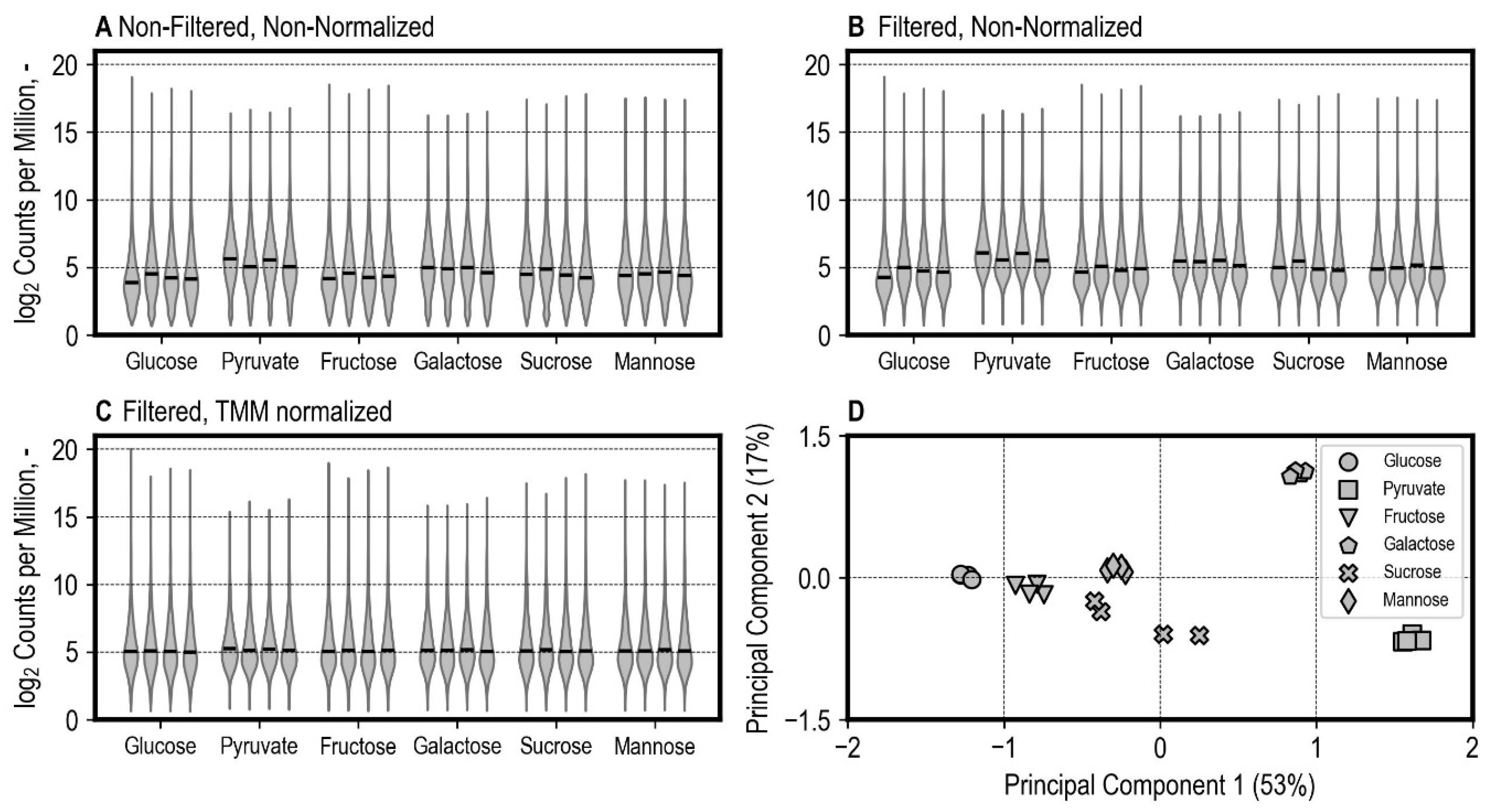
3.2. Automated High-Throughput RNA-Seq Sample Preparation
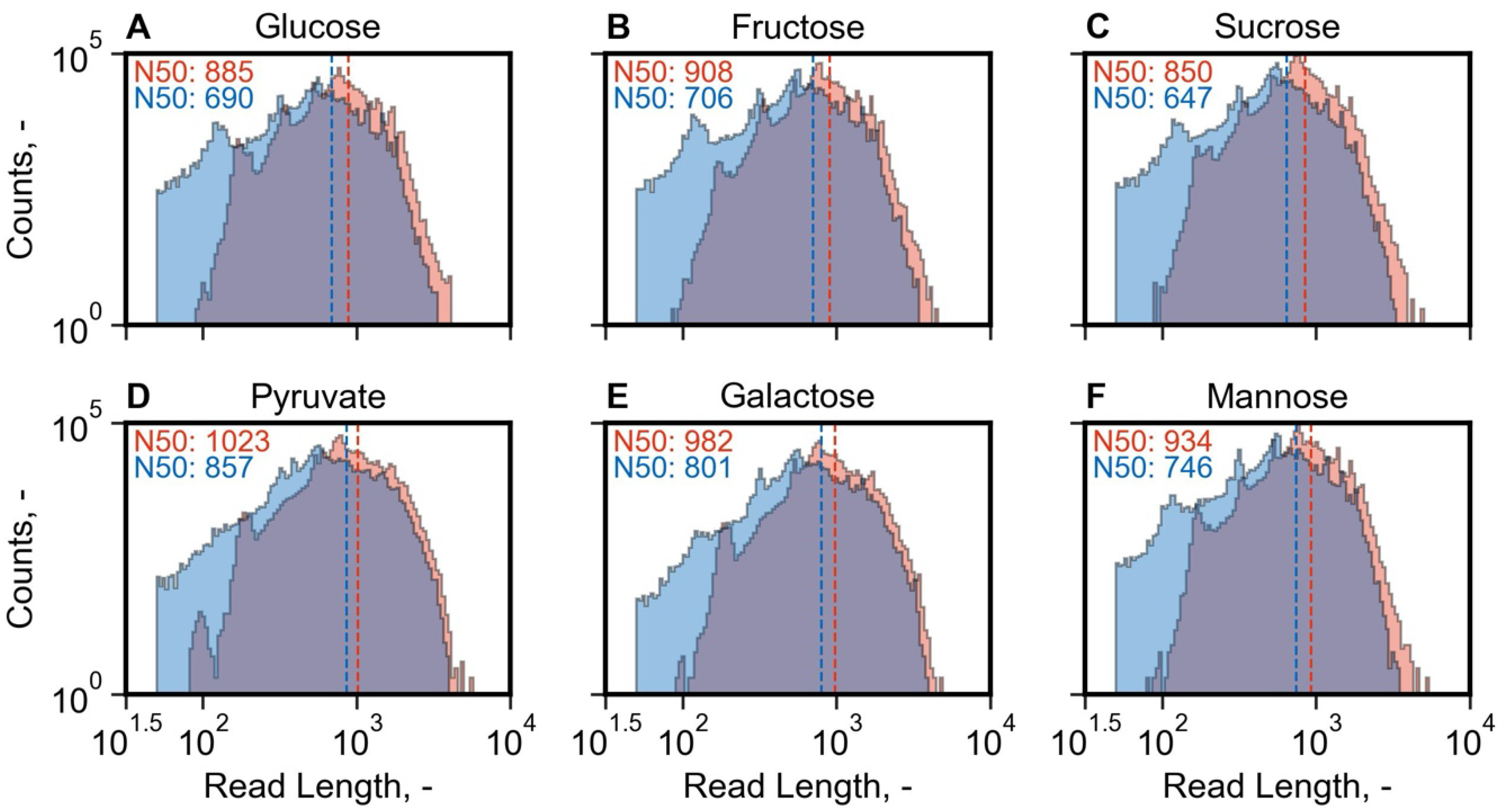
3.3. Off-Line Nanopore Sequencing
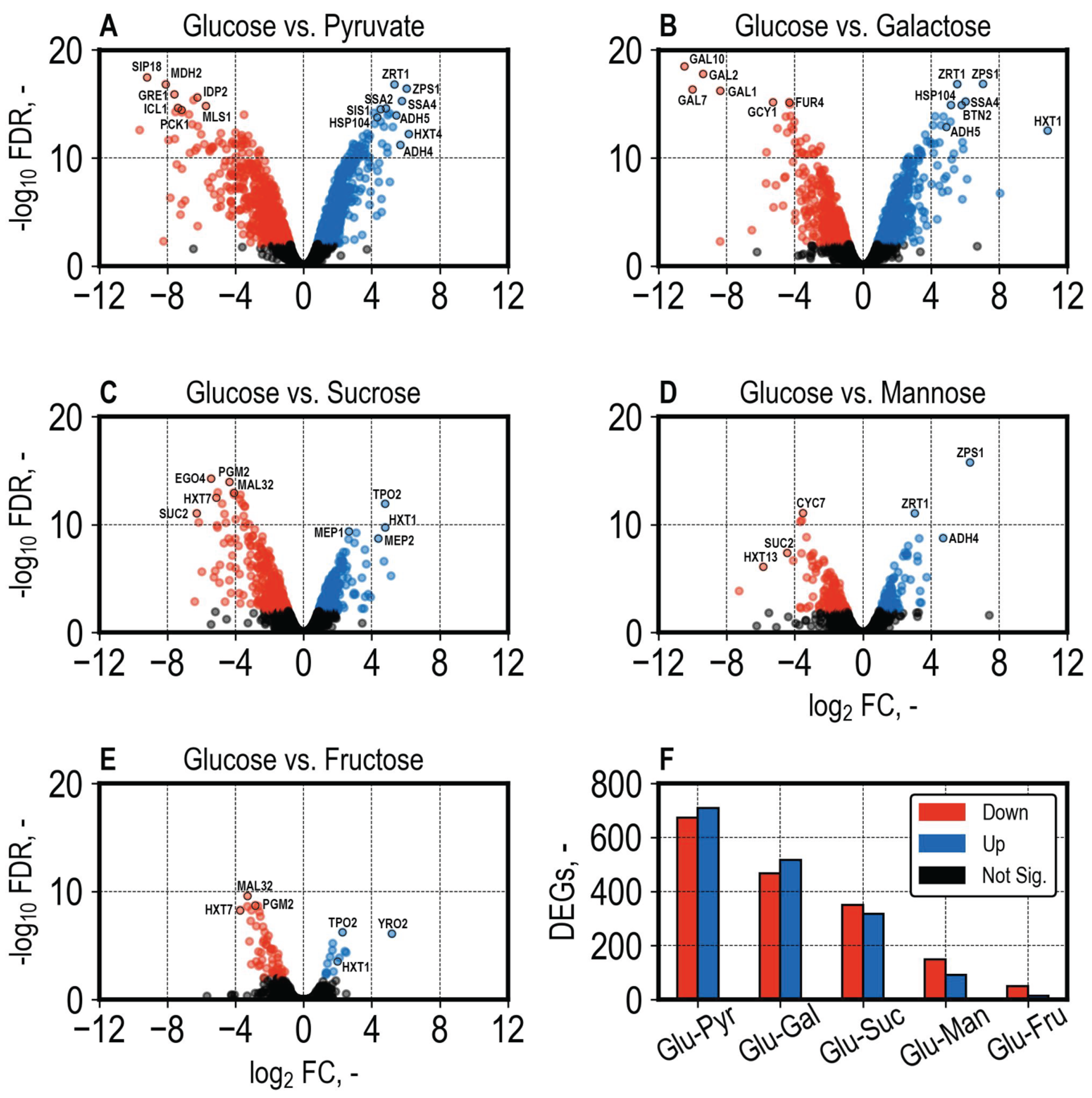
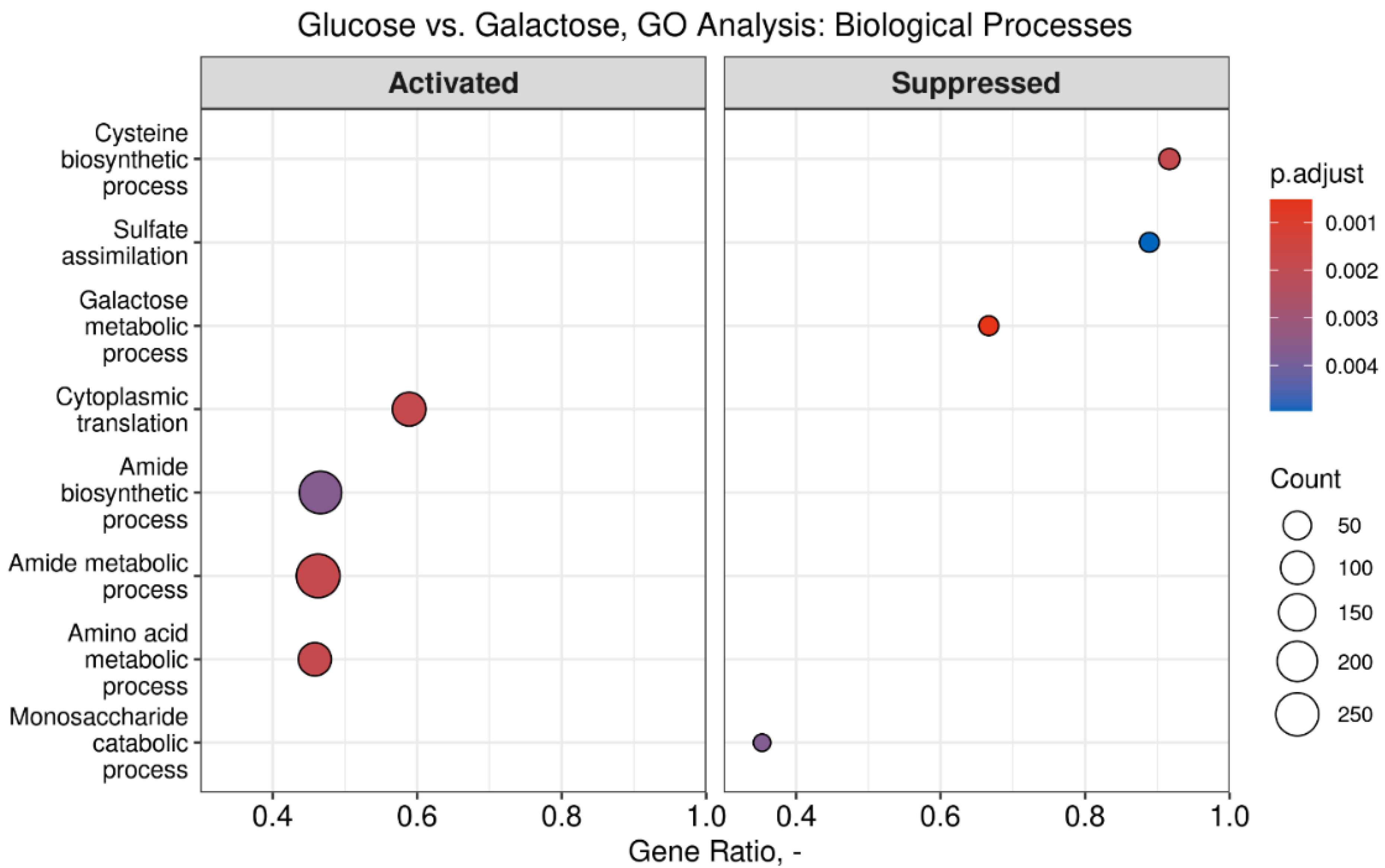
3.4. Differential Gene Expression Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hemmerich, J.; Noack, S.; Wiechert, W.; Oldiges, M. Microbioreactor Systems for Accelerated Bioprocess Development. Biotechnol. J. 2018, 13, e1700141. [Google Scholar] [CrossRef] [PubMed]
- Von den Eichen, N.; Bromig, L.; Sidarava, V.; Marienberg, H.; Weuster-Botz, D. Automated Multi-Scale Cascade of Parallel Stirred-Tank Bioreactors for Fast Protein Expression Studies. J. Biotechnol. 2021, 332, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Puskeiler, R.; Kaufmann, K.; Weuster-Botz, D. Development, Parallelization, and Automation of a Gas-Inducing Milliliter-Scale Bioreactor for High-Throughput Bioprocess Design (HTBD). Biotechnol. Bioeng. 2005, 89, 512–523. [Google Scholar] [CrossRef] [PubMed]
- Kusterer, A.; Krause, C.; Kaufmann, K.; Arnold, M.; Weuster-Botz, D. Fully Automated Single-Use Stirred-Tank Bioreactors for Parallel Microbial Cultivations. Bioprocess. Biosyst. Eng. 2008, 31, 207–215. [Google Scholar] [CrossRef]
- Schmideder, A.; Hensler, S.; Lang, M.; Stratmann, A.; Giesecke, U.; Weuster-Botz, D. High-Cell-Density Cultivation and Recombinant Protein Production with Komagataella pastoris in Stirred-Tank Bioreactors from Milliliter to Cubic Meter Scale. Process Biochem. 2016, 51, 177–184. [Google Scholar] [CrossRef]
- von den Eichen, N.; Osthege, M.; Dolle, M.; Bromig, L.; Wiechert, W.; Oldiges, M.; Weuster-Botz, D. Control of Parallelized Bioreactors II: Probabilistic Quantification of Carboxylic Acid Reductase Activity for Bioprocess Optimization. Bioprocess. Biosyst. Eng. 2022, 45, 1939–1954. [Google Scholar] [CrossRef]
- Janzen, N.H.; Schmidt, M.; Krause, C.; Weuster-Botz, D. Evaluation of Fluorimetric pH Sensors for Bioprocess Monitoring at Low pH. Bioprocess. Biosyst. Eng. 2015, 38, 1685–1692. [Google Scholar] [CrossRef]
- Haby, B.; Hans, S.; Anane, E.; Sawatzki, A.; Krausch, N.; Neubauer, P.; Bournazou, M.N.C. Integrated Robotic Mini Bioreactor Platform for Automated, Parallel Microbial Cultivation with Online Data Handling and Process Control. SLAS Technol. 2019, 24, 569–582. [Google Scholar] [CrossRef]
- Benner, P.; Effenberger, S.; Franzgrote, L.; Kurzrock-Wolf, T.; Kress, K.; Weuster-Botz, D. Contact-Free Infrared OD Measurement for Online Monitoring of Parallel Stirred-Tank Bioreactors up to High Cell Densities. Biochem. Eng. J. 2020, 164, 107749. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A Revolutionary Tool for Transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Park, S.T.; Kim, J. Trends in Next-Generation Sequencing and a New Era for Whole Genome Sequencing. Int. Neurourol. J. 2016, 20 (Suppl. S2), S76–S83. [Google Scholar] [CrossRef] [PubMed]
- Levy, S.E.; Myers, R.M. Advancements in Next-Generation Sequencing. Annu. Rev. Genom. Hum. Genet. 2016, 17, 95–115. [Google Scholar] [CrossRef] [PubMed]
- Hör, J.; Gorski, S.A.; Vogel, J. Bacterial RNA Biology on a Genome Scale. Mol. Cell 2018, 70, 785–799. [Google Scholar] [CrossRef] [PubMed]
- Stark, R.; Grzelak, M.; Hadfield, J. RNA Sequencing: The Teenage Years. Nat. Rev. Genet. 2019, 20, 631–656. [Google Scholar] [CrossRef]
- Jain, M.; Olsen, H.E.; Paten, B.; Akeson, M. The Oxford Nanopore Minion: Delivery of Nanopore Sequencing to the Genomics Community. Genome Biol. 2016, 17, 239. [Google Scholar]
- Jenjaroenpun, P.; Wongsurawat, T.; Pereira, R.; Patumcharoenpol, P.; Ussery, D.W.; Nielsen, J.; Nookaew, I. Complete Genomic and Transcriptional Landscape Analysis Using Third-Generation Sequencing: A Case Study of Saccharomyces cerevisiae CEN.PK113-7D. Nucleic Acids Res. 2018, 46, e38. [Google Scholar] [CrossRef]
- Grünberger, F.; Ferreira-Cerca, S.; Grohmann, D. Nanopore Sequencing of RNA and cDNA Molecules in Escherichia coli. RNA 2022, 28, 400–417. [Google Scholar] [CrossRef]
- Rodger, G.; Lipworth, S.; Barrett, L.; Oakley, S.; Crook, D.W.; Eyre, D.W.; Stoesser, N. Comparison of Direct cDNA and PCR-cDNA Nanopore Sequencing of RNA from Escherichia coli Isolates. Microb. Genom. 2024, 10, 001296. [Google Scholar] [CrossRef]
- Parapouli, M.; Vasileiadis, A.; Afendra, A.S.; Hatziloukas, E. Saccharomyces cerevisiae and Its Industrial Applications. AIMS Microbiol. 2020, 6, 1–31. [Google Scholar] [CrossRef]
- Mohd Azhar, S.H.; Abdulla, R.; Jambo, S.A.; Marbawi, H.; Gansau, J.A.; Faik, A.A.M.; Rodrigues, K.F. Yeasts in Sustainable Bioethanol Production: A Review. Biochem. Biophys. Rep. 2017, 10, 52–61. [Google Scholar] [CrossRef]
- Nielsen, J. Production of Biopharmaceutical Proteins by Yeast. Bioengineered 2013, 4, 207–211. [Google Scholar] [CrossRef]
- Vieira Gomes, A.M.; Carmo, T.S.; Carvalho, L.S.; Bahia, F.M.; Parachin, N.S. Comparison of Yeasts as Hosts for Recombinant Protein Production. Microorganisms 2018, 6, 38. [Google Scholar] [CrossRef] [PubMed]
- Baptista, S.L.; Costa, C.E.; Cunha, J.T.; Soares, P.O.; Domingues, L. Metabolic Engineering of Saccharomyces cerevisiae for the Production of Top Value Chemicals from Biorefinery Carbohydrates. Biotechnol. Adv. 2021, 47, 107697. [Google Scholar] [CrossRef] [PubMed]
- Bozell, J.J.; Petersen, G.R. Technology Development for the Production of Biobased Products from Biorefinery Carbohydrates—The US Department of Energy’s “Top 10” Revisited. Green. Chem. 2010, 12, 539–554. [Google Scholar] [CrossRef]
- Overkamp, K.M.; Bakker, B.M.; Kötter, P.; Luttik, M.A.; Van Dijken, J.P.; Pronk, J.T. Metabolic Engineering of Glycerol Production in Saccharomyces cerevisiae. Appl. Env. Microbiol. 2002, 68, 2814–2821. [Google Scholar] [CrossRef]
- Santos, L.O.; Silva, P.G.P.; Lemos Junior, W.J.F.; de Oliveira, V.S.; Anschau, A. Glutathione Production by Saccharomyces cerevisiae: Current State and Perspectives. Appl. Microbiol. Biotechnol. 2022, 106, 1879–1894. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, R.; Yang, Q.; Zhang, J.; Zhao, Y.; Zheng, Y.; Yang, J. Chapter One-Recent Advances in the Biosynthesis of Isoprenoids in Engineered Saccharomyces cerevisiae. In Advances in Applied Microbiology; Gadd, G.M., Sariaslani, S., Eds.; Academic Press: Cambridge, MA, USA, 2021; pp. 1–35. [Google Scholar]
- Gancedo, J.M. Yeast Carbon Catabolite Repression. Microbiol. Mol. Biol. Rev. 1998, 62, 334–361. [Google Scholar] [CrossRef]
- Frey, P.A. The Leloir Pathway: A Mechanistic Imperative for Three Enzymes to Change the Stereochemical Configuration of a Single Carbon in Galactose. FASEB J. 1996, 10, 461–470. [Google Scholar] [CrossRef]
- Dynesen, J.; Smits, H.P.; Olsson, L.; Nielsen, J. Carbon Catabolite Repression of Invertase during Batch Cultivations of Saccharomyces cerevisiae: The Role of Glucose, Fructose, and Mannose. Appl. Microbiol. Biotechnol. 1998, 50, 579–582. [Google Scholar] [CrossRef]
- Marques, W.L.; Raghavendran, V.; Stambuk, B.U.; Gombert, A.K. Sucrose and Saccharomyces cerevisiae: A Relationship Most Sweet. FEMS Yeast Res. 2015, 16, fov107. [Google Scholar] [CrossRef]
- Hortsch, R.; Weuster-Botz, D. Power Consumption and Maximum Energy Dissipation in a Milliliter-Scale Bioreactor. Biotechnol. Prog. 2010, 26, 595–599. [Google Scholar] [PubMed]
- Bromig, L.; von den Eichen, N.; Weuster-Botz, D. Control of Parallelized Bioreactors I: Dynamic Scheduling Software for Efficient Bioprocess Management in High-Throughput Systems. Bioprocess. Biosyst. Eng. 2022, 45, 1927–1937. [Google Scholar] [PubMed]
- Dyer, S.C.; Austine-Orimoloye, O.; Azov, A.G.; Barba, M.; Barnes, I.; Barrera-Enriquez, V.P.; Becker, A.; Bennett, R.; Beracochea, M.; Berry, A.; et al. Ensembl 2025. Nucleic Acids Res. 2024, 53, D948–D957. [Google Scholar]
- Li, H. Minimap2: Pairwise Alignment for Nucleotide Sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar]
- Li, H. New Strategies to Improve Minimap2 Alignment Accuracy. Bioinformatics 2021, 37, 4572–4574. [Google Scholar]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon Provides Fast and Bias-Aware Quantification of Transcript Expression. Nat. Methods 2017, 14, 417–419. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Subgroup Genome Project Data Processing. The Sequence Alignment/Map Format and Samtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Shen, W.; Le, S.; Li, Y.; Hu, F. Seqkit: A Cross-Platform and Ultrafast Toolkit for Fasta/Q File Manipulation. PLoS ONE 2016, 11, e0163962. [Google Scholar]
- Soneson, C.; Love, M.I.; Robinson, M.D. Differential Analyses for RNA-Seq: Transcript-Level Estimates Improve Gene-Level Inferences. F1000Research 2015, 4, 1521. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, L.; Lun, A.T.; Baldoni, P.L.; Smyth, G.K. edgeR V4: Powerful Differential Analysis of Sequencing Data with Expanded Functionality and Improved Support for Small Counts and Larger Datasets. Nucleic Acids Res. 2025, 53, gkaf018. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Statist. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- McCarthy, D.J.; Smyth, G.K. Testing Significance Relative to a Fold-Change Threshold Is a Treat. Bioinformatics 2009, 25, 765–771. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. Clusterprofiler: An R Package for Comparing Biological Themes among Gene Clusters. Omics 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Szcześniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X.; et al. A Survey of Best Practices for RNA-Seq Data Analysis. Genome Biol. 2016, 17, 13. [Google Scholar] [CrossRef]
- Robinson, M.D.; Oshlack, A. A Scaling Normalization Method for Differential Expression Analysis of RNA-Seq Data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef]
- Turcotte, B.; Liang, X.B.; Robert, F.; Soontorngun, N. Transcriptional Regulation of Nonfermentable Carbon Utilization in Budding Yeast. FEMS Yeast Res. 2010, 10, 2–13. [Google Scholar] [CrossRef]
- Stanley, D.; Bandara, A.; Fraser, S.; Chambers, P.J.; Stanley, G.A. The Ethanol Stress Response and Ethanol Tolerance of Saccharomyces cerevisiae. J. Appl. Microbiol. 2010, 109, 13–24. [Google Scholar] [CrossRef]
- Kato, S.; Yoshida, M.; Izawa, S. Btn2 Is Involved in the Clearance of Denatured Proteins Caused by Severe Ethanol Stress in Saccharomyces cerevisiae. FEMS Yeast Res. 2019, 19, foz079. [Google Scholar] [CrossRef]
- De Smidt, O.; Du Preez, J.C.; Albertyn, J. The Alcohol Dehydrogenases of Saccharomyces cerevisiae: A Comprehensive Review. FEMS Yeast Res. 2008, 8, 967–978. [Google Scholar] [CrossRef]
- Zhao, X.-Q.; Bai, F.-W. Zinc and Yeast Stress Tolerance: Micronutrient Plays a Big Role. J. Biotechnol. 2012, 158, 176–183. [Google Scholar] [CrossRef]
- Tegally, H.; San, J.E.; Giandhari, J.; de Oliveira, T. Unlocking the Efficiency of Genomics Laboratories with Robotic Liquid-Handling. BMC Genom. 2020, 21, 729. [Google Scholar] [CrossRef] [PubMed]
- Kong, N.; Ng, W.; Thao, K.; Agulto, R.; Weis, A.; Kim, K.S.; Korlach, J.; Hickey, L.; Kelly, L.; Lappin, S.; et al. Automation of PacBio SMRTbell NGS Library Preparation for Bacterial Genome Sequencing. Stand. Genom. Sci. 2017, 12, 27. [Google Scholar] [CrossRef] [PubMed]
- Holland, I.; Davies, J.A. Automation in the Life Science Research Laboratory. Front. Bioeng. Biotechnol. 2020, 8, 571777. [Google Scholar] [CrossRef] [PubMed]
- Annona, G.; Liberti, A.; Pollastro, C.; Spagnuolo, A.; Sordino, P.; De Luca, P. Reaping the Benefits of Liquid Handlers for High-Throughput Gene Expression Profiling in a Marine Model Invertebrate. BMC Biotechnol. 2024, 24, 4. [Google Scholar] [CrossRef]
- Berglund, E.; Saarenpää, S.; Jemt, A.; Gruselius, J.; Larsson, L.; Bergenstråhle, L.; Lundeberg, J.; Giacomello, S. Automation of Spatial Transcriptomics Library Preparation to Enable Rapid and Robust Insights into Spatial Organization of Tissues. BMC Genom. 2020, 21, 298. [Google Scholar] [CrossRef]
- Jemt, A.; Salmén, F.; Lundmark, A.; Mollbrink, A.; Fernández Navarro, J.; Ståhl, P.L.; Yucel-Lindberg, T.; Lundeberg, J. An Automated Approach to Prepare Tissue-Derived Spatially Barcoded RNA-Sequencing Libraries. Sci. Rep. 2016, 6, 37137. [Google Scholar] [CrossRef]
- Schurch, N.J.; Schofield, P.; Gierliński, M.; Cole, C.; Sherstnev, A.; Singh, V.; Wrobel, N.; Gharbi, K.; Simpson, G.G.; Owen-Hughes, T.; et al. How Many Biological Replicates Are Needed in an RNA-Seq Experiment and Which Differential Expression Tool Should You Use? RNA 2016, 22, 839–851. [Google Scholar] [CrossRef]
- Ueffing, K.; Hug, N.; Niggli, H.; Tran, T.-T.-T. Hamilton’s Magpip Technology Allows Automation of Pipetting Low Volume Reactions at High Speed and Quality. 2023. Available online: https://www.hamiltoncompany.com/automated-liquid-handling (accessed on 8 January 2025).
- Hadimioglu, B.; Stearns, R.; Ellson, R. Moving Liquids with Sound: The Physics of Acoustic Droplet Ejection for Robust Laboratory Automation in Life Sciences. J. Lab. Autom. 2016, 21, 4–18. [Google Scholar] [CrossRef]
- Schroeder, A.; Mueller, O.; Stocker, S.; Salowsky, R.; Leiber, M.; Gassmann, M.; Lightfoot, S.; Menzel, W.; Granzow, M.; Ragg, T. The RIN: An RNA Integrity Number for Assigning Integrity Values to RNA Measurements. BMC Mol. Biol. 2006, 7, 3. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blums, K.; Herzog, J.; Costa, J.; Quirico, L.; Turber, J.; Weuster-Botz, D. Automation of RNA-Seq Sample Preparation and Miniaturized Parallel Bioreactors Enable High-Throughput Differential Gene Expression Studies. Microorganisms 2025, 13, 849. https://doi.org/10.3390/microorganisms13040849
Blums K, Herzog J, Costa J, Quirico L, Turber J, Weuster-Botz D. Automation of RNA-Seq Sample Preparation and Miniaturized Parallel Bioreactors Enable High-Throughput Differential Gene Expression Studies. Microorganisms. 2025; 13(4):849. https://doi.org/10.3390/microorganisms13040849
Chicago/Turabian StyleBlums, Karlis, Josha Herzog, Jonathan Costa, Lara Quirico, Jonas Turber, and Dirk Weuster-Botz. 2025. "Automation of RNA-Seq Sample Preparation and Miniaturized Parallel Bioreactors Enable High-Throughput Differential Gene Expression Studies" Microorganisms 13, no. 4: 849. https://doi.org/10.3390/microorganisms13040849
APA StyleBlums, K., Herzog, J., Costa, J., Quirico, L., Turber, J., & Weuster-Botz, D. (2025). Automation of RNA-Seq Sample Preparation and Miniaturized Parallel Bioreactors Enable High-Throughput Differential Gene Expression Studies. Microorganisms, 13(4), 849. https://doi.org/10.3390/microorganisms13040849