
Development and Validation of Early Alert Model for Diabetes Mellitus–Tuberculosis Comorbidity



Abstract
1. Introduction
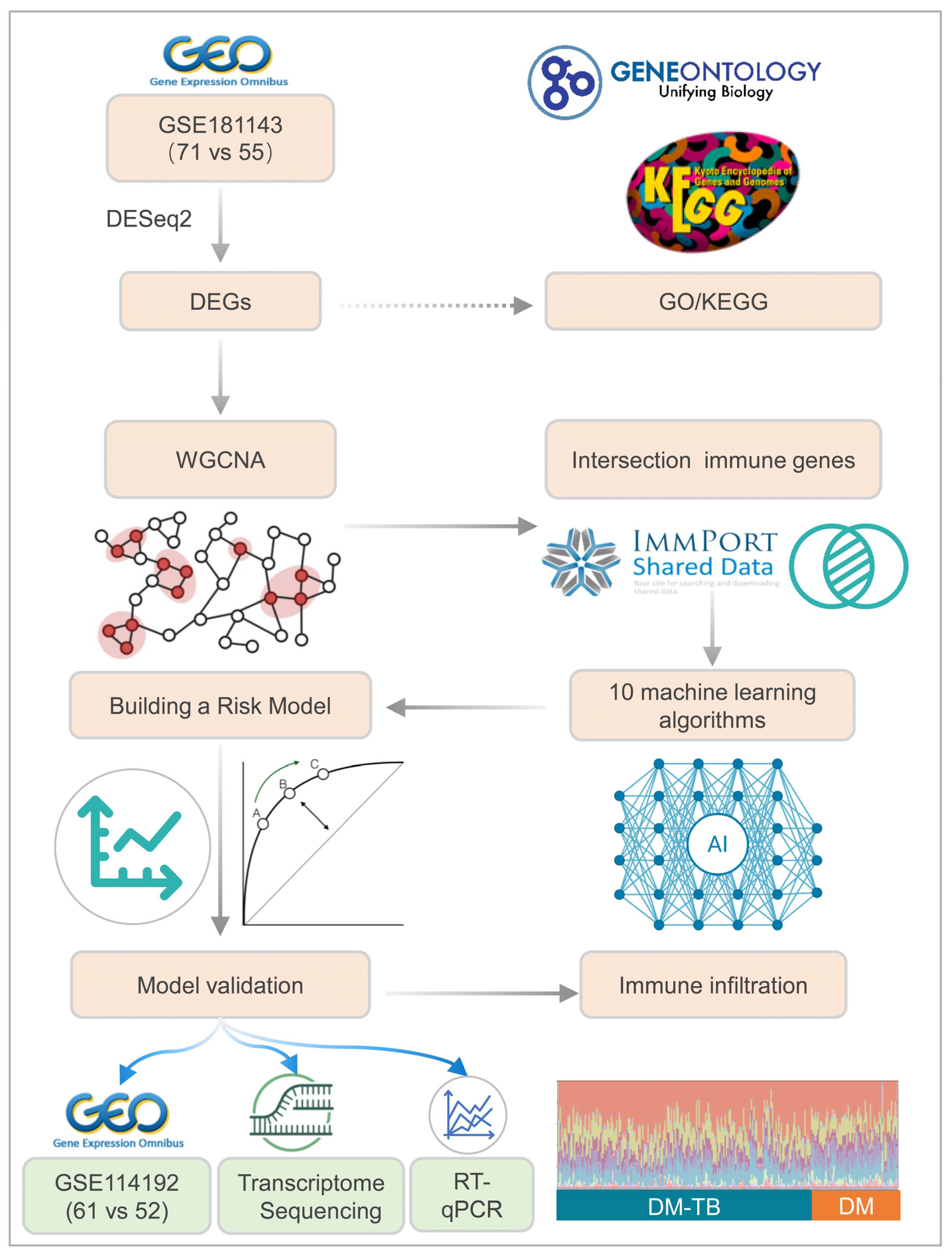
2. Materials and Methods
2.1. Data Acquisition and Initial Processing
2.2. Differential Gene Expression Analysis
2.3. Functional Enrichment Analysis
2.4. Weighted Gene Co-Expression Network Analysis (WGCNA)
2.5. Identification of Immune-Related Genes Utilizing ImmPort
2.6. Machine Learning for Immune-Related Biomarker Selection
2.7. Prospective Cohort and Transcriptome Sequencing
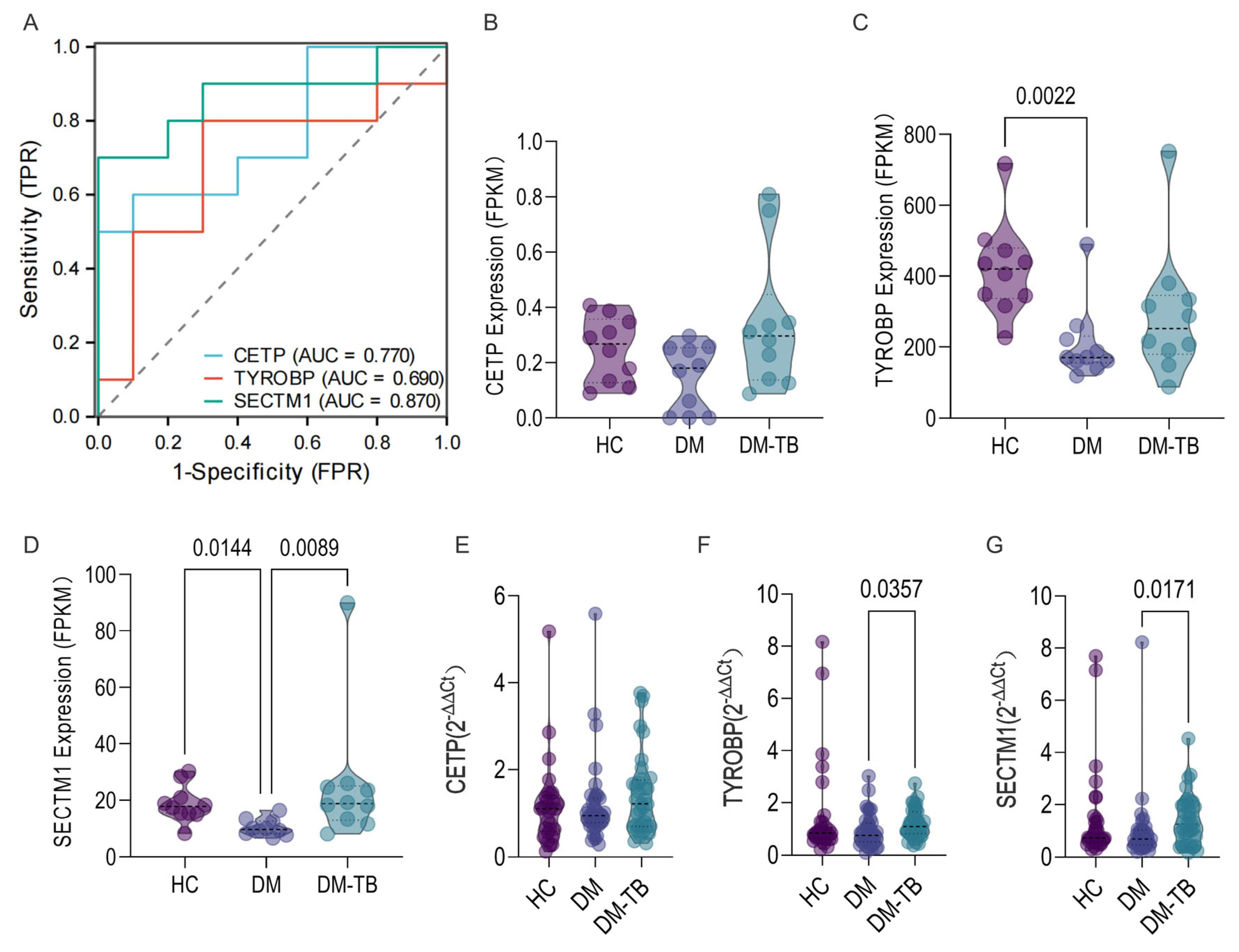
2.8. Retrospective Cohort and RT-qPCR
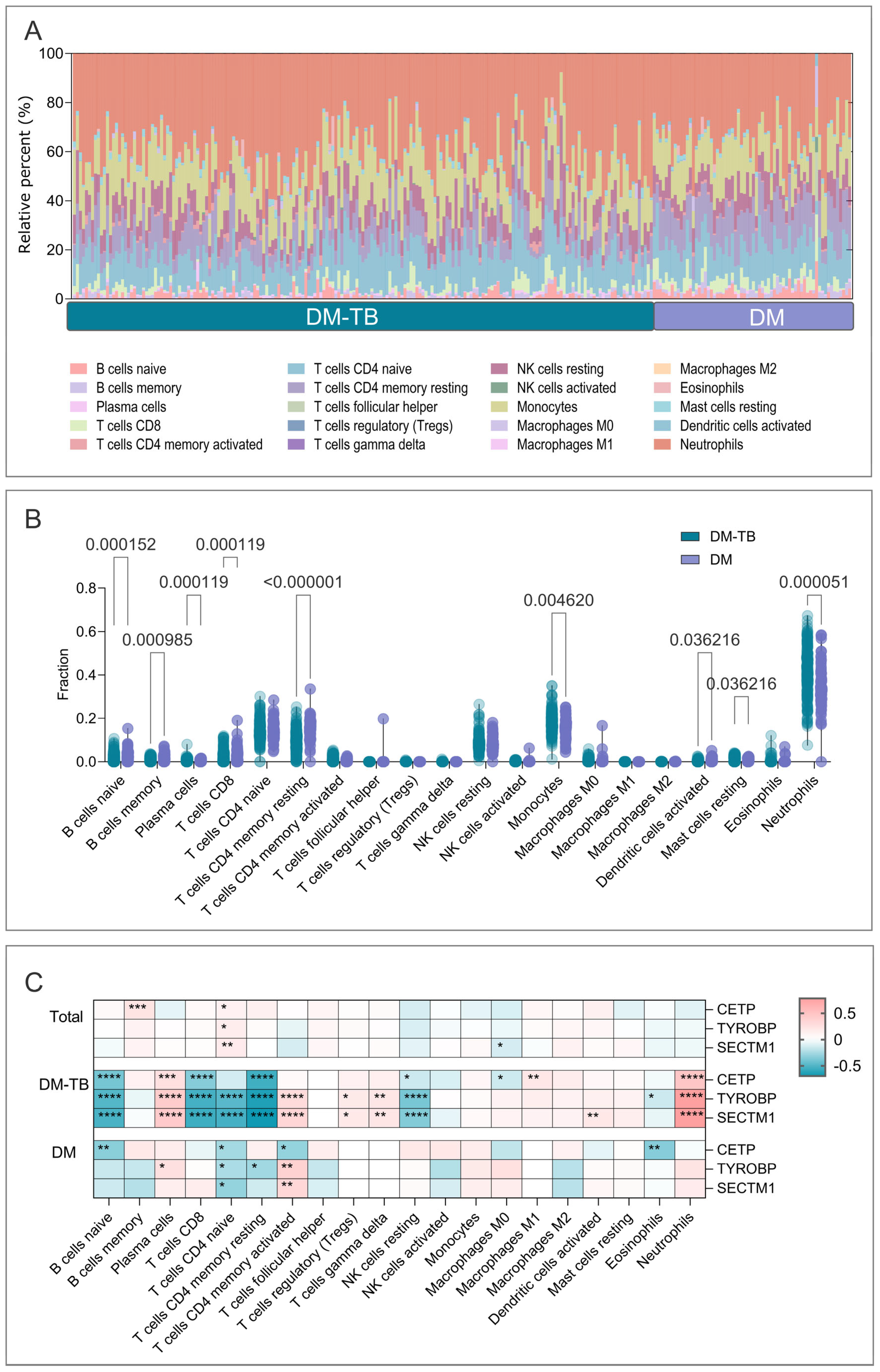
2.9. CIBERSORT Analysis of Immune Cell Infiltration
2.10. Statistical Analysis
3. Results
3.1. Comparison of Clinical Characteristics Among (HCs, DM, and DM–TB Cohorts from Brazil and India (GSE181143 Dataset)
3.2. Identification of DEGs in DM–TB Patients
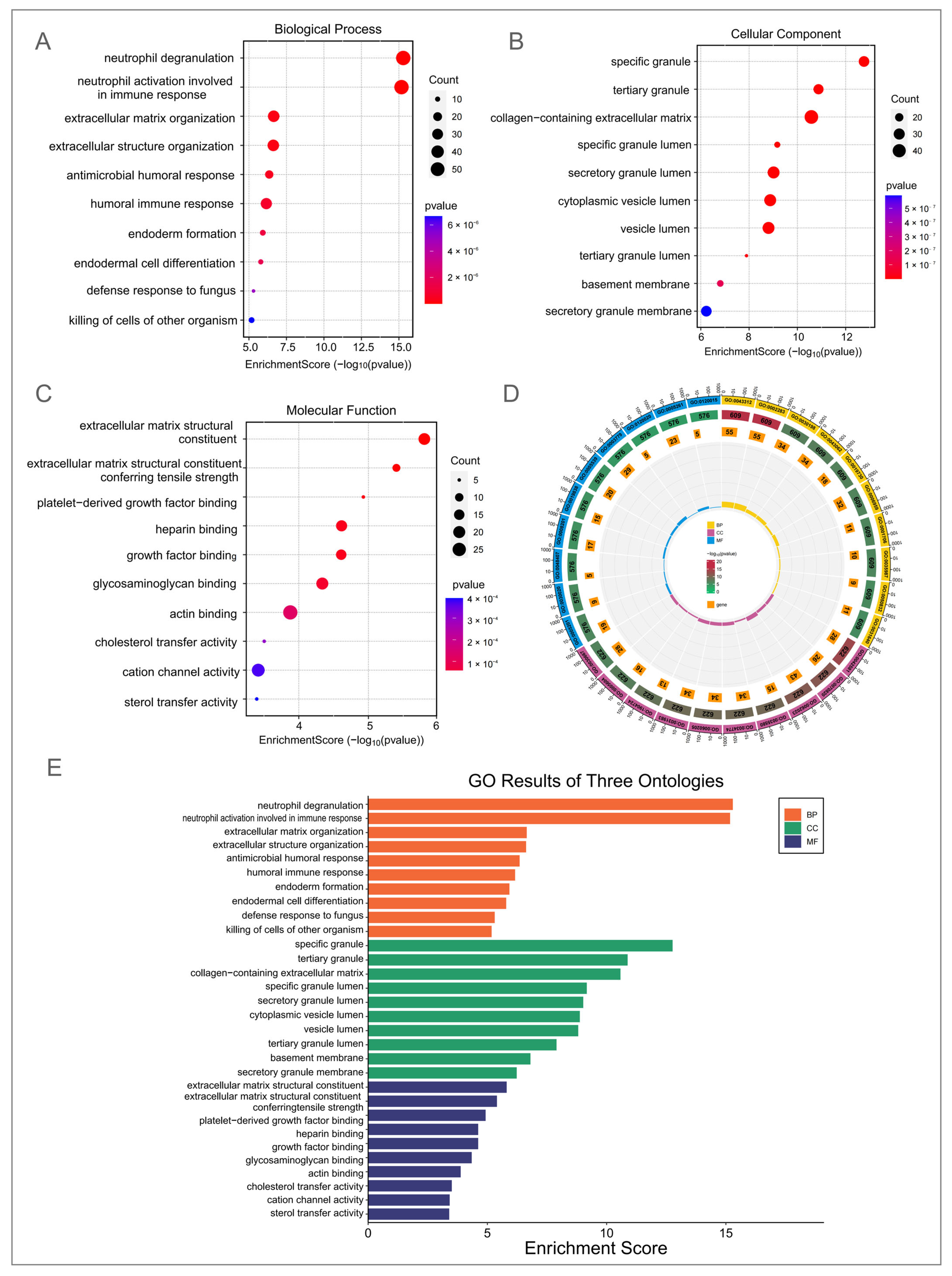
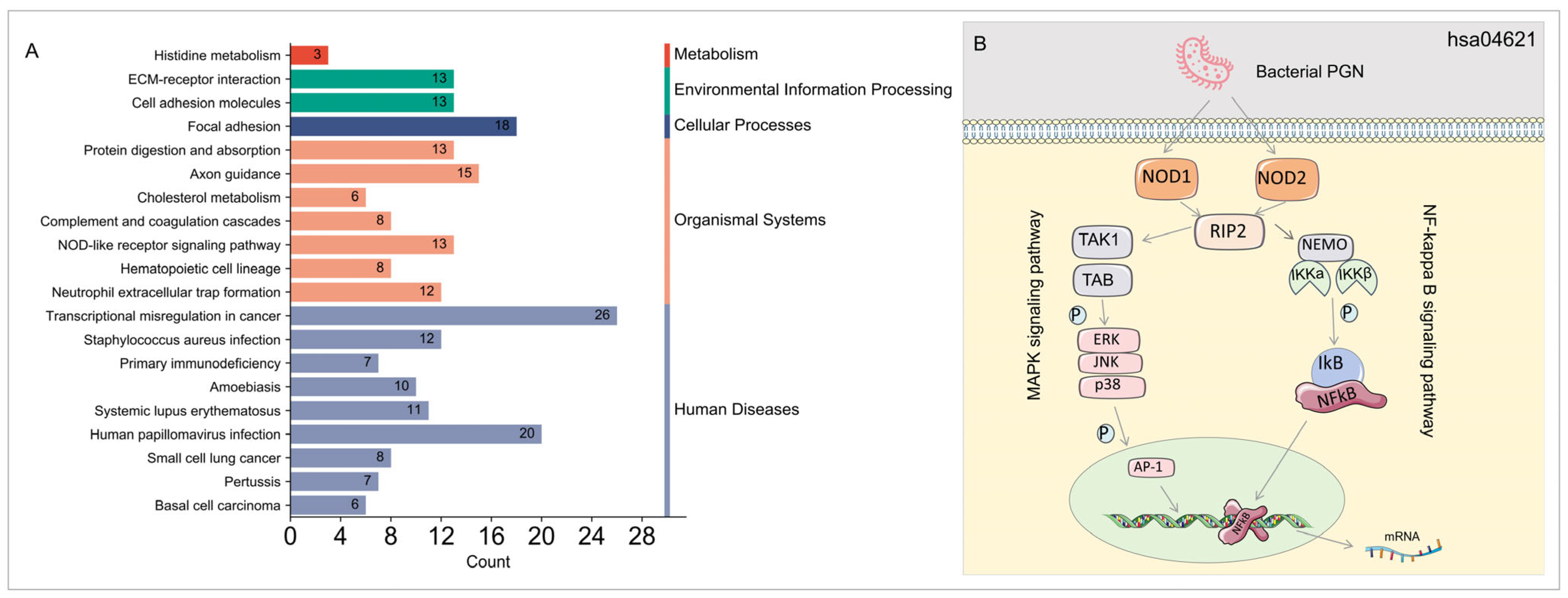
3.3. GO and KEGG Enrichment Analysis of DEGs in DM–TB Patients
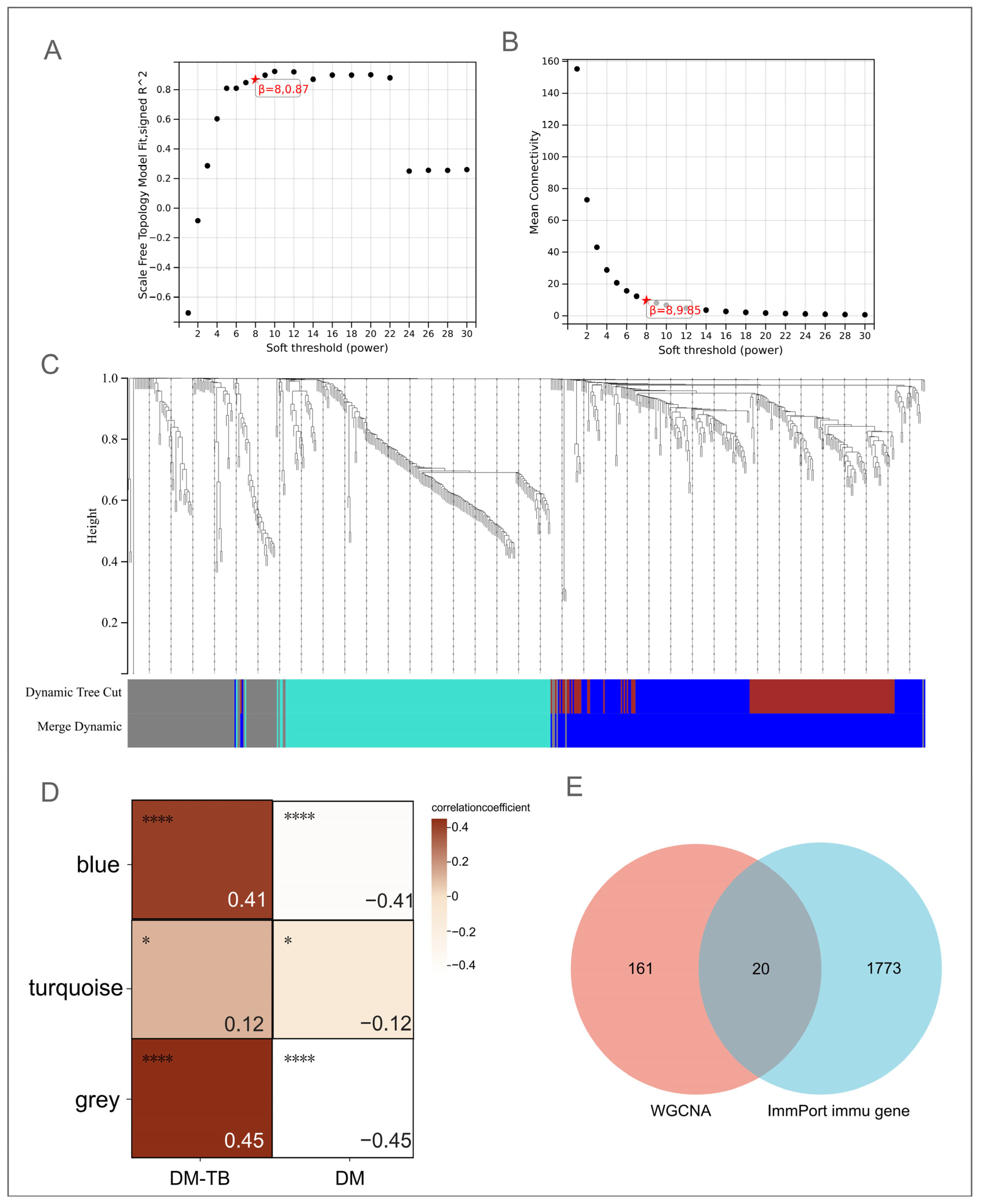
3.4. Identification of Core Genes in DM–TB Using WGCNA
3.5. ImmPort Identification of Immune-Related Genes in DM–TB
3.6. Machine Learning Identification of Candidate Biomarkers for DM–TB
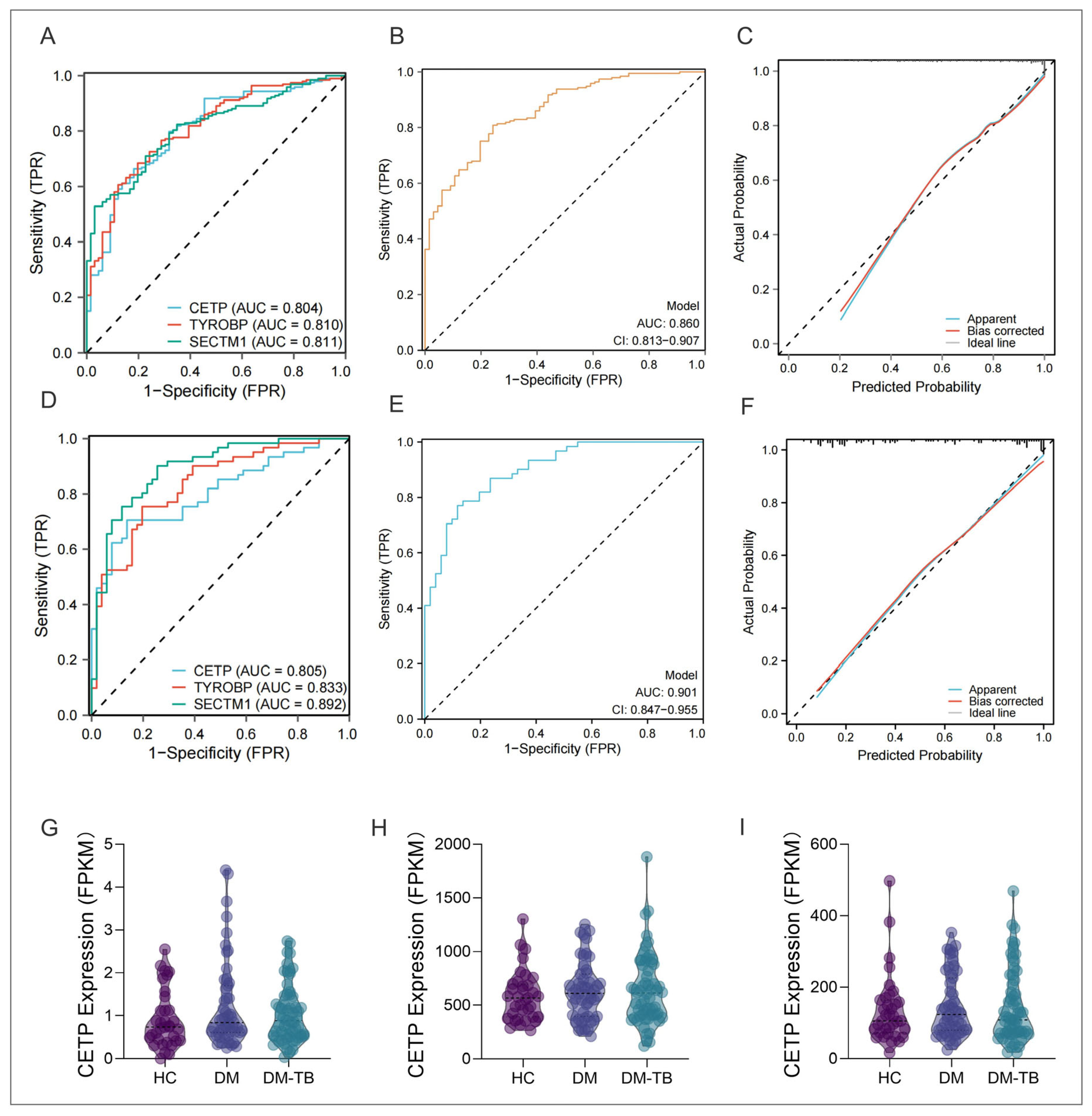
3.7. Logistic Regression Prediction of DM–TB Risk: Construction of a Nomogram
3.8. External Dataset and Population Cohort Validation of the DM–TB Early Risk Alert Model
3.9. Immune Infiltration Analysis in DM–TB and DM Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- IDF. IDF Diabetes Atlas, 11th ed.; International Diabetes Federation: Brussels, Belgium, 2025. [Google Scholar]
- El Tawil, E.H.; Saliby, R.; Halabi, R.; El Khoury, J.; Assaf, S.; Hamdan, M.; Abou Nader, G.; Abou Jaoude, E. Prevalence and associations of asymptomatic left ventricular systolic dysfunction in Lebanese patients with type 2 diabetes mellitus. PLoS ONE 2024, 19, e0304801. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Li, L.; Yang, L.; Zhuang, L.; Aspatwar, A.; Wang, L.; Gong, W. Impact of diabetes mellitus on tuberculosis prevention, diagnosis, and treatment from an immunologic perspective. Exploration 2024, 4, 20230138. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wang, T.; Du, J.; Sun, L.; Wang, G.; Ni, R.; An, Y.; Fan, X.; Li, Y.; Guo, R.; et al. Decoding the WHO Global Tuberculosis Report 2024: A Critical Analysis of Global and Chinese Key Data. Zoonoses 2025, 5, 1. [Google Scholar] [CrossRef]
- Boadu, A.A.; Yeboah-Manu, M.; Osei-Wusu, S.; Yeboah-Manu, D. Tuberculosis and diabetes mellitus: The complexity of the comorbid interactions. Int. J. Infect. Dis. 2024, 146, 107140. [Google Scholar] [CrossRef]
- Antonio-Arques, V.; Franch-Nadal, J.; Caylà, J.A. Diabetes and tuberculosis: A syndemic complicated by COVID-19. Med. Clin. 2021, 157, 288–293. [Google Scholar] [CrossRef]
- Wei, R.; Li, P.; Xue, Y.; Liu, Y.; Gong, W.; Zhao, W. Impact of Diabetes Mellitus on the Immunity of Tuberculosis Patients: A Retrospective, Cross-Sectional Study. Risk Manag. Health Policy 2022, 15, 611–627. [Google Scholar] [CrossRef]
- Baker, M.A.; Harries, A.D.; Jeon, C.Y.; Hart, J.E.; Kapur, A.; Lönnroth, K.; Ottmani, S.E.; Goonesekera, S.D.; Murray, M.B. The impact of diabetes on tuberculosis treatment outcomes: A systematic review. BMC Med. 2011, 9, 81. [Google Scholar] [CrossRef]
- Jeon, C.Y.; Murray, M.B. Diabetes mellitus increases the risk of active tuberculosis: A systematic review of 13 observational studies. PLoS Med. 2008, 5, e152. [Google Scholar] [CrossRef]
- Zhang, M.; He, J.Q. Impacts of metformin on tuberculosis incidence and clinical outcomes in patients with diabetes: A systematic review and meta-analysis. Eur. J. Clin. Pharmacol. 2020, 76, 149–159. [Google Scholar] [CrossRef]
- Al-Rifai, R.H.; Pearson, F.; Critchley, J.A.; Abu-Raddad, L.J. Association between diabetes mellitus and active tuberculosis: A systematic review and meta-analysis. PLoS ONE 2017, 12, e0187967. [Google Scholar] [CrossRef]
- Kornfeld, H.; West, K.; Kane, K.; Kumpatla, S.; Zacharias, R.R.; Martinez-Balzano, C.; Li, W.; Viswanathan, V. High Prevalence and Heterogeneity of Diabetes in Patients with TB in South India: A Report from the Effects of Diabetes on Tuberculosis Severity (EDOTS) Study. Chest 2016, 149, 1501–1508. [Google Scholar] [CrossRef] [PubMed]
- Gilardini Montani, M.S.; Granato, M.; Cuomo, L.; Valia, S.; Di Renzo, L.; D’Orazi, G.; Faggioni, A.; Cirone, M. High glucose and hyperglycemic sera from type 2 diabetic patients impair DC differentiation by inducing ROS and activating Wnt/β-catenin and p38 MAPK. Biochim. Biophys. Acta 2016, 1862, 805–813. [Google Scholar] [CrossRef] [PubMed]
- Ngo, M.D.; Bartlett, S.; Bielefeldt-Ohmann, H.; Foo, C.X.; Sinha, R.; Arachchige, B.J.; Reed, S.; Mandrup-Poulsen, T.; Rosenkilde, M.M.; Ronacher, K. A Blunted GPR183/Oxysterol Axis During Dysglycemia Results in Delayed Recruitment of Macrophages to the Lung During Mycobacterium tuberculosis Infection. J. Infect. Dis. 2022, 225, 2219–2228. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Martínez, A.; Casals, M.; Orcau, À.; Gorrindo, P.; Masdeu, E.; Caylà, J.A. Factors associated with diabetes mellitus among adults with tuberculosis in a large European city, 2000–2013. Int. J. Tuberc. Lung Dis. 2015, 19, 1507–1512. [Google Scholar] [CrossRef]
- Ugarte-Gil, C.; Alisjahbana, B.; Ronacher, K.; Riza, A.L.; Koesoemadinata, R.C.; Malherbe, S.T.; Cioboata, R.; Llontop, J.C.; Kleynhans, L.; Lopez, S.; et al. Diabetes Mellitus Among Pulmonary Tuberculosis Patients From 4 Tuberculosis-endemic Countries: The TANDEM Study. Clin. Infect. Dis. 2020, 70, 780–788. [Google Scholar] [CrossRef]
- Du, J.; Su, Y.; Qiao, J.; Gao, S.; Dong, E.; Wang, R.; Nie, Y.; Ji, J.; Wang, Z.; Liang, J.; et al. Application of artificial intelligence in diagnosis of pulmonary tuberculosis. Chin. Med. J. 2024, 137, 559–561. [Google Scholar] [CrossRef]
- Li, L.; Yang, L.; Zhuang, L.; Ye, Z.; Zhao, W.; Gong, W. From immunology to artificial intelligence: Revolutionizing latent tuberculosis infection diagnosis with machine learning. Mil. Med. Res. 2023, 10, 58. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef]
- Bu, D.; Luo, H.; Huo, P.; Wang, Z.; Zhang, S.; He, Z.; Wu, Y.; Zhao, L.; Liu, J.; Guo, J.; et al. KOBAS-i: Intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Res. 2021, 49, W317–W325. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed]
- Su, C.; Zhang, J.; Zhao, H. Estimating cell-type-specific gene co-expression networks from bulk gene expression data with an application to Alzheimer’s disease. J. Am. Stat. Assoc. 2024, 119, 811–824. [Google Scholar] [CrossRef] [PubMed]
- Fuller, T.F.; Ghazalpour, A.; Aten, J.E.; Drake, T.A.; Lusis, A.J.; Horvath, S. Weighted gene coexpression network analysis strategies applied to mouse weight. Mamm. Genome 2007, 18, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Ravasz, E.; Somera, A.L.; Mongru, D.A.; Oltvai, Z.N.; Barabási, A.L. Hierarchical organization of modularity in metabolic networks. Science 2002, 297, 1551–1555. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Andorf, S.; Gomes, L.; Dunn, P.; Schaefer, H.; Pontius, J.; Berger, P.; Desborough, V.; Smith, T.; Campbell, J.; et al. ImmPort: Disseminating data to the public for the future of immunology. Immunol. Res. 2014, 58, 234–239. [Google Scholar] [CrossRef]
- Wang, J.; Jiang, F.; Cheng, P.; Ye, Z.; Li, L.; Yang, L.; Zhuang, L.; Gong, W. Construction of novel multi-epitope-based diagnostic biomarker HP16118P and its application in the differential diagnosis of Mycobacterium tuberculosis latent infection. Mol. Biomed. 2024, 5, 15. [Google Scholar] [CrossRef]
- Peng, C.; Jiang, F.; Liu, Y.; Xue, Y.; Cheng, P.; Wang, J.; Wang, L.; Gong, W. Development and Evaluation of a Promising Biomarker for Diagnosis of Latent and Active Tuberculosis Infection. Infect. Dis. Immun. 2024, 4, 10–24. [Google Scholar] [CrossRef]
- Craven, K.E.; Gökmen-Polar, Y.; Badve, S.S. CIBERSORT analysis of TCGA and METABRIC identifies subgroups with better outcomes in triple negative breast cancer. Sci. Rep. 2021, 11, 4691. [Google Scholar] [CrossRef]
- Song, C.; Zhou, D.; Cheng, K.; Liu, F.; Cai, W.; Mei, Y.; Chen, J.; Huang, C.; Liu, Z. Bioinformatics-based discovery of intervertebral disc degeneration biomarkers and immune-inflammatory infiltrates. JOR Spine 2024, 7, e1311. [Google Scholar] [CrossRef]
- Vinhaes, C.L.; Fukutani, E.R.; Santana, G.C.; Arriaga, M.B.; Barreto-Duarte, B.; Araújo-Pereira, M.; Maggitti-Bezerril, M.; Andrade, A.M.S.; Figueiredo, M.C.; Milne, G.L.; et al. An integrative multi-omics approach to characterize interactions between tuberculosis and diabetes mellitus. iScience 2024, 27, 109135. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.K.; Khan, A.; Xu, Y.; Mai, S.; Zhang, L.; Mishra, A.; Restrepo, B.I.; Pan, P.Y.; Chen, S.H.; Jagannath, C. Antibody-Mediated LILRB2-Receptor Antagonism Induces Human Myeloid-Derived Suppressor Cells to Kill Mycobacterium tuberculosis. Front. Immunol. 2022, 13, 865503. [Google Scholar] [CrossRef] [PubMed]
- Rani, J.; Bhargav, A.; Seth, S.; Datta, M.; Bajpai, U.; Ramachandran, S. Identification of perturbed pathways rendering susceptibility to tuberculosis in type 2 diabetes mellitus patients using BioNSi simulation of integrated networks of implicated human genes. J. Biosci. 2022, 47, 69. [Google Scholar] [CrossRef]
- Athari, S.Z.; Mirzaei Bavil, F.; Keyhanmanesh, R.; Lotfi, H.; Sajed, Y.; Delkhosh, A.; Ghiasi, F. Voluntary exercise improves pulmonary inflammation through NF-κB and Nrf2 in type 2 diabetic male rats. Iran. J. Basic Med. Sci. 2024, 27, 74–80. [Google Scholar] [CrossRef]
- Martinez, N.; Vallerskog, T.; West, K.; Nunes-Alves, C.; Lee, J.; Martens, G.W.; Behar, S.M.; Kornfeld, H. Chromatin decondensation and T cell hyperresponsiveness in diabetes-associated hyperglycemia. J. Immunol. 2014, 193, 4457–4468. [Google Scholar] [CrossRef]
- Peng, Y.; Tang, T.; Li, Q.; Zhou, S.; Sun, Q.; Zhou, X.; Zhu, Y.; Wang, C.; Bermudez, L.E.; Liu, H.; et al. Mycobacterium tuberculosis FadD18 Promotes Proinflammatory Cytokine Secretion to Inhibit the Intracellular Survival of Bacillus Calmette-Guérin. Cells 2024, 13, 1019. [Google Scholar] [CrossRef]
- Poladian, N.; Orujyan, D.; Narinyan, W.; Oganyan, A.K.; Navasardyan, I.; Velpuri, P.; Chorbajian, A.; Venketaraman, V. Role of NF-κB during Mycobacterium tuberculosis Infection. Int. J. Mol. Sci. 2023, 24, 1772. [Google Scholar] [CrossRef]
- Peng, Z.; Yue, Y.; Xiong, S. Mycobacterial PPE36 Modulates Host Inflammation by Promoting E3 Ligase Smurf1-Mediated MyD88 Degradation. Front. Immunol. 2022, 13, 690667. [Google Scholar] [CrossRef]
- Noyes, D.; Bag, A.; Oseni, S.; Semidey-Hurtado, J.; Cen, L.; Sarnaik, A.A.; Sondak, V.K.; Adeegbe, D. Tumor-associated Tregs obstruct antitumor immunity by promoting T cell dysfunction and restricting clonal diversity in tumor-infiltrating CD8+ T cells. J. Immunother. Cancer 2022, 10, e004605. [Google Scholar] [CrossRef]
- Cao, C.; Zhang, Y.; Chai, Y.; Wang, L.; Yin, C.; Shou, S.; Jin, H. Attenuation of Sepsis-Induced Cardiomyopathy by Regulation of MicroRNA-23b Is Mediated Through Targeting of MyD88-Mediated NF-κB Activation. Inflammation 2019, 42, 973–986. [Google Scholar] [CrossRef]
- Zhou, X.; Fang, D.; Liu, H.; Ou, X.; Zhang, C.; Zhao, Z.; Zhao, S.; Peng, J.; Cai, S.; He, Y.; et al. PMN-MDSCs accumulation induced by CXCL1 promotes CD8(+) T cells exhaustion in gastric cancer. Cancer Lett. 2022, 532, 215598. [Google Scholar] [CrossRef]
- Bi, W.; Li, X.; Jiang, Y.; Gao, T.; Zhao, H.; Han, Q.; Zhang, J. Tumor-derived exosomes induce neutrophil infiltration and reprogramming to promote T-cell exhaustion in hepatocellular carcinoma. Theranostics 2025, 15, 2852–2869. [Google Scholar] [CrossRef] [PubMed]
- Prasad, G.; Bandesh, K.; Giri, A.K.; Kauser, Y.; Chanda, P.; Parekatt, V.; Mathur, S.; Madhu, S.V.; Venkatesh, P.; Bhansali, A.; et al. Genome-Wide Association Study of Metabolic Syndrome Reveals Primary Genetic Variants at CETP Locus in Indians. Biomolecules 2019, 9, 321. [Google Scholar] [CrossRef] [PubMed]
- Rebhan, M.; Chalifa-Caspi, V.; Prilusky, J.; Lancet, D. GeneCards: Integrating information about genes, proteins and diseases. Trends Genet. 1997, 13, 163. [Google Scholar] [CrossRef] [PubMed]
- Xin, G.; Yang, G.; Hui, L. Study to assess whether waist circumference and changes in serum glucose and lipid profile are independent variables for the CETP gene. Diabetes Res. Clin. Pract. 2014, 106, 95–100. [Google Scholar] [CrossRef]
- Ramón-Arbués, E.; Martínez-Abadía, B.; Gracia-Tabuenca, T.; Yuste-Gran, C.; Pellicer-García, B.; Juárez-Vela, R.; Guerrero-Portillo, S.; Sáez-Guinoa, M. Prevalence of overweight/obesity and its association with diabetes, hypertension, dyslipidemia and metabolic syndrome: A cross-sectional study of a sample of workers in Aragón, Spain. Nutr. Hosp. 2019, 36, 51–59. [Google Scholar] [CrossRef]
- Paublini, H.; López González, A.A.; Busquets-Cortés, C.; Tomas-Gil, P.; Riutord-Sbert, P.; Ramírez-Manent, J.I. Relationship between Atherogenic Dyslipidaemia and Lipid Triad and Scales That Assess Insulin Resistance. Nutrients 2023, 15, 2105. [Google Scholar] [CrossRef]
- Auguet, T.; Terra, X.; Hernández, M.; Sabench, F.; Porras, J.A.; Orellana-Gavaldà, J.M.; Llutart, J.; Guiu-Jurado, E.; Berlanga, A.; Martinez, S.; et al. Clinical and adipocytokine changes after bariatric surgery in morbidly obese women. Obesity 2014, 22, 188–194. [Google Scholar] [CrossRef]
- AlSaeed, H.; Haider, M.J.A.; Alzaid, F.; Al-Mulla, F.; Ahmad, R.; Al-Rashed, F. PPARdelta: A key modulator in the pathogenesis of diabetes mellitus and Mycobacterium tuberculosis co-morbidity. iScience 2024, 27, 110046. [Google Scholar] [CrossRef]
- Korb, V.C.; Chuturgoon, A.A.; Moodley, D. Mycobacterium tuberculosis: Manipulator of Protective Immunity. Int. J. Mol. Sci. 2016, 17, 131. [Google Scholar] [CrossRef]
- Kim, H.; Shin, S.J. Revolutionizing control strategies against Mycobacterium tuberculosis infection through selected targeting of lipid metabolism. Cell Mol. Life Sci. 2023, 80, 291. [Google Scholar] [CrossRef] [PubMed]
- Jeyanathan, M.; McCormick, S.; Lai, R.; Afkhami, S.; Shaler, C.R.; Horvath, C.N.; Damjanovic, D.; Zganiacz, A.; Barra, N.; Ashkar, A.; et al. Pulmonary M. tuberculosis infection delays Th1 immunity via immunoadaptor DAP12-regulated IRAK-M and IL-10 expression in antigen-presenting cells. Mucosal Immunol. 2014, 7, 670–683. [Google Scholar] [CrossRef] [PubMed]
- Urdahl, K.B.; Shafiani, S.; Ernst, J.D. Initiation and regulation of T-cell responses in tuberculosis. Mucosal Immunol. 2011, 4, 288–293. [Google Scholar] [CrossRef] [PubMed]
- Chackerian Alissa, A.; Perera Thushara, V.; Behar Samuel, M. Gamma Interferon-Producing CD4+ T Lymphocytes in the Lung Correlate with Resistance to Infection withMycobacterium tuberculosis. Infect. Immun. 2001, 69, 2666–2674. [Google Scholar] [CrossRef]
- Divangahi, M.; Yang, T.; Kugathasan, K.; McCormick, S.; Takenaka, S.; Gaschler, G.; Ashkar, A.; Stampfli, M.; Gauldie, J.; Bramson, J.; et al. Critical negative regulation of type 1 T cell immunity and immunopathology by signaling adaptor DAP12 during intracellular infection. J. Immunol. 2007, 179, 4015–4026. [Google Scholar] [CrossRef]
- Almeida, A.S.; Lago, P.M.; Boechat, N.; Huard, R.C.; Lazzarini, L.C.; Santos, A.R.; Nociari, M.; Zhu, H.; Perez-Sweeney, B.M.; Bang, H.; et al. Tuberculosis is associated with a down-modulatory lung immune response that impairs Th1-type immunity. J. Immunol. 2009, 183, 718–731. [Google Scholar] [CrossRef]
- Iizasa, E.; Chuma, Y.; Uematsu, T.; Kubota, M.; Kawaguchi, H.; Umemura, M.; Toyonaga, K.; Kiyohara, H.; Yano, I.; Colonna, M.; et al. TREM2 is a receptor for non-glycosylated mycolic acids of mycobacteria that limits anti-mycobacterial macrophage activation. Nat. Commun. 2021, 12, 2299. [Google Scholar] [CrossRef]
- Voss, O.H.; Murakami, Y.; Pena, M.Y.; Lee, H.N.; Tian, L.; Margulies, D.H.; Street, J.M.; Yuen, P.S.; Qi, C.F.; Krzewski, K.; et al. Lipopolysaccharide-Induced CD300b Receptor Binding to Toll-like Receptor 4 Alters Signaling to Drive Cytokine Responses that Enhance Septic Shock. Immunity 2016, 44, 1365–1378. [Google Scholar] [CrossRef]
- Samuel, V.T.; Shulman, G.I. Mechanisms for insulin resistance: Common threads and missing links. Cell 2012, 148, 852–871. [Google Scholar] [CrossRef]
- Yuan, L.; Zhang, L.; Liu, X.; Li, S.; Zou, J. Identification of differential immune cells and related diagnostic genes in patients with diabetic retinopathy. Medicine 2023, 102, e35331. [Google Scholar] [CrossRef]
- Li, B.; Zhao, X.; Xie, W.; Hong, Z.; Zhang, Y. Integrative analyses of biomarkers and pathways for diabetic nephropathy. Front. Genet. 2023, 14, 1128136. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Ma, L.; Deng, D.; Tian, D.; Liu, W.; Diao, Z. Title: Bioinformatic Identification of Genes Involved in Diabetic Nephropathy Fibrosis and their Clinical Relevance. Biochem. Genet. 2023, 61, 1567–1584. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, R.; Liu, T.; Jia, Y.; Gao, X.; Zhang, X. CD163 in Macrophages: A Potential Biomarker for Predicting the Progression of Diabetic Nephropathy based on Bioinformatics Analysis. Endocr. Metab. Immune Disord. Drug Targets 2023, 23, 294–303. [Google Scholar] [CrossRef]
- Wang, T.; Huang, C.; Lopez-Coral, A.; Slentz-Kesler, K.A.; Xiao, M.; Wherry, E.J.; Kaufman, R.E. K12/SECTM1, an interferon-γ regulated molecule, synergizes with CD28 to costimulate human T cell proliferation. J. Leukoc. Biol. 2012, 91, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Khan, T.A.; Mazhar, H.; Saleha, S.; Tipu, H.N.; Muhammad, N.; Abbas, M.N. Interferon-Gamma Improves Macrophages Function against M. tuberculosis in Multidrug-Resistant Tuberculosis Patients. Chemother. Res. Pract. 2016, 2016, 7295390. [Google Scholar] [CrossRef]
- Kumar, N.P.; Moideen, K.; Dhakshinraj, S.D.; Banurekha, V.V.; Nair, D.; Dolla, C.; Kumaran, P.; Babu, S. Profiling leucocyte subsets in tuberculosis-diabetes co-morbidity. Immunology 2015, 146, 243–250. [Google Scholar] [CrossRef]
- Zhuang, L.; Yang, L.; Li, L.; Ye, Z.; Gong, W. Mycobacterium tuberculosis: Immune response, biomarkers, and therapeutic intervention. MedComm 2024, 5, e419. [Google Scholar] [CrossRef]
- Gong, W.; Pan, C.; Cheng, P.; Wang, J.; Zhao, G.; Wu, X. Peptide-Based Vaccines for Tuberculosis. Front. Immunol. 2022, 13, 830497. [Google Scholar] [CrossRef]
- Gong, W.; Liang, Y.; Wang, J.; Liu, Y.; Xue, Y.; Mi, J.; Li, P.; Wang, X.; Wang, L.; Wu, X. Prediction of Th1 and cytotoxic T lymphocyte epitopes of Mycobacterium tuberculosis and evaluation of their potential in the diagnosis of tuberculosis in a mouse model and in humans. Microbiol. Spectr. 2022, 10, e0143822. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Year | Accession | Platform | Sequencing Type | Sample (n) | Sample Source Country (n) | Species | Tissue | |
|---|---|---|---|---|---|---|---|---|
| 1 | 2021 | GSE181143 | GPL20795 | RNA seq | 201 (DM–TB: 71, DM: 55, HCs: 75) | Brazil (61) and India (140) | Homo sapiens | Whole blood |
| 2 | 2020 | GSE114192 | GPL18573 | RNA seq | 149 (DM–TB: 61, DM: 52, HCs: 36) | Romania (44), Indonesia (19), South Africa (72), and Peru (12) | Homo sapiens | Whole blood |
| Gene Symbol | Primer Sequence |
|---|---|
| CETP | Forward: 5′-GGCCAAGTCAAGTATGGGTTG-3′ Reverse: 5′-ACAGACACGTTCTGAATGGAGA-3′ |
| TYROBP | Forward: 5′-ACTGAGACCGAGTCGCCTTAT-3′ Reverse: 5′-ATACGGCCTCTGTGTGTTGAG-3′ |
| SECTM1 | Forward: 5′-GGGACACCAGAGAAATAACAGACAAG-3′ Reverse: 5′-AGAGCGACCAAGAGGATGAAGAC-3′ |
| GAPDH | Forward: 5′-CTCTGGTAAAGTGGATATTGT-3′ Reverse: 5′-GGTGGAATCATATTGGAACA-3′ |
| Variables * | HCs | DM | DM–TB | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Brazil | India | p | Brazil | India | p | Brazil | India | p | |
| N | 15 | 60 | 15 | 40 | 31 | 40 | |||
| Age | 35 (28–51) | 35 (31–39) | 0.64 | 56 (51–57) | 52.5 (42–78) | 0.46 | 48 (38–67) | 48 (40–65) | 0.7 |
| Female no. (%) | 9 (60%) | 32 (53%) | 0.6 | 8 (57%) | 22 (55%) | 0.88 | 9 (29%) | 10 (25%) | 0.7 |
| BMI (kg/m2) | 24.9 (20.5–28.9) | 16.9 (16–20) | 0.2 | 30.3 (26–32) | 25.4 (23.6–27.7) | <0.001 | 22.5 (20–25.7) | 21.9 (18–28.9) | 0.79 |
| Smoking (current) | 5 (33.3%) | 2 (3.3%) | 0.004 | 3 (20%) | 10 (25%) | 0.6 | 12 (38.7%) | 6 (15%) | 0.02 |
| Alcohol (current) | 13 (86.7%) | 11 (18.4%) | <0.001 | 14 (93.4%) | 9 (22.5%) | <0.001 | 28 (90.3%) | 5 (12.5%) | <0.001 |
| Metformin | N/A | N/A | N/A | Not assessed | 26 (65%) | Not assessed | 6 (19.4%) | 27 (87%) | <0.001 |
| Statin | N/A | N/A | N/A | Not assessed | 3 (7.5%) | Not assessed | Not assessed | 9(22.5%) | <0.001 |
| Cavitary TB | N/A | N/A | N/A | N/A | N/A | N/A | 9 (29%) | 26 (65%) | <0.001 |
| HbA1c (%) | 5.1 (4.9–5.2) | 5 (5–5.5) | 0.25 | 6.1 (5.9–7.4) | 9.4 (8.4–11.1) | <0.001 | 8.5 (6.8–11.4) | 11.7 (10–12.5) | 0.001 |
| Characteristics | OR | 95% CI | p-Value |
|---|---|---|---|
| CETP | 1.051 | 1.033–1.068 | <0.001 |
| TYROBP | 1.004 | 1.003–1.006 | <0.001 |
| SECTM1 | 1.005 | 1.003–1.007 | <0.001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ye, Z.; Bai, G.; Yang, L.; Zhuang, L.; Li, L.; Li, Y.; Ni, R.; An, Y.; Wang, L.; Gong, W. Development and Validation of Early Alert Model for Diabetes Mellitus–Tuberculosis Comorbidity. Microorganisms 2025, 13, 919. https://doi.org/10.3390/microorganisms13040919
Ye Z, Bai G, Yang L, Zhuang L, Li L, Li Y, Ni R, An Y, Wang L, Gong W. Development and Validation of Early Alert Model for Diabetes Mellitus–Tuberculosis Comorbidity. Microorganisms. 2025; 13(4):919. https://doi.org/10.3390/microorganisms13040919
Chicago/Turabian StyleYe, Zhaoyang, Guangliang Bai, Ling Yang, Li Zhuang, Linsheng Li, Yufeng Li, Ruizi Ni, Yajing An, Liang Wang, and Wenping Gong. 2025. "Development and Validation of Early Alert Model for Diabetes Mellitus–Tuberculosis Comorbidity" Microorganisms 13, no. 4: 919. https://doi.org/10.3390/microorganisms13040919
APA StyleYe, Z., Bai, G., Yang, L., Zhuang, L., Li, L., Li, Y., Ni, R., An, Y., Wang, L., & Gong, W. (2025). Development and Validation of Early Alert Model for Diabetes Mellitus–Tuberculosis Comorbidity. Microorganisms, 13(4), 919. https://doi.org/10.3390/microorganisms13040919